
Abstract
Verona Integron-encoded Metallo-beta-lactamase (VIM) is the most frequently-encountered carbapenemase in the healthcare-related pathogen Pseudomonas aeruginosa. In the Netherlands, a low-endemic country for antibiotic-resistant bacteria, no national surveillance data on the prevalence of carbapenemase-producing P. aeruginosa (CPPA) was available. Therefore, in 2016, a national surveillance pilot study was initiated to investigate the occurrence, molecular epidemiology, genetic characterization, and resistomes of CPPA among P. aeruginosa isolates submitted by medical microbiology laboratories (MMLs) throughout the country. From 1221 isolates included in the study, 124 (10%) produced carbapenemase (CIM-positive); of these, the majority (95, 77%) were positive for the blaVIM gene using PCR. Sequencing was performed on 112 CIM-positive and 56 CIM-negative isolates (n = 168), and genetic clustering revealed that 75/168 (45%) isolates were highly similar. This genetic cluster, designated Group 1, comprised isolates that belonged to high-risk sequence type ST111/serotype O12, had similar resistomes, and all but two carried the blaVIM-2 allele on an identical class 1 integron. Additionally, Group 1 isolates originated from around the country (i.e. seven provinces) and from multiple MMLs. In conclusion, the Netherlands had experienced a nationwide, inter-institutional, clonal outbreak of VIM-2-producing P. aeruginosa for at least three years, which this pilot study was crucial in identifying. A structured, national surveillance program is strongly advised to monitor the spread of Group 1 CPPA, to identify emerging clones/carbapenemase genes, and to detect transmission in and especially between hospitals in order to control current and future outbreaks.
Similar content being viewed by others

Introduction
Carbapenemase-producing Pseudomonas aeruginosa (CPPA) is an emerging pathogen responsible for many serious healthcare-related infections worldwide1,2 Carbapenemases hydrolyze important antibiotics for treating P. aeruginosa infections, and frequently co-occur with other resistance mechanisms, resulting in a multidrug-resistant (MDR) or extensively drug-resistant (XDR) phenotype1,3,4. In 2017, the World Health Organization prioritized carbapenem-resistant P. aeruginosa among the top three antibiotic-resistant bacteria in “critical” need of new antibiotics5. Verona Integron-encoded Metallo-beta-lactamase (VIM) is the most frequently-encountered carbapenemase in P. aeruginosa3. Among variants, the VIM-2 metallo-beta-lactamase exhibits the broadest geographical distribution, including on the European continent6,7.
In the Netherlands, there is low endemicity for antibiotic-resistant bacteria8. In 2018, only 2% of Dutch clinical P. aeruginosa isolates were MDR (i.e. resistant to ≥ 3 antimicrobial groups), but among MDR isolates, around 50% were phenotypically resistant to carbapenems8. The first Dutch CPPA outbreak was reported in 2011 by one tertiary-care hospital; an investigation into isolates obtained between 2008 and 2009 that were resistant to imipenem revealed that 33% contained the blaVIM gene, and most belonged to a single genetic cluster9. A surveillance study in 2012 based on convenience sampling, incorporating isolates from 2009 to 2011 and 21 medical microbiology laboratories (MMLs), similarly unveiled one large genetic cluster of VIM-2-producing P. aeruginosa belonging to sequence type ST111/serotype O1210. This high-risk, epidemic lineage is one of the most globally-widespread MDR/XDR P. aeruginosa lineages, and is associated with high morbidity and mortality in patients6,10,11. Since then, ST111/O12 has continued to be isolated from patients and hospital environments, and based on sporadic studies, appeared to be the most frequently-encountered CPPA sequence type in the Netherlands10,12,13. The outcomes of these studies also suggested that CPPA may be distributed nationwide in the Netherlands, but systematically-collected, national surveillance data was not available.
In 2016, a national surveillance pilot study was initiated by the National Institute for Public Health and the Environment (RIVM) in the Netherlands to determine the necessity of a structured surveillance program for CPPA. This was in addition to an existing surveillance program on carbapenemase-producing Enterobacterales that began in 2011; during that time, the RIVM had also coincidentally received P. aeruginosa isolates from several participating MMLs. In this study, P. aeruginosa isolates collected by the RIVM between 2015 and 2017 were analyzed to estimate the occurrence of CPPA among submitted isolates, characterize their genetic environment and molecular epidemiology using a whole-genome multilocus sequence typing (wgMLST) scheme, and determine their antibiotic resistance gene profiles.
Results
CPPA occurrence among submitted isolates
From January 2015 until December 2017, 39 Dutch MMLs submitted 1445 confirmed P. aeruginosa isolates with reduced sensitivity to meropenem and/or imipenem. These isolates were subjected to the carbapenemase inactivation method (CIM) to assess possible carbapenemase production. Only the first carbapenemase-producing (CIM-positive) and first non-carbapenemase-producing (CIM-negative) isolate per patient submitted during the study period were included, resulting in a total of 1221 isolates from 1216 patients, five of whom carried both a CIM-positive and CIM-negative P. aeruginosa isolate. For NGS, only a single isolate per patient was used.
Carbapenemase production was observed in 124 (10%) isolates using the CIM test; of these, 107 (86%) isolates were also positive for a carbapenemase gene using PCR (Table 1). All 1097 non-carbapenemase-producing isolates were also carbapenemase-PCR negative. The majority of CIM-positive isolates (77%, 95/124) carried a gene belonging to the blaVIM family. Analysis of next-generation sequencing (NGS) data on 83 CIM-positive, blaVIM-PCR-positive isolates revealed that they all carried the blaVIM-2 allele. Nine isolates carried a blaIMP gene comprising five different blaIMP alleles. Three isolates carried a blaNDM-1 allele. Seventeen CIM-positive isolates did not yield a PCR product, and were sequenced; three carried a blaGES-5 allele, but a carbapenemase-encoding gene could not be found in the remaining 14 isolates.
Sequencing revealed a large genetic cluster of blaVIM-2-containing CPPA
NGS was performed on 112 CIM-positive isolates, and on 56 CIM-negative isolates that were matched to CIM-positive isolates based on MML and sampling year. Demographic data provided by MMLs on the patients from which these isolates derived is available in Supplementary Table S1. In some regions, P. aeruginosa was not submitted or found, so isolates from these regions were not sequenced. A complete geographical overview of CIM-positive and CIM-negative isolates that were included is available in Supplementary Figure S1.
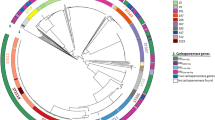
Genotypic relationships between isolates were determined using wgMLST (Fig. 1). There was a high degree of genotypic diversity, with large allelic distances often exceeding > 3500 alleles between isolates. However, 75/168 (45%) isolates partitioned closely together with a maximum distance of 35 alleles. This group, designated Group 1, comprised isolates that were all CIM-positive (with the exception of one isolate), all belonged to sequence type ST111 and serotype O12, and all but two isolates carried the blaVIM-2 allele; one isolate carried blaIMP-13, and in the other, a CIM-negative isolate, no carbapenemase-encoding gene could be identified.

Minimum-spanning tree of 112 CIM-positive P. aeruginosa isolates and 56 CIM-negative P. aeruginosa isolates based on wgMLST analysis. Each circle represents an isolate. Colors indicate the carbapenemase-encoding gene. Yellow circles (CIM+, no carba allele) indicate CIM-positive isolates in which no carbapenemase-encoding gene could be identified. White circles indicate CIM-negative isolates. Numbers on the lines between circles indicate the number of allelic differences between isolates. To avoid long branches, lines connecting circles are logarithmic representations of allelic distances. For allelic distances larger than 3500 genes, a dashed line is used and distances are not shown. All minimum spanning trees were created in BioNumerics v7.6, exported as metafiles, and imported into Adobe Illustrator (Adobe Creative Cloud 2020, www.adobe.com).
Within Group 1, several genetic clusters could be seen with few allelic differences between isolates. One CIM-negative isolate (also belonging to ST111/O12) was separated from Group 1 by only 95 allelic differences. All other isolates differed from Group 1 by > 3500 allelic differences. Group 1 also comprised isolates from seven Dutch provinces (Fig. 2).

Geographical distribution of Group 1 isolates in the Netherlands. Above, Minimum-spanning tree of Group 1 isolates based on wgMLST analysis. Each circle represents an isolate. Colors indicate the region where the isolate was obtained. Dutch provinces are given in italics. The red circle with a thick border represents the CIM-positive Group 1 isolate that carried a blaIMP-13 gene. The beige circle with a thick border represents the CIM-negative Group 1 isolate. The white circle represents the CIM-negative ST111/O12 isolate most closely related to Group 1. For understanding distances between isolates, refer to Fig. 1. Below, Map of the Netherlands showing the distribution of 112 CIM-positive P. aeruginosa isolates that were sequenced in this study, and the number of those isolates that were Group 1, by province. Map was created by importing a screen capture into Adobe Illustrator 2020 and plotting the origin of isolates using the Type-Ned MRSA website (www.typened-mrsa.rivm.nl).
The composition of the integron regions of six Group 1 isolates was reconstructed by combined short- and long-read sequencing. This showed that all four VIM-2-encoding Group 1 isolates contained an identical class 1 integron (Type A) carrying the intI integrase gene, the blaVIM-2 gene flanked by the aminoglycoside resistance genes aac(6′)-29a and aac(6′)-29b, an incomplete qacE gene involved in quaternary ammonium compound resistance, and the sulfonamide resistance gene sul1 (Fig. 3). In the CIM-negative Group 1 isolate, the blaVIM-2 and aac(6′)-29b genes were lost from this integron by deletion (Type B). The IMP-13-encoding Group 1 isolate was also shown to carry an integron (Type C) similar to the Type A integron, but contained the blaIMP-13 gene and the aminoglycoside resistance gene aac(6′)-Ib3. Mapping the Illumina reads of the other VIM-2-encoding Group 1 isolates against these reconstructed integrons showed that all carried the Type A integron.

Class 1 integron compositions of Group 1 isolates. All Group 1 isolates except two carried a 3554 bp Type A integron containing the blaVIM-2 gene cassette flanked by aac(6′)-29a and aac(6′)-29b genes. Yellow and green squares flanking the aac(6′)-29a/b genes denote identical sequences. The integron structure of the CIM-negative Group 1 isolate was identical to a Type A integron, but the blaVIM-2 and aac(6′)-29b genes had been deleted (Type B). The IMP-13-encoding Group 1 isolate carried a different gene cassette composition (Type C). P, promoter located within the integrase gene (intI1); aac(6′)-29a, aac(6′)-29b, and aac(6′)-Ib3, aminoglycoside resistance genes; blaVIM-2 and blaIMP-13, carbapenem resistance genes; qacEΔ, incomplete quaternary ammonium compound resistance gene; sul1, sulfonamide resistance gene. Figure was created using BioNumerics v7.6 and Adobe Illustrator 2020.
Group 1 isolates were distinct from internationally-derived CPPA isolates
The wgMLST profiles of the 168 sequenced isolates in this study were compared to 260 complete, annotated P. aeruginosa chromosomal sequences from the National Center for Biotechnology Information’s GenBank database (Fig. 4). Group 1 is shown in the zoomed in panel; notably, the isolates sequenced in this study (blue circles) were interconnected and not interrupted by a different P. aeruginosa sequence (white circles). However, four isolates with the blaVIM-2 gene were closely related to Group 1, separated by only 3–66 allelic differences. The first strain (accession no. CP016955) was RIVM-EMC4982 from the Erasmus MC University Medical Center Rotterdam, which was the reference isolate used to design the wgMLST scheme for this study. The second strain, Carb01 63 (accession no. CP011317), originated from Maasstad Hospital, another hospital in Rotterdam, the Netherlands. The third strain, PA38182 (accession no. HG530068), originated from a hospital in the United Kingdom, and has been involved in several major outbreaks14,15. The fourth strain, PaeAG1 (accession no. CP045739), was a MDR strain isolated from a patient with pneumonia admitted to intensive care in Costa Rica, and was the first strain reported to carry two carbapenemase-encoding genes (blaVIM-2 and blaIMP-18)16. Five other GenBank isolates partitioned with Group 1 at larger distances (separated by 77–400 allelic differences). These isolates did not carry the blaVIM-2 gene, but one (accession no. LS998783) carried the carbapenemase-encoding gene blaGES-5.

Minimum-spanning tree of the 168 P. aeruginosa isolates sequenced in this study (blue circles), and of 260 P. aeruginosa chromosomal sequences obtained from the NCBI GenBank database (white circles) based on wgMLST analysis. The enlarged portion of the tree displays Group 1 isolates, and the closely-related NCBI sequences with their accession numbers. The teal circle denotes the CIM-negative ST111/O12 isolate from this study that partitioned the closest to Group 1. The red circle indicates the Erasmus MC reference isolate that was used to design the wgMLST scheme. For understanding distances between isolates, refer to Fig. 1.
Antibiotic resistance gene profiles and QRDR analysis
ResFinder analyses showed that all 168 P. aeruginosa sequenced isolates carried the beta-lactamase gene blaPAO, the aminoglycoside resistance gene aph(3′)-IIb, and the fosfomycin resistance gene fosA (Supplementary Table S2). All but four isolates carried the chloramphenicol transferase gene catB7. Twenty-six other genes encoding beta-lactamase production, and 27 other genes associated with aminoglycoside resistance, were also found among the 168 isolates. The most prevalent beta-lactamase gene (56%, 94/168) was the blaOXA-50-like gene blaOXA-395. This gene was present in all Group 1 isolates, in 29% (11/38) of CIM-positive isolates that did not belong to Group 1, and in 15% (8/55) of CIM-negative isolates that did not belong to Group 1. Both aac(6')-29a and aac(6′)-29b genes were found in all 73 VIM-2-encoding Group 1 isolates; only aac(6')-29a was present in the CIM-negative Group 1 isolate, and neither gene was present in the IMP-13-encoding Group 1 isolate, confirming the integron compositions of Group 1 isolates after read-mapping. Among other isolates, one of either gene was found in three of the 38 CIM-positive isolates that did not belong to Group 1, but neither gene was found in any CIM-negative isolate that did not belong to Group 1.
An analysis of quinolone resistance-determining regions (QRDR) for genes gyrA, gyrB, parC, and parE in all sequenced isolates revealed that the gyrA T83I and the parC S87L mutations were found in 100% of ST111 isolates, but were also common among other sequence types; therefore, no unique mutation pattern could be determined for Group 1 isolates (Supplementary Table S3). The combination of T83I and D87N mutations in gyrA were only found in 60% (3/5) of ST175 isolates. The S87W mutation in parC was exclusively found in ST175 isolates.
Discussion
This national surveillance pilot study unveiled that the Netherlands had experienced an ongoing, nationwide, inter-institutional outbreak of a single, clonal genetic cluster of VIM-2-producing P. aeruginosa over a period of at least three years. It was clear from previous reports that several individual hospitals had already recognized this outbreak within their own settings. After the single-center CPPA outbreak reported by van der Bij et al., a surveillance study into 11 hospitals in the Netherlands in 2012 found that the outbreak by CPPA belonging to ST111 was widespread10. The surprising results of that study, however, were not followed by the implementation of a structured, national surveillance program. The current study revealed that ST111/O12 has continued to prevail in the Netherlands, and that the outbreak has involved multiple MMLs distributed over a large part of the country.
Genetic clustering of 168 sequenced isolates revealed that almost half (45%, 75/168) showed high genetic similarity, and, with the exception of two isolates, carried the blaVIM-2 allele. Few differences (≤ 35 alleles) were seen between isolates in this genetic cluster, designated Group 1. All VIM-2-encoding Group 1 isolates possessed identical integron structures; coupled with the fact that Group 1 isolates originated from around the country and were submitted by multiple MMLs, we could confirm that Group 1 CPPA have clonally spread throughout the Netherlands. Notably, Group 1 isolates were genetically distinct compared to the other sequenced isolates, exceeding 3500 allelic differences, and to most publicly-available sequences on GenBank. As P. aeruginosa strain Carb01 63 originated from the Netherlands and was isolated in 2012, the authors suspect that this strain also belonged to the outbreak described by this study. Like Carb01 63, Group 1 isolates belonged to sequence type ST111/O12, the predominant P. aeruginosa lineage in Europe6,7,10.
P. aeruginosa ST111/O12 clones exhibit high morbidity and mortality in infected patients6,11,15. ST111/O12 was first reported in the Netherlands in 2003, more than a decade before the inclusion period of this study, and then again in 20059,13. It is reasonable to suspect that the inter-institutional outbreak described by this study, and the multicenter outbreak described in 2012 by van der Bij et al., are linked, and may have started much earlier than previously anticipated. It is unclear when ST111/O12 was first introduced to the Netherlands, or how country-wide transmission could have occurred. Since it is known that patient referral networks can contribute to the spread of high-risk clones within a country, the transfer of patients between Dutch healthcare institutions most likely played a role in transmission, especially in cases of unnoticed colonization17. To date, there have been no studies analyzing the impact of patient transfers between healthcare institutions in the Netherlands. It is highly recommended that patient transfers include accompanying reports on colonization by highly-resistant microorganisms to limit potential inter-institutional transmission. In case of increasing prevalence rates encountered via a national surveillance system, an additional measure could be a national policy to screen patients for CPPA on admission. Screening should especially be performed in patients with a history of recent hospitalization in another healthcare center reporting a CPPA outbreak. Furthermore, CPPA, including ST111/O12 clones, have been shown to reside in the wet niches of hospitals through the formation of biofilm reservoirs12,14,18. These reservoirs are persistent, may resist disinfection, and can disperse CPPA to vulnerable patient populations19,20, so identifying and limiting environmental sources of ST111/O12 clones in hospitals are of particular interest. ST111/O12 clones have also been found outside of hospitals in wastewater effluents21,22; as the presence of CPPA reservoirs outside of healthcare institutions was not investigated during this study, community-acquired CPPA infections cannot be ruled out.
This study has some limitations. Firstly, no epidemiological data were collected, so no risk factors could be conclusively linked to the outbreak, and transmission events could not be identified. Secondly, MML compliance with submitting isolates was not checked. Thirdly, not all CPPA could be sequenced with the Illumina platform due to budgetary reasons, so it is possible that additional emerging genetic clusters had been missed among the other included isolates; selection bias was limited by sequencing all CPPA isolates received between 2016 and 2017, and sequencing a random selection of CPPA isolates that had been voluntarily submitted in 2015. Finally, long-read sequencing was only performed for six Group 1 isolates.
As a result of this national surveillance pilot study, MMLs have been advised to continue submitting P. aeruginosa isolates to the RIVM for NGS and surveillance. Only isolates with demonstrable carbapenemase production and/or a carbapenemase-encoding gene(s) have been requested. However, structured, national surveillance is still lacking in the Netherlands, since results from current CPPA surveillance are only reported in the Type-Ned database. National surveillance should include alerting the MMLs involved, and supporting epidemiological investigations into possible transmission routes; this is especially important for new, emerging clones, such as a clone with blaGES-5. National surveillance should also collect epidemiological data so that risk factors can be assessed. Additionally, the RIVM developed an in-house wgMLST scheme for P. aeruginosa to investigate the clonality of submitted isolates for this study, but recently several other validated schemes were published that may aid surveillance and outbreak investigations23,24,25.
In conclusion, the widespread distribution of Group 1 CPPA throughout the Netherlands went unnoticed for a period of at least three years, and this national surveillance pilot study was crucial in identifying the outbreak. Based on previous reports, it is likely that this inter-institutional outbreak started even earlier than previously anticipated. Therefore, the authors strongly recommend the implementation of a structured, national surveillance program in the Netherlands that incorporates wgMLST to monitor the spread of Group 1 CPPA, to identify emerging clones/carbapenemase genes, and to detect transmission in and especially between hospitals to control current and future outbreaks.
Methods
Inclusion of isolates
In 2016, all Dutch MMLs were sent a letter requesting that P. aeruginosa isolates be sent to the RIVM for a national surveillance pilot study. Criteria for submission were that isolates had a minimum inhibitory concentration of > 2 µg/ml for meropenem or > 4 µg/ml for imipenem (as determined by the MML’s preferred antimicrobial susceptibility method), and that one isolate per-person-per-year-per-lab was submitted. For each submitted isolate, MMLs were also requested to provide patient age, patient sex, sampling year, sampling site, and MML location in a secured, web-based database called Type-Ned26. During an existing surveillance program on carbapenemase-producing Enterobacterales that began before this study, the RIVM had also received P. aeruginosa isolates from several MMLs, so isolates received in 2015 were also considered.
P. aeruginosa isolates submitted between January 1, 2015 to December 31, 2017 were characterized as follows: species level identification was confirmed using MALDI-TOF MS (Bruker Daltonik, Bremen, Germany), carbapenemase production was assessed using the CIM test27, and the detection of blaVIM, blaIMP, blaKPC, blaOXA-48, and blaNDM genes was performed using multiplex PCR with primers and conditions as previously described26,27. CIM-positive P. aeruginosa isolates, and a subset of CIM-negative P. aeruginosa isolates matched by sampling year and MML, were subjected to sequencing.
Whole-genome sequence analyses
NGS was performed using Illumina HiSeq 2500 (Illumina, San Diego, CA, USA), resulting in reads with 125 bases length. De novo assembly was performed using CLC Genomics Workbench v9.5.3 (Qiagen Bioinformatics, Aarhus, Denmark), and contig sequences with a minimum length of 500 bp and at least 30× average read coverage per contig were used for further analyses. QRDR analysis was performed using the sequence extraction tool in BioNumerics v7.6 (Applied Maths, Sint-Martens-Latem, Belgium), in which extracted sequences were translated and aligned to identify coding sequence changes. Sequence types and serotypes were inferred from NGS data in SeqSphere v3.5.0 (Ridom, Münster, Germany) as well as PAst software from the Center for Genomic Epidemiology28. Identification of wgMLST alleles was performed in SeqSphere using an in-house wgMLST scheme comprising 6117 core genes and 325 accessory genes based on the fully sequenced and annotated P. aeruginosa strain RIVM-EMC4982 (accession no. CP016955). Allelic distances between isolates were calculated using BioNumerics v7.6. Genes absent in the sequenced isolates were ignored and not counted as allelic differences.
For long-read sequencing, the Oxford Nanopore protocol SQK-LSK108 (https://community.nanoporetech.com) and the expansion kit for native barcoding EXP-NBD104 was used. DNA was repaired using FFPE and end-repair kits (New England BioLabs, Ipswich, MA, USA), followed by ligation of barcodes with bead cleanup using AMPure XP (Beckman Coulter, Brea, CA, USA) after each step. Barcoded isolates were pooled, and sequencing adapters were added by ligation. The final library was loaded onto a MinION flow cell (MIN-106 R9.4.1). After a 48-h sequence run, base calling and de-multiplexing was performed using Albacore 2.3.1, and a single FASTA file per isolate was extracted from the FAST5 files using Poretools 0.5.1 (https://www.github.com/arq5x/poretools)29. Illumina and Nanopore data were used in a hybrid assembly performed by Unicycler v0.4.4 (https://github.com/rrwick/Unicycler)30.
Antibiotic resistance gene profiles were generated by using the ResFinder program v3.2, and the database available from the Center for Genomic Epidemiology website (https://bitbucket.org/genomicepidemiology/resfinder/src/master; accessed 08-05-2020)31. For resistance gene identification, a 90% identity threshold and a minimum length of 60% were used as criteria.
Ethical statement
The standard administrative procedure for carbapenemase-producing Enterobacterales was used to collect strains and demographic data26. Patient identifiers provided by MMLs were encrypted and then stored in the Type-Ned database, ensuring patient privacy in accordance with General Data Protection Regulation. Ethical approval was not required.
Data availability
Sequence data on the isolates in this paper have been deposited in the European Nucleotide Archive under study accession number PRJEB39528 (https://www.ebi.ac.uk/ena/browser/view/PRJEB39528).
References
Mesaros, N. et al. Pseudomonas aeruginosa: Resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 13, 560–578. https://doi.org/10.1111/j.1469-0691.2007.01681.x (2007).
Persoon, M. C. et al. Mortality related to Verona Integron-encoded Metallo-β-lactamase-positive Pseudomonas aeruginosa: Assessment by a novel clinical tool. Antimicrob. Resist. Infect. Control. 8, 107. https://doi.org/10.1186/s13756-019-0556-9 (2019).
Hong, D. J. et al. Epidemiology and characteristics of metallo-β-lactamase-producing Pseudomonas aeruginosa. Infect. Chemother. 47, 81–97. https://doi.org/10.3947/ic.2015.47.2.81 (2015).
Magiorakos, A.-P. et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. https://doi.org/10.1111/j.1469-0691.2011.03570.x (2012).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. https://doi.org/10.1016/S1473-3099(17)30753-3 (2018).
Oliver, A., Mulet, X., López-Causapé, C. & Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug. Resist. Updat. 21–22, 41–59. https://doi.org/10.1016/j.drup.2015.08.002 (2015).
Karampatakis, T., Antachopoulos, C., Tsakris, A. & Roilides, E. Molecular epidemiology of carbapenem-resistant Pseudomonas aeruginosa in an endemic area: Comparison with global data. Eur. J. Clin. Microbiol. Infect. Dis. 37, 1211–1220. https://doi.org/10.1007/s10096-018-3244-4 (2018).
NethMap. Consumption of antimicrobial agents and antimicrobial resistance among medically important bacteria in the Netherlands in 2018. Date accessed: 20-02-2020. https://www.rivm.nl/documenten/nethmap-2019 (2019).
van der Bij, A. K. et al. First outbreak of VIM-2 metallo-β-lactamase-producing Pseudomonas aeruginosa in the Netherlands: microbiology, epidemiology and clinical outcomes. Int. J. Antimicrob. Agents. 37, 513–518. https://doi.org/10.1016/j.ijantimicag.2011.02.010 (2011).
van der Bij, A. K. et al. Metallo-β-lactamase-producing Pseudomonas aeruginosa in the Netherlands: the nationwide emergence of a single sequence type. Clin. Microbiol. Infect. 18, E369–E372. https://doi.org/10.1111/j.1469-0691.2012.03969.x (2012).
Thrane, S. W. et al. The widespread multidrug-resistant serotype O12 Pseudomonas aeruginosa clone emerged through concomitant horizontal transfer of serotype antigen and antibiotic resistance gene clusters. MBio 6, E01396-15. https://doi.org/10.1128/mBio.01396-15 (2015).
Knoester, M. et al. An integrated approach to control a prolonged outbreak of multidrug-resistant Pseudomonas aeruginosa in an intensive care unit. Clin. Microbiol. Infect. 20, O207–O215. https://doi.org/10.1111/1469-0691.12372 (2014).
Croughs, P. D. et al. Unexpected mechanisms of resistance in Dutch Pseudomonas aeruginosa isolates collected during 14 years of surveillance. Int. J. Antimicrob. Agents. 52, 407–410. https://doi.org/10.1016/j.ijantimicag.2018.05.009 (2018).
Breathnach, A. S., Cubbon, M. D., Karunaharan, R. N., Pope, C. F. & Planche, T. D. Multidrug-resistant Pseudomonas aeruginosa outbreaks in two hospitals: Association with contaminated hospital waste-water systems. J. Hosp. Infect. 82, 19–24. https://doi.org/10.1016/j.jhin.2012.06.007 (2012).
Witney, A. A. et al. Genome sequencing and characterization of an extensively drug-resistant sequence type 111 serotype O12 hospital outbreak strain of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 20, O609–O618. https://doi.org/10.1111/1469-0691.12528 (2014).
Molina-Mora, J. A., Campos-Sánchez, R., Rodríguez, C., Shi, L. & García, F. High quality 3C de novo assembly and annotation of a multidrug resistant ST-111 Pseudomonas aeruginosa genome: Benchmark of hybrid and non-hybrid assemblers. Sci. Rep. 10, 1392. https://doi.org/10.1038/s41598-020-58319-6 (2020).
Donker, T., Wallinga, J. & Grundmann, H. Dispersal of antibiotic-resistant high-risk clones by hospital networks: Changing the patient direction can make all the difference. J. Hosp. Infect. 86, 34–41. https://doi.org/10.1016/j.jhin.2013.06.021 (2014).
Pirzadian, J. et al. Novel use of culturomics to identify the microbiota in hospital sink drains with and without persistent VIM-positive Pseudomonas aeruginosa. Sci. Rep. 10, 17052. https://doi.org/10.1038/s41598-020-73650-8 (2020).
Hopman, J. et al. Risk assessment after a severe hospital-acquired infection associated with carbapenemase-producing Pseudomonas aeruginosa. JAMA Netw. Open. 2, E187665. https://doi.org/10.1001/jamanetworkopen.2018.7665 (2019).
Hota, S. et al. Outbreak of multidrug-resistant Pseudomonas aeruginosa colonization and infection secondary to imperfect intensive care unit room design. Infect. Control Hosp. Epidemiol. 30, 25–33. https://doi.org/10.1086/592700 (2009).
Slekovec, C. et al. Tracking down antibiotic-resistant Pseudomonas aeruginosa isolates in a wastewater network. PLoS ONE 7, E49300. https://doi.org/10.1371/journal.pone.0049300 (2012).
Golle, A., Janezic, S. & Rupnik, M. Low overlap between carbapenem resistant Pseudomonas aeruginosa genotypes isolated from hospitalized patients and wastewater treatment plants. PLoS ONE 12, E0186736. https://doi.org/10.1371/journal.pone.0186736 (2017).
Stanton, R. A. et al. Development and application of a core genome multilocus sequence typing scheme for the health care-associated pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 58, E00214-E220. https://doi.org/10.1128/JCM.00214-20 (2020).
de Sales, R. O., Migliorini, L. B., Puga, R., Kocsis, B. & Severino, P. A core genome multilocus sequence typing scheme for Pseudomonas aeruginosa. Front. Microbiol. 11, 1049. https://doi.org/10.3389/fmicb.2020.01049 (2020).
Tönnies, H., Prior, K., Harmsen, D. & Mellmann, A. Establishment and evaluation of a core genome multilocus sequence typing scheme for whole-genome sequence-based typing of Pseudomonas aeruginosa. J. Clin. Microbiol. 59, E01987-E2020. https://doi.org/10.1128/JCM.01987-20 (2021).
van der Zwaluw, K. et al. Molecular characteristics of carbapenemase-producing Enterobacterales in the Netherlands; results of the 2014–2018 national laboratory surveillance. Clin. Microbiol. Infect. 26(1412), E7-12. https://doi.org/10.1016/j.cmi.2020.01.027 (2020).
van der Zwaluw, K. et al. The Carbapenem Inactivation Method (CIM), a simple and low-cost alternative for the Carba NP test to assess phenotypic carbapenemase activity in Gram-negative rods. PLoS ONE 10, E0123690. https://doi.org/10.1371/journal.pone.0123690 (2015).
Thrane, S. W., Taylor, V. L., Lund, O., Lam, J. S. & Jelsbak, L. Application of whole-genome sequencing data for O-specific antigen analysis and in silico serotyping of Pseudomonas aeruginosa isolates. J. Clin. Microbiol. 54, 1782–1788. https://doi.org/10.1128/JCM.00349-16 (2016).
Loman, N. J. & Quinlan, A. R. Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401. https://doi.org/10.1093/bioinformatics/btu555 (2014).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595. https://doi.org/10.1371/journal.pcbi.1005595 (2017).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. https://doi.org/10.1093/jac/dks261 (2012).
Acknowledgements
We are indebted to Prof. Jaap van Dissel, director of the Center for Disease Control, for supporting and stimulating this work. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Consortia
Contributions
L.S. and M.V. designed and initiated the study. S.W. and M.S.V. conducted the experimental procedures. S.W., M.S.V., and L.S. analyzed sequencing data. J.P., M.P., J.S., C.K., S.G., M.M., A.S., C.W., L.S. and M.V. analyzed and interpreted all other data. J.P. wrote the manuscript. All study authors read and approved the final manuscript. Members of the Dutch CPE surveillance Study Group submitted isolates to the RIVM and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
JS and CK received non-financial support from bioMérieux outside of this work. All other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pirzadian, J., Persoon, M.C., Severin, J.A. et al. National surveillance pilot study unveils a multicenter, clonal outbreak of VIM-2-producing Pseudomonas aeruginosa ST111 in the Netherlands between 2015 and 2017. Sci Rep 11, 21015 (2021). https://doi.org/10.1038/s41598-021-00205-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-00205-w
This article is cited by
-
Tracing the origin of NDM-1-producing and extensively drug-resistant Pseudomonas aeruginosa ST357 in the Netherlands
BMC Infectious Diseases (2024)
-
In search of the best method to detect carriage of carbapenem-resistant Pseudomonas aeruginosa in humans: a systematic review
Annals of Clinical Microbiology and Antimicrobials (2024)
-
ESKAPE pathogens: antimicrobial resistance, epidemiology, clinical impact and therapeutics
Nature Reviews Microbiology (2024)
-
Impact of multidrug resistance on the virulence and fitness of Pseudomonas aeruginosa: a microbiological and clinical perspective
Infection (2024)
-
Description of a nationwide structure for monitoring nosocomial outbreaks of (highly resistant) microorganisms in the Netherlands: characteristics of outbreaks in 2012–2021
Antimicrobial Resistance & Infection Control (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
