
Abstract
The isolation and sequencing of new strains of Pseudomonas aeruginosa created an extensive dataset of closed genomes. Many of the publicly available genomes are only used in their original publication while additional in silico information, based on comparison to previously published genomes, is not being explored. In this study, we defined and investigated the genome of the environmental isolate P. aeruginosa KRP1 and compared it to more than 100 publicly available closed P. aeruginosa genomes. By using different genomic island prediction programs, we could identify a total of 17 genomic islands and 8 genomic islets, marking the majority of the accessory genome that covers ~ 12% of the total genome. Based on intra-strain comparisons, we are able to predict the pathogenic potential of this environmental isolate. It shares a substantial amount of genomic information with the highly virulent PSE9 and LESB58 strains. For both of these, the increased virulence has been directly linked to their accessory genome before. Hence, the integrated use of previously published data can help to minimize expensive and time consuming wetlab work to determine the pathogenetic potential.
Similar content being viewed by others


Introduction
Pseudomonas aeruginosa has been isolated from terrestrial and marine soil, fresh and salt water, sewage, plants, animals, and humans1. For the latter habitats, it is known as an opportunistic pathogen, which usually spreads to already vulnerable patients, causing ~ 10% of all nosocomial infections in most European Union hospitals2. Its combinatory virulence is transmitted through the action of a myriad of virulence factors. Not every P. aeruginosa isolate conveys an equal level of virulence to a given infection model and a strain that is effective in infecting a plant does not necessarily show an equal amount of virulence towards an animal3,4. For the frequently researched P. aeruginosa PA14 strain this increased virulence, as compared to the type strain PAO1, is mainly due to the presence of additional virulence factors. Their genes are predominantly clustered on two genomic islands (GIs) termed P. aeruginosa pathogenicity islands (PAPI)5.
Due to short generation times, mutations are frequently observed in bacterial genomes, which makes them a dynamic rather than a static gene collection6. For P. aeruginosa, numerous studies have proven that the pan genome can be viewed as a mosaic of a conserved core (~ 90% of a specific genome) and variable accessory Sets. 7,8,9. Core genes are defined as genes with orthologues in nearly all strains, which show a conserved synteny and a low average nucleotide substitution rate7. One study suggests the core genome of the P. aeruginosa species, which makes up the smallest fraction of the pan genome, to consist of 4000–5000 open reading frames (ORFs)4. The second fraction is the accessory genome with about 10,000 genes. It can be grouped according to general features like the means of inter- and intrachromosomal relocation. By assigning different functional modules, it can be sorted into (i) integrative and conjugative elements (ICEs), (ii) replacement islands, (iii) prophages and phage-like elements, and (iv) transposons, insertion sequences (ISs) and integrons7. These genes are only shared by certain, but not all strains of the species and are mainly located in GIs and genomic islets (GIts). By definition, GIs have a size of at least 10 kb, while GIts are smaller than 10 kb. Both types of elements have been acquired via horizontal gene transfer7. They are the cause for alterations in the genome size of P. aeruginosa, which has been reported to range from 5.2 to 7.4 Mb4,8. By prokaryotic standards, this is considered rather big, encoding genes from numerous and distinct gene families. This highlights the great genetic and functional diversity of this species7. Depending on the encoded genes, GIs can be classified into four functional categories: (i) pathogenicity islands (PIs; predominantly encoding pathogenicity factors), (ii) resistance islands (RIs; predominantly encoding resistance functions), (iii) metabolic islands (MIs; predominantly encoding biosynthesis of (secondary) metabolites), and (iv) symbiotic islands (SIs; predominantly encoding genes related to a host-bacterium symbiotic relationship)19. The by far largest fractions of the pan genome are singletons and rare genes that are only shared by very few strains. Their estimated number is at least 30,000 for the P. aeruginosa species4.
Over the years, a different nomenclature was established naming the islands PAPI-X (P. aeruginosa pathogenicity island), PAGI-X (P. aeruginosa genomic island) and LESGI-X (Liverpool Epidemic Strain genomic island). It is important to note that no direct correlation between PAGI and LESGI exists and that the respective islands are not exclusive to the PA or LES strains of P. aeruginosa. Besides PAPI-I and PAPI-II of P. aeruginosa PA14, 42 other GI have been previously described in the P. aeruginosa species9,10,11,12, of which multiple have been directly linked to an increased pathogenicity of the harboring strains12,13,14,15. Different detection software packages are available to help identifying regions of foreign DNA within a given genome. As the algorithms use different characteristics, have a different degree of sensitivity, and different shortcomings, usually not one program is able to identify all GIs and GIts. Hence, a combination of multiple complementary tools should be applied to get a thorough detection. Here, we used the established SIGI-HMM16, IslandPath-DIMOB17, PHASTER18 and GIPSy19 bioinformatic tools.
In this study, we describe how the abundantly available sequencing information of a species like P. aeruginosa can be used to characterize a newly sequenced strain. To this end, we sequenced the KRP1 environmental isolate and characterized its phylogenetic relationship by using more than 100 previously published closed P. aeruginosa genomes. We further employed different GI detection software programs and manual mining, to investigate the genome composition of this exemplary strain. The strain KRP1 was first isolated from a microbial fuel cell as one of the dominating bacterial species responsible for the high electron transfer efficiency of the mixed community20. Our previous study has shown that this strain shows a remarkably different behavior in lab operated bioelectrochemical systems, as compared to other P. aeruginosa variants21, including an increased production of the redox-active pathogenicity factors phenazines. For deeper investigations into the reasons behind this phenomenon in the future, knowledge of the genomic make-up of this strain is needed. By comparing the genomic content with other highly virulent P. aeruginosa variants, we are able to make educated predictions of the strains pathogenetic potential, without having to perform time- as well as money consuming animal experiments. While these findings are only predictions and may not be considered proven until actual wet lab testing was performed, they can still be of substantial aid for the Pseudomonas community and the labs working with the strain in question.
Results and discussion
Pseudomonas sp. KRP1 belongs to the P. aeruginosa species
The in silico hybrid approach assembly of the de novo sequenced KRP1 strain resulted in two circular contigs of 6,162,740 bps and 575,136 bps. As a recent study points out, the choice of the assembly algorithm can have a profound impact on all subsequent analysis22. We therefore employed a combination of a short and long read assembler, followed by a manual curation to ensure fulfillment of the suggested 3 C criteria (contiguity, correctness and completeness)22. Synteny comparisons between this initial in silico assembly and closely related P. aeruginosa strains showed multiple rearrangements of the ORFs encoded on the putative mega plasmid. In P. aeruginosa PA14, the corresponding sequence is located between two large homologous ribosomal RNA clusters. These clusters are known to be spots of inner genome rearrangements within the P. aeruginosa species3,23. Therefore, PCR was used to investigate the DNA sequence surrounding the ribosomal RNA clusters on the main chromosome and on the potential mega plasmid. This resulted in a redefined genome structure of KRP1, with one circular chromosome, containing 6,301 annotated protein-coding genes (Table 1).
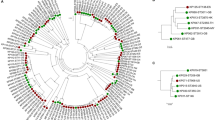
In the original study20, isolate KRP1 showed the highest similarity BLAST hit with Pseudomonas aeruginosa ATCC 27853 at 95% identity along a 197 bp fragment of the 16S rRNA gene. To re-evaluate its phylogenetic relationship within the Pseudomonas genus the average nucleotide identity (ANI) percentage of the KRP1 genome was calculated with respect to 105 fully sequenced P. aeruginosa strains and 8 other Pseudomonas species (Table S2). When compared to the P. aeruginosa species, all ANI values are well above the accepted species threshold of 95–96%. For the eight other closely related Pseudomonas species, ANI values range between 80.4% (P. citronellolis P3B5) and 74.4% (P. psychrotolerans PRS08). Besides this nucleotide based comparison, a phylogenetic tree was built based on a core of 1,537 genes per genome, each comprised of 532,537 amino acid residues (Figure S1). For better visualization, a reduced version of the tree containing only the eight non-aeruginosa species and six P. aeruginosa strains is shown (Fig. 1). The phylogenetic analysis clearly marks the strain KRP1 as a representative of the species P. aeruginosa and shows a clear distinction of the strain towards other members of the same genus.

P. aeruginosa KRP1 relation to closely related P. aeruginosa strains
The phylogenetic trees in Figs. 1 and S1 are based on amino acid-sequences, and therefore present only non-synonymous nucleotide substitutions. For a more in depth investigation of KRP1, its genome was compared to the type strain PAO1, the frequently researched strain PA14, the highly virulent LESB58 strain and the two strains FA-HZ1 and W45909, to which KRP1 clusters most closely in the phylogenetic analyses (Table 1). They also show the same Multilocus sequence type (MLST) as KRP1, as they encode perfect homologues of all seven housekeeping genes used for the profiling by the MLST 2.0 software26 (acsA, aroE, guaA, mutL, nuoD, ppsA and trpE; Table S1). The other strains (PAO1, PA14 and LESB58) differ in at least four out of the seven genes. For FA-HZ1 and W45909, only their sample origins and genomes are known so far. FA-HZ1 is an environmental isolate from China, which was characterized for its dibenzofuran-degrading ability27, while W45909 is a clinical isolate from the USA28.
When looking at the overall genome arrangement, KRP1 shows a high degree of synteny throughout the whole genome with the strains FA-HZ1, W45909, LESB58 and PA14. Only with respect to the type strain P. aeruginosa PAO1 the known large-scale inversion of 70% of the genome is apparent3,23 (Fig. 2).

Synteny plot of the P. aeruginosa KRP1 strain and five other P. aeruginosa strains. Each dot represents a gene of KRP1 and its corresponding homologue in the respective comparative strain. x-axis shows the position within the chromosome of KRP1 and y-axis shows the relative position within the compared genome. Analysis was performed with the EDGAR platform24,25.
The genome of P. aeruginosa has a mosaic-like structure, built of a conserved core, which is interrupted by genomic islands containing variable accessory genes7. The numerical distribution between genes belonging to the core- and the accessory genome of the six P. aeruginosa strains (KRP1, PAO1, PA14, LESB58, FA-HZ1 & W45909) was analyzed using EDGAR (Fig. 3). These six strains share a common core genome of 4,978 genes, which corresponds to 76.9% (W45909)—87.3% (PAO1) of all genes annotated in the respective genomes (79% for KRP1). The core predominantly includes primary metabolism related genes, as well as genes involved in transcription and translation29. The core genome shared by KRP1 and the two predominantly researched strains PAO1 and PA14 consists of 5,278 genes (Fig. 3A). This is equivalent to 83.8% (KRP1)—92.6% (PAO1) of all genes annotated in the respective genomes (Table 1). There are 583 genes in KRP1, for which orthologues are not found in either of the two other strains (area I, Fig. 3A). Thus, the environmental isolate KRP1 encodes for a substantially higher number of singletons than PAO1 or PA14. As a species, P. aeruginosa contains another 10,000 genes, which make up the accessory genome. The overlap of genes belonging to this genome fragment in KRP1 is more pronounced with the FA-HZ1 and W45909 strains of P. aeruginosa (area II, Fig. 3B), which also cluster as the closest relatives of KRP1 during the phylogenetic evaluation (Figs. 1, S1). The three strains share a total of 5,667 genes, which corresponds to 89.94% of all KRP1 predicted ORFs (core + in common accessory genes). This is interesting, since all three strains originate from three different habitats and continents. This combination of core and accessory genes seems to enable the strains to thrive in a pathogenic (W45909) as well as an environmental (KRP1 and FA-HZ1) setting.

Venn diagrams showing the number of genes shared as orthologues in all possible logical combinations between different strains of P. aeruginosa. A: PAO1 [red], PA14 [green], KRP1 [yellow]; B: LESB58 [red], FA-HZ1 [green], KRP1 [blue], W45909 [yellow]. For further information regarding individual areas marked with roman numbers see text. Analysis was performed with the EDGAR platform24,25.
With the highly virulent LESB58 strain, KRP1 shares a total of 5,503 genes (core + area III & IV, Fig. 3B). In an inter-species comparison of these four strains (LESB58, FA-HZ1, KRP1 & W45909; Fig. 3B), the KRP1 genome encodes the lowest number of singletons (area V, Fig. 3B). Of these 102 genes, ~ 78% did not yield a BLAST hit within the COG database, highlighting that most of the genes of this area are novel or hypothetical proteins (Fig. 4; Table S3). This high portion of unclassified genes was typical for all closer investigated overlap areas, except for the overlap of the KRP1 strain with LESB58 and W45909 (area III, Fig. 3B). Here, the majority of the genes have a metabolic function and ~ 27% are related to cellular processes and signaling, which gives a hint that the biological niches occupied by these strains seems to be similar (Fig. 4; Table S4). The strain KRP1 contains 65 singletons with respect to the other five strains. The majority of them are not classified within the COG database (Fig. 4), but are recognized as phage related proteins by the PHASTER software and are located within the identified GIs of KRP1 (Table S5).

ORFs of areas I to V (groups of genes, which are singletons to KRP1 or shared by KRP1 and up to five other P. aeruginosa strains; see Fig. 3) classified by Clusters of Orthologous Groups (COGs) database. Category designations are as follows: [R]—General function prediction only; [S]—Function unknown; [D]—Cell cycle control, cell division, chromosome partitioning; [M]—Cell wall/membrane/envelope biogenesis; [N]—Cell motility; [O]—Post-translational modification, protein turnover, and chaperones; [T]—Signal transduction mechanisms; [U]—Intracellular trafficking, secretion, and vesicular transport; [V]—Defense mechanisms; [W]—Extracellular structures; [A]—RNA processing and modification; [J]—Translation, ribosomal structure and biogenesis; [K]—Transcription; [L]—Replication, recombination and repair; [C]—Energy production and conversion; [E]—Amino acid transport and metabolism; [F]—Nucleotide transport and metabolism; [G]—Carbohydrate transport and metabolism; [H]—Coenzyme transport and metabolism; [I]—Lipid transport and metabolism; [P]—Inorganic ion transport and metabolism; [Q]—Secondary metabolites biosynthesis, transport, and catabolism; [X]—Phage-derived proteins, transposases and other mobilome components.
The accessory genome of P. aeruginosa KRP1
The majority of genes belonging to the accessory genome are not scattered randomly throughout the P. aeruginosa KRP1 genome, but are mainly clustered in 17 GIs and 8 GIts throughout the KRP1 genome (Table 2; Fig. 5) as detected with different bioinformatics tools (SIGI-HMM16, IslandPath-DIMOB17, PHASTER18 and GIPSy19). For some islands only different subparts were detected by the programs. If the subparts were confirmed via manual inspection to be part of the same island, they were numbered with a-e. This means that also the area in between the different sub-islands can be considered part of the accessory genome of P. aeruginosa KRP1. Multiple known GIs of P. aeruginosa were not detected by any of the used software tools but instead were determined via manual scanning of the genome. This highlights on the one hand, the usefulness of the multiple program approach for detection of putative genomic islands within a novel sequenced strain. On the other hand, it shows that the detection algorithms of the programs are not perfect and by just relying on them, relevant information might be overlooked. It is therefore crucial to complement the in silico analysis by implementing previously reported results to obtain a comprehensive view of the genomic structure of a newly sequenced strain.

Visualization of genome plasticity in the P. aeruginosa KRP1 genome detected with different prediction programs. KRP1 main chromosome in comparison to selected P. aeruginosa genomes. Starting from the innermost circle going outwards: major- (500 kb) and minor tick (100 kb) measurements of the KRP1 genome; G + C content (black traces); BLAST comparisons of PAO1 genome against the KRP1 genome (red ring); BLAST comparisons of PA14 genome against the KRP1 genome (blue ring); BLAST comparisons of LESB58 genome against the KRP1 genome (ocher ring); BLAST comparisons of FA-HZ1 genome against the KRP1 genome (green ring); BLAST comparisons of W45909 genome against the KRP1 genome (magenta ring); combined genome plasticity prediction of SIGI-HMM16, IslandPath-DIMOB17, PHASTER18 and GIPSy19, when comparing KRP1 to PA14 as a reference (red segments: uncategorized genomic islets [GIts]; aqua segments: uncategorized genomic islands [GIs]; blue segments: pathogenicity islands [PIs]; green segments: pathogenicity/resistance islands [PI/RIs]; purple segments: pathogenicity/symbiotic islands [PI/SIs]; orange segments: resistance/symbiotic islands [RI/SIs]). Whole genome BLAST comparison and image generation was performed with BRIG31.
Since the overall average G + C content of P. aeruginosa KRP1 is at 66.3% (Table 1) and therefore considered G + C-rich, genes acquired through horizontal gene transfer usually have a lower G + C content (black ring in Fig. 5). After integration of the foreign DNA into the chromosome, it is subject to the same selective evolutionary pressure as the rest of the host chromosome. Thus, over time it is likely to lose the sequence compositional differences, making it undistinguishable from genomic material originating from P. aeruginosa7. These regions are therefore not detected by GI prediction software targeting differences in sequence composition. In the case of the 17 putative GIs and 8 putative GIts in KRP1, most have a notably lower G + C content compared to the surrounding core genome and are therefore of rather young evolutionary origin. Several of the homologous PAGI and LESGI GIs in KRP1, in contrast, were not detected by any of the used algorithms, which might point to an evolutionary older event of acquisition of these GIs and GIts (Tables 2, 4).
GIs and GIts tend to integrate in certain genomic loci termed “regions of genomic plasticity (RGPs)”30, which mark locations where integration of foreign DNA into the P. aeruginosa genome have been previously reported to happen with increased frequency. For the majority of GIs and GIts, a specific RGP could be assigned (Table 2). In P. aeruginosa KRP1 all functional classes of GIs19 are found, except for MIs (Table 2; Fig. 5). Since it is not necessary that each single gene of the respective GI falls into the respective category, some GIs are placed in more than one category.
The genome of KRP1 was also analyzed to identify which version of the four known replacement islands (pilin/pilin modification, flagellin glycosylation island, O-antigen gene cluster, and pyoverdine production) are encoded, as these traits represent critical determinants for the fitness and virulence of an individual P. aeruginosa strain7 (Table 3). A replacement island contains the same functional content and occupies nearly always the same genomic loci within the P. aeruginosa core genome. Intriguingly, the specific genetic sequence of each island is highly diverse between strains34,35. The gene loci of the O-antigen gene cluster and the flagellin glycosylation replacement island are part of the PI/SI 7 and the RI/SI 16, respectively. The pyoverdine locus is located between PI/RI 12 and GI 13, while the pilin modification genes are situated between PI 19 and GI 20. Both are not identified by the different genomic island detection programs. It is remarkable that KRP1 shares all four replacement islands subgroups with strains FA-HZ1 and W45909. In contrast, it only shares the pyoverdine subgroup with PAO1 and PA14. Variations in the pyoverdine locus have been mainly associated with different environmental fitness, as they are an entry target for pyocins, bacterially produced phage-like molecules with antibacterial properties36. The other three loci (pilin/pilin modification, flagellin glycosylation and O-antigen modification) have been linked to virulence properties of strains before37,38,39,40,41,42,43,44. The common group-I pilin variant expressed by KRP1 has been linked increasingly to cystic fibrosis environments37. As O-antigens, pili and flagella are recognized targets for phage entry and the host immune system, keeping different varieties of the same gene locus is thought to be a defense mechanism of P. aeruginosa7. In the case of KRP1, the intact JBD93 bacteriophage, which was detected as GI 23 (92% identity over 86% of the query length with the PHASTER software), uses O-antigen mediated infection45. Since PAO1 and PA14 encode the genes for different O-antigen serotypes (Table 3), they are likely no targets for JBD93. Therefore, almost all of the 51 ORFs encoded in GI 23 are unique to KRP1 in the inter-strain comparison (area I; Fig. 3A). Even though the closely related FA-HZ1 and W45909 strains also have the O1-serotype, the prophage is not encoded in their genome. Further, its integration disrupts the MdlC benzoylformate decarboxylase locus (PA14_64770), which has not been recognized as a RGP in P. aeruginosa before. This leads us to believe that this prophage integration into the KRP1 genome is a recent evolutionary event. Besides GI 23, the PHASTER software18 identifies and annotates six more prophages throughout the KRP1 genome (Table S5). All of the detected sequences can be assigned to specific GIs/GIts and were also recognized by the other genomic island detection programs tested. In general, PHASTER is not a classical GI detection software, but as the integration of a phage into a host genome is a form of horizontal gene transfer, they are part of the accessory genome of the host organism7. Usually other GI prediction tools will also recognize the GIs containing the putative prophage sequences, as their G + C content often differs from the one of the host, which software like GIPSY19 will detect. At the same time, prophages might go undetected, if by chance their G + C content is close to the nucleotide usage of the host. In these cases, PHASTER can lead to additional, otherwise undetected hits, since it utilizes a BLASTP comparison of the query genome with a frequently updated prophage sequence database18,46. Hence, phage related ORFs and proteins will be recognized on the basis of their sequence rather than their properties, like codon usage or G + C content by PHASTER. The software classified four out of the seven prophages of KRP1 as intact, hence their genome contains all the necessary parts to be a complete phage and therefore to also leave the genome again. It will be interesting to see what the functional role of these prophages in the lifestyle of P. aeruginosa KRP1 is, as they are known to be crucial for the fitness of P. aeruginosa under certain conditions15,47. These prophages might also relate to the absence of a detectable intact CRISPR-Cas defense system in the KRP1 strain22,48. This phenomenon has been previously recognized in other P. aeruginosa strains and likely relates to the increased ability of the strains to acquire antibiotic resistances through mobile elements49. For KRP1, the CRISPRCasFinder software detected two sets of one spacer sequence each surrounded by direct repeats. These putative spacers are not located within any of the detected GIs/GIts.
Of the GIs recognized by the prediction software packages, PI/RI 1, GI 3, PI/RI 12 and GI 17 share a large portion of their nucleotide sequence with the other investigated P. aeruginosa genomes (e.g., with PA14: 50%, 80%, 80% and 90%, respectively). On the other hand, unique putative genes within these islands are assigned to only one of the analyzed strains and their integration into the core genome could be traced to a specific known RGP (Table 2). This classifies them as valid regions of the accessory genome of P. aeruginosa.
Frequently, GI integration is observed downstream of a tRNA57,58. The 3′-ends of tRNAs carry attB sites, which are recognized and used for site-specific recombination between an integrative and conjugative element (ICE) and the main chromosome. Overall, the integration of PI 8, GI 11, RI/SI 16, GI 17, PI 19, GI 20 and PI 24b&d occurred just downstream of specific tRNAs within the KRP1 genome. Of these islands, GI 11, PI 19 and GI 20 belong to the same family of P. aeruginosa GIs, which are marked by their bipartite structure. While the first segment, downstream of the tRNA, contains strain-specific cargo ORFs, the second part shows a high degree of sequence similarity between the strains15,57 and mainly encodes structural and mobility-related genes, as well as genes for conjugal transfer9. Cargo genes of GI 11 include heavy metal resistance genes, genes for metabolic enzymes and enzymes used for the formation and altering of nucleic acids, transcription regulators, a two-component system, as well as an antibiotic resistance gene. While the here analyzed cargo genes are KRP1-specific with respect to detected and analyzed GIs (i.e., PAGI-2, PAGI-3 and LESGI-315,57), they share 99% sequence identity with 13 of the 105 P. aeruginosa isolates used for phylogenetic comparison (Table S2). Hence, the entire genomic island is part of the genomic make-up of multiple previously sequenced P. aeruginosa cultures. These include the previously mentioned FA-HZ1 and W45909 strains. We hypothesize that this set of cargo genes form a unit, which contributes to the successful survival of P. aeruginosa in certain habitats.
Genomic resemblance of KRP1 to highly virulent P. aeruginosa strains
The production of many known important virulence factors of P. aeruginosa is encoded within the core genome59. While no apathogenic variants of the species have been reported so far, a strong intraspecies gradient of virulence is observed, ranging from highly infective to only mellow virulent strains4,13. This phenomenon is likely linked to the varying accessory genome of the variants. Based on the genome analysis presented here, overall predictions of the virulence of KRP1 are possible, which can be used as a guidance for further experiments involving this organism. P. aeruginosa KRP1 contains an array of genomic elements that are found in the highly virulent strains PSE913,33 and LESB5812,15,60 (Table 4). Unfortunately, no complete genome sequence is available for PES9 yet, so it could not be included in the full genome comparison. However, some of the shared GIs have been shown to be the source of the strain dependent virulence within the P. aeruginosa species13,14,15. KRP1 encodes all seven genomic islands found in the clinical isolate PSE913,33 (Table 4). The PSE9 strain originated from a patient with ventilator-associated pneumonia isolated at a hospital in Barcelona, Spain in the mid-1990s61. It was found to be the most virulent out of 35 strains in a mouse model of acute pneumonia62. So far, two studies were able to link the increased virulence of PSE9 directly to PAGI-5 and PAGI-913,14. Since KRP1 contains both of the mentioned islands, an increased virulence similar to the levels of PSE9 can be anticipated. PAGI-9 of PSE9 and GIt 5 of KRP1, respectively, consist of 6581 bps and one large ORF, which was identified as a Rhs (rearrangement hot spot) element33. Similarly, PAGI-10 is a Rhs element of PSE9, which is also found within KRP1 (PI/RI 9). The nucleotide sequence of these proteins generally has a bipartite structure composed of a long G + C rich core and a relatively G + C poor tip sequence. While the core sequence is intra- and interspecies highly conserved, the tip is rather variable. The fact that the strains PSE9 and KRP1 show sequence identity over the entire length of the ORFs and not only in the conserved core shows the close genomic relationship between the hyper virulent PSE9 and KRP1.
PAGI-11 of PSE9 (GIt 14 in KRP1) is only 2003 bps long and located at RGP 52 (Table 4) and while Battle et al.33 did not find any ORFs contained, the Prokka pipeline63, applied to the KRP1 genome, predicts the hypothetical protein KRP1_16515. The G + C content of just 43.19% is far below the average of the KRP1 genome (i.e. 66.3%). Other strains are known to contain larger GIs encoding mobile element related genes at this specific genomic locus30. Therefore, PAGI-11 might have been a larger genomic island in the past, which was partially lost over time in PSE9 and KRP1.
Further, PSE9 and KRP1 share the same O-antigen type O1 (Table 3). The O-antigen type of the outer membrane lipopolysaccharide (LPS) layer has been previously linked to the virulence of P. aeruginosa, but most studies consider the serotype of the type strain PAO1 (type O5)43. Both strains are also exoS positive and exoU negative, a genotype that has been linked to an invasive phenotype64. Since no full genome sequence of PSE9 is available so far, a deeper in silico comparison between both strains is currently impossible.
Besides PSE9, the P. aeruginosa strain KRP1 shows substantial similarities in its accessory genome with the LESB58 strain, an aggressive pathogen of a cystic fibrosis patient from Liverpool in 198812,15,60 (Table 4). The strain is beta-lactam-resistant60, exhibits enhanced survival on dry surfaces65, shows an increased patient morbidity66, and overexpression of parts of the quorum sensing regulon during early growth phases (e.g., LasA, elastase, and pyocyanin)67,68. It is also known to replace previously established P. aeruginosa strains due to its aggressive nature, thereby causing a superinfection69. A LES isolate has even been reported to have infected the non-CF parents of a CF patient70. While the complete reasons for its increased virulence are still partially unknown, a lot of the responsible factors are thought to be driven by the accessory genome of the strain12,15. These LESGI termed genomic islands differentiate the LES strain from other P. aeruginosa strains. Of the 17 known LESGIs and six LESGI-prophages, the genome of KRP1 contains 12 LESGIs and one prophage (Table 4). The majority of the shared GIs and GIts were found via manual search rather than by the applied software programs (Table 2). LESGI-6 to LESGI-17 were first detected by Jani et al.12. The authors used a genome segmentation approach to identify genomic regions of foreign origin within the LESB58 strain. This technique varies from the ones used in this study and therefore different putative GIs and GIts were detected. The authors could show that these GI encode for additional virulence factors (LESGI-6, -8, -13, and -15) as well as drug and metal resistance cassettes (LESGI-12 and -17). LESGI-9, -16, and -17 add additional versatility to the LESB58 metabolic repertoire12. Since KRP1 encodes all of these GIs as well, it is very likely that it employs their functions and therefore shows an increased virulence potential, similar to the LESB58 strain.
In contrast, the two strains showing the closest ANI identity and phylogenetic relationship with KRP1 are P. aeruginosa strain FA-HZ1 and W45909 (Fig. 1). FA-HZ1 is a dibenzofuran-degrading isolate from China27 while W45909 is a clinical isolate from the USA28. All but three identified GIs in KRP1 are also present in these two most related strains (PI 8, PI 19 and GI 23 for W45909 and GI 23 for FA-HZ1). This provides circumstantial evidence that the genomic repertoire of P. aeruginosa KRP1 is likely to sustain a pathogenic as well as an apathogenic lifestyle in nature. While their genetic information is available, no further studies have been performed with either of these strains but we stand to believe that they will also show an increased virulence like PSE9, LESB58 and likely KRP1.
Conclusion
The genome of the BES isolate Pseudomonas sp. KRP1 was de novo sequenced and analyzed in depth for its phylogenetic relationship within the Pseudomonas clade. Due to the sequence composition of its core genome, it could clearly be assigned to belong to the P. aeruginosa species. Its closest relatives are two recently sequenced strains from China (FA-HZ1)27 and the USA (W45909)28.
The accessory genome of KRP1 was thoroughly analyzed. Using four different prediction programs, 17 putative genomic islands and 8 putative genomic islets were detected. This analysis was extended by mining for the 44 GI complexes previously described in P. aeruginosa9,10,11,12. Most of the GIs and GIts could clearly be assigned to a known RGP (Table 2). The majority of the KRP1 singletons, with respect to the strains PAO1, PA14, FA-HZ-1, W45909 and LESB58, are contained in these islands, marking them as the main source of genome divergence between the strains.
Utilizing the increased amount of sequencing data made publicly available in the past decade, it is possible to make in silico based educated prediction towards the virulence potential of a newly isolated strain of P. aeruginosa. Hence, it decreases the need for laborious trial and error type wet lab experiments and animal testing. The hurdle to get permission to do animal experiments, for example in Germany, is fairly high and not every lab facility has the necessary infrastructure for this type of investigations. With an in silico investigation, like the one presented in the manuscript, also these labs have the option to easily obtain valuable information on the strain they investigate. This kind of educated knowledge about the expected pathogenicity of an isolate can as well help in the daily handling of the organism in the labs itself. As every P. aeruginosa has a certain pathogenic potential, they are all classified as risk group two and should all be handled with the same caution in the lab. But the degree of virulence actually varies substantially between strains4. For species isolated from e.g., infection scenarios, a high virulence is obvious. Instead, KRP1 is an environmental isolate that was spotted because it dominated in a natural mixed culture biofilm20. By being aware of the potentially high virulence of the organisms, personal safety measurements can be increased to avoid an accidental exposition of the organism. Using publicly available data and their integration with own research data can help to substantially speed up research in the future and to draw wider, more general conclusions. The true degree to which the individual GIs and GIts contribute to virulence of the strain is still to be determined and proves to be a rather difficult task since virulence in P. aeruginosa is known to be combinatorial3,71.
Methods
Strain and medium
P. aeruginosa KRP1 was isolated from a microbial fuel cell setup at the Laboratory of Microbial Ecology and Technology (LabMET) at Ghent University (deposited into the Belgian Co-ordinated Collections of Microorganisms, BCCM; strain number LMG 23,160)20. Cultures were grown in shake flasks in Luria Broth medium at 37 °C, 200 rpm shaking.
DNA sequencing
Genomic DNA of P. aeruginosa KRP1 was isolated via phenol–chloroform extraction72; mod.. Besides a purity check on a NanoDrop One/OneC Microvolume-UV–Vis-Spectrophotometer (Thermo Fisher Scientific) and an integrity check via agarose gel electrophoresis, the concentration of isolated genomic DNA was estimated via a PicoGreen dsDNA quantification assay (Quant-iT PicoGreen dsDNA Assay Kit, Thermo Fisher Scientific). The measurement for this assay was done with a Synergy Mx microplate reader (BioTek) in 96-well plates using an excitation wavelength of 480 nm, an emission wavelength of 520 nm, a scan width of 9.0 and an overflow value of 80.
For shotgun library preparation, 1 µg of chromosomal DNA was used (TruSeq DNA PCR-Free Library Preparation Kit, Illumina). Samples were sequenced on an Illumina MiSeq system using the MiSeq Reagent Kit v3 for 600 cycles. The data (542.3 Mb equaling ~ 81.3 × coverage) were assembled using Newbler v.2.8 (Roche), which resulted in 58 scaffolds containing 94 contigs. Gap closure was conducted with a MinION Mk1B Sequencer from Oxford Nanopore Technologies. For this second shotgun library, 2 μg of genomic DNA was used as starting material. Size-selected DNA-fragments of 5 to 50 kb were used to create a 1D2 sequencing library according to the manufacturer’s instructions (1D2 Sequencing Kit (R9.5), Oxford Nanopore Technologies). The sequencing library was run on a R9.5 flowcell for 3 h. Base calling and data conversion to FastQ was performed using Albacore v1.2.473. The resulting 72.4 Mb (12 × coverage) sequencing data were assembled with Canu v1.574. After assembly, the resulting 23 contigs were polished with the short Illumina reads using PILON75. The final assembly was done manually using Consed76 to combine the contigs of the Newbler and Canu assemblies, as well as to resolve any discrepancies between the two different assemblies. This hybrid approach of a short read- (Newbler) and long read assembler (Canu) followed by manual curation, was done to fulfill the 3C criteria of genome assembly (contiguity, correctness and completeness)22 to a sufficient degree. Gene prediction and annotation of the finished genome were performed using the Prokka pipeline63. Visualization and inspection of the annotated sequence was done in Artemis77.
To clarify the existence of a potential mega plasmid, a PCR using EconoTaq PLUS GREEN DNA polymerase (Lucigen) was performed. The PCR fragments were sequenced by Eurofins Genomics (Germany).
Comparative genome analysis
For the analysis of the assembled KRP1 genome in respect to other Pseudomonas genomes and to find orthologous genes in related genomes, the EDGAR ("Efficient Database framework for comparative genome analyses using BLAST score Ratios")24,25 platform was used. Via the platform, the synteny analysis, the distribution of gene sets into core genome, accessory genome and singletons, the ANI calculations, and the phylogenetic tree generation were performed. For the phylogenetic trees, EDGAR utilizes an alignment of all core genes of every genome included in the comparison via MUSCLE78. This compiled alignment is the input for the neighbor-joining algorithm used by the PHYLIP package (https://evolution.genetics.washington.edu/phylip.html) to construct the phylogenetic tree. Hence, rather than being based on the 16S RNA sequence or the MLST sequences, the here presented trees are based on the entire core genome of the analyzed strains.
For functional gene classification, ORFs were checked against the Clusters of Orthologous Groups (COG) database79. Parameters were set to an e-value of < 1e−10 and 80% identity. MLST profiling was done using MLST 2.0 v2.0.426. The genome of KRP1 was compared to 105 fully sequenced P. aeruginosa strains and 8 other Pseudomonas species. These represent all publicly available fully finished and closed P. aeruginosa genomes available from the NCBI website at the time of conducting this study. More in depth analyses were performed with the type strain PAO1 (AE004091; NC_002516.2;23), the frequently researched strain PA14 (UCBPP-PA14; NC_008463.13), the highly virulent strain LESB58 (NC_011770.115) and the two phylogenetically closest strains FA-HZ1 (NZ_CP017353.127) and W45909 (NZ_CP008871.228). The accession numbers of the other ~ 100 Pseudomonas strains used for the ANI and phylogenetic comparison can be found in Table S2.
Multiple genomic island prediction software packages were applied to analyze the KRP1 genome with respect to its genome plasticity. For GI and GIt detection the following programs were used: IslandViewer80,81, which incorporates the SIGI-HMM16 and the IslandPath-DIMOB17 software, and GIPSy (Genomic island prediction software)19. PHASTER (PHAge Search Tool—Enhanced Release) was used for identification and annotation of prophage sequences within the KRP1 genome18,46. Spine and AGEnt were applied for prediction of the accessory genome in its entirety82. Results were imaged with BRIG (BLAST Ring Image Generator)31. This automated GI detection was complemented by manual curration of the precise starting and stopping position of each detected island via different blast comparisons. Additionally, the genome was manually mined for any of the 44 GI complexes previously described in P. aeruginosa9,10,11,12. To evaluate the relationship of the GI content with a potential CRISPR-Cas systems in the strain, CRISPRCasFinder v1.1.283 was used.
The ACT (Artemis Comparison Tool)84 was used for manual detection of regions of genomic plasticity (RGPs). It was also the visualization method of choice for partial and whole genome comparisons of KRP1 with different P. aeruginosa strains.
Accession code
The dataset (full genome data of P. aeruginosa KRP1) generated and analysed during the current study is available in the NCBI BioProject database repository, (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number CP046069. It is part of the ElectricMicrobe100 Umbrella BioProject, which can be accessed via PRJNA417841.
References
Silby, M. W., Winstanley, C., Godfrey, S. A., Levy, S. B. & Jackson, R. W. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680 (2011).
de Bentzmann, S. & Plesiat, P. The Pseudomonas aeruginosa opportunistic pathogen and human infections. Environ. Microbiol. 13, 1655–1665 (2011).
Lee, D. G. et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7, R90 (2006).
Hilker, R. et al. Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 17, 29–46 (2015).
He, J. et al. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. USA 101, 2530–2535 (2004).
Bennett, P. M. Genome plasticity: insertion sequence elements, transposons and integrons, and DNA rearrangement. Methods Mol. Biol. 266, 71–113 (2004).
Kung, V. L., Ozer, E. A. & Hauser, A. R. The Accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641 (2010).
Tümmler, B. In Pseudomonas: Volume 4 Molecular Biology of Emerging Issues (eds J.-L. Ramos & R. C. Levesque) 35–68 (Springer US, 2006).
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F. & Tummler, B. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2, 150 (2011).
Silveira, M. C., Albano, R. M., Asensi, M. D. & Carvalho-Assef, A. P. D. A. Description of genomic islands associated to the multidrug-resistant Pseudomonas aeruginosa clone ST277. Infect. Genet. Evol. 42, 60–65 (2016).
Hong, J. S., Yoon, E. J., Lee, H., Jeong, S. H. & Lee, K. Clonal dissemination of Pseudomonas aeruginosa sequence type 235 isolates carrying blaIMP-6 and emergence of blaGES-24 and blaIMP-10 on novel genomic islands PAGI-15 and -16 in South Korea. Antimicrob. Agents Chemother. 60, 7216–7223 (2016).
Jani, M., Mathee, K. & Azad, R. K. Identification of novel genomic islands in liverpool epidemic strain of Pseudomonas aeruginosa using segmentation and clustering. Front. Microbiol. 7, 1210 (2016).
Battle, S. E., Meyer, F., Rello, J., Kung, V. L. & Hauser, A. R. Hybrid pathogenicity island PAGI-5 contributes to the highly virulent phenotype of a Pseudomonas aeruginosa isolate in mammals. J. Bacteriol. 190, 7130–7140 (2008).
Kung, V. L. et al. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc. Natl. Acad. Sci. USA 109, 1275–1280 (2012).
Winstanley, C. et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 19, 12–23 (2009).
Waack, S. et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 7, 142 (2006).
Hsiao, W., Wan, I., Jones, S. J. & Brinkman, F. S. L. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420 (2003).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Soares, S. C. et al. GIPSy: Genomic island prediction software. J. Biotechnol. 232, 2–11 (2016).
Rabaey, K., Boon, N., Siciliano, S. D., Verhaege, M. & Verstraete, W. Biofuel cells select for microbial consortia that self-mediate electron transfer. Appl. Environ. Microbiol. 70, 5373–5382 (2004).
Bosire, E. M., Blank, L. M. & Rosenbaum, M. A. Strain- and substrate-dependent redox mediator and electricity production by Pseudomonas aeruginosa. Appl. Environ. Microbiol. 82, 5026–5038 (2016).
Molina-Mora, J. A., Campos-Sánchez, R., Rodríguez, C., Shi, L. & García, F. High quality 3C de novo assembly and annotation of a multidrug resistant ST-111 Pseudomonas aeruginosa genome: benchmark of hybrid and non-hybrid assemblers. Sci. Rep. 10, 1392 (2020).
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 (2000).
Blom, J. et al. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 10, 154 (2009).
Blom, J. et al. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22-W28, (2016).
Larsen, M. V. et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361 (2012).
Ali, F., Hu, H., Xu, P. & Tang, H. Complete genome sequence of Pseudomonas aeruginosa FA-HZ1, an efficient dibenzofuran-degrading bacterium. Genome Announc. 5, e01634-e1616 (2017).
Yan, J. et al. Bow-tie signaling in c-di-GMP: Machine learning in a simple biochemical network. PLoS Comput. Biol. 13, e1005677 (2017).
Valot, B. et al. What it takes to be a Pseudomonas aeruginosa? The core genome of the opportunistic pathogen updated. PLoS ONE 10, e0126468 (2015).
Mathee, K. et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 105, 3100–3105 (2008).
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402 (2011).
Roy, P. H. et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 5, e8842 (2010).
Battle, S. E., Rello, J. & Hauser, A. R. Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 290, 70–78 (2009).
Smith, E. E., Sims, E. H., Spencer, D. H., Kaul, R. & Olson, M. V. Evidence for diversifying selection at the pyoverdine locus of Pseudomonas aeruginosa. J. Bacteriol. 187, 2138–2147 (2005).
Subedi, D., Kohli, G. S., Vijay, A. K., Willcox, M. & Rice, S. A. Accessory genome of the multi-drug resistant ocular isolate of Pseudomonas aeruginosa PA34. PLoS ONE 14, e0215038 (2019).
Baysse, C. et al. Uptake of pyocin S3 occurs through the outer membrane ferripyoverdine type II receptor of Pseudomonas aeruginosa. J. Bacteriol. 181, 3849–3851 (1999).
Kus, J. V., Tullis, E., Cvitkovitch, D. G. & Burrows, L. L. Significant differences in type IV pilin allele distribution among Pseudomonas aeruginosa isolates from cystic fibrosis (CF) versus non-CF patients. Microbiology 150, 1315–1326 (2004).
Arora, S. K., Neely, A. N., Blair, B., Lory, S. & Ramphal, R. Role of motility and flagellin glycosylation in the pathogenesis of Pseudomonas aeruginosa burn wound infections. Infect. Immunol. 73, 4395–4398 (2005).
Kuang, Z. et al. The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol. Microbiol. 79, 1220–1235 (2011).
Verma, A. et al. Glycosylation of b-type flagellin of Pseudomonas aeruginosa: structural and genetic basis. J. Bacteriol. 188, 4395–4403 (2006).
Meyer, J. M., Neely, A., Stintzi, A., Georges, C. & Holder, I. A. Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect. Immunol. 64, 518–523 (1996).
Cornelis, P. & Dingemans, J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front. Cell Infect. Microbiol. 3, (2013).
Pier, G. B. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol 297, 277–295 (2007).
Augustin, D. K. et al. Presence or absence of lipopolysaccharide O antigens affects type III secretion by Pseudomonas aeruginosa. J. Bacteriol. 189, 2203–2209 (2007).
Bondy-Denomy, J. et al. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10, 2854–2866 (2016).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352 (2011).
Molina-Mora, J. A. et al. Transcriptomic determinants of the response of ST-111 Pseudomonas aeruginosa AG1 to ciprofloxacin identified by a top-down systems biology approach. Sci. Rep. 10, 13717 (2020).
van der Zee, A. et al. Spread of carbapenem resistance by transposition and conjugation among Pseudomonas aeruginosa. Front. Microbiol. 9, 2057–2057 (2018).
Pawluk, A., Bondy-Denomy, J., Cheung, V. H., Maxwell, K. L. & Davidson, A. R. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. mBio 5, e00896, (2014).
Liu, P. V. & Wang, S. Three new major somatic antigens of Pseudomonas aeruginosa. J. Clin. Microbiol. 28, 922–925 (1990).
Burrows, L. L., Charter, D. F. & Lam, J. S. Molecular characterization of the Pseudomonas aeruginosa serotype O5 (PAO1) B-band lipopolysaccharide gene cluster. Mol. Microbiol. 22, 481–495 (1996).
Lam, J. S., Taylor, V. L., Islam, S. T., Hao, Y. & Kocíncová, D. Genetic and functional diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front. Microbiol. 2, 118–118 (2011).
Meyer, J. M. et al. Use of siderophores to type pseudomonads: the three Pseudomonas aeruginosa pyoverdine systems. Microbiology 143(Pt 1), 35–43 (1997).
CL Giltner N Rana MN Lunardo AQ Hussain LL Burrows 2011 Evolutionary and functional diversity of the Pseudomonas type IVa pilin island Environ. Microbiol. 13 250 264
Arora, S. K., Bangera, M., Lory, S. & Ramphal, R. A genomic island in Pseudomonas aeruginosa carries the determinants of flagellin glycosylation. Proc. Natl. Acad. Sci. USA 98, 9342–9347 (2001).
Varga, J. J. et al. Genotypic and phenotypic analyses of a Pseudomonas aeruginosa chronic bronchiectasis isolate reveal differences from cystic fibrosis and laboratory strains. BMC Genomics 16, 883 (2015).
Larbig, K. D. et al. Gene Islands Integrated into tRNA(Gly) Genes confer genome diversity on a Pseudomonas aeruginosa clone. J. Bacteriol. 184, 6665–6680 (2002).
Kiewitz, C., Larbig, K., Klockgether, J., Weinel, C. & Tümmler, B. Monitoring genome evolution ex vivo: reversible chromosomal integration of a 106 kb plasmid at two tRNALys gene loci in sequential Pseudomonas aeruginosa airway isolates. Microbiology 146, 2365–2373 (2000).
Wolfgang, M. C. et al. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 100, 8484–8489 (2003).
Cheng, K. et al. Spread of beta-lactam-resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 348, 639–642 (1996).
Hauser, A. R. et al. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 30, 521–528 (2002).
Schulert, G. S. et al. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa Isolates obtained from patients with hospital-acquired pneumonia. J. Infect. Dis. 188, 1695–1706 (2003).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Juan, C., Peña, C. & Oliver, A. Host and Pathogen Biomarkers for Severe Pseudomonas aeruginosa Infections. J. Infect. Dis. 215, S44–S51 (2017).
Panagea, S., Winstanley, C., Walshaw, M. J., Ledson, M. J. & Hart, C. A. Environmental contamination with an epidemic strain of Pseudomonas aeruginosa in a Liverpool cystic fibrosis centre, and study of its survival on dry surfaces. J. Hosp. Infect. 59, 102–107 (2005).
Al-Aloul, M. et al. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax 59, 334–336 (2004).
Salunkhe, P. et al. A cystic fibrosis epidemic strain of Pseudomonas aeruginosa displays enhanced virulence and antimicrobial resistance. J. Bacteriol. 187, 4908–4920 (2005).
Fothergill, J. L. et al. Widespread pyocyanin over-production among isolates of a cystic fibrosis epidemic strain. BMC Microbiol. 7, 45 (2007).
McCallum, S. J. et al. Superinfection with a transmissible strain of Pseudomonas aeruginosa in adults with cystic fibrosis chronically colonised by P. aeruginosa. Lancet 358, 558–560, (2001).
McCallum, S. et al. Spread of an epidemic Pseudomonas aeruginosa strain from a patient with cystic fibrosis (CF) to non-CF relatives. Thorax 57, 559–560 (2002).
Harrison, E. M. et al. Pathogenicity islands PAPI-1 and PAPI-2 contribute Individually And Synergistically To The Virulence Of Pseudomonas aeruginosa strain PA14. Infect. Immunol. 78, 1437–1446 (2010).
Altenbuchner, J. & Cullum, J. DNA amplification and an unstable arginine gene in Streptomyces lividans 66. Mol. Gen. Genet. 195, 134–138 (1984).
Albacore v1.2.4 tool. https: //github.com/Albacore/albacore.
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Gordon, D. & Green, P. Consed: a graphical editor for next-generation sequencing. Bioinformatics 29, 2936–2937 (2013).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics (Oxford, England) 16, 944–945 (2000).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Langille, M. G. I. & Brinkman, F. S. L. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665 (2009).
Bertelli, C. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30-w35 (2017).
Ozer, E. A., Allen, J. P. & Hauser, A. R. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15, 737 (2014).
Couvin, D. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246-w251 (2018).
Carver, T. J. et al. ACT: the Artemis Comparison Tool. Bioinformatics 21, 3422–3423 (2005).
Acknowledgements
We thank Prof. Lars Blank for providing us access to his laboratory facilities and the helpful discussions at the iAMB in Aachen. We also want to express our gratitude to Dr. Tobias Busche and Anika Winkler from the CeBiTec in Bielefeld for generating the MinION library, performing the sequencing and in general for the excellent technical assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG). The research grant AG156/1-1 from the DFG was awarded to MAR, CB was financed through this grant. The funding body did not play any roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. MAR is further supported by an ERC consolidator grant e-MICROBe, grant no. 864669.
Author information
Authors and Affiliations
Contributions
C.B. designed, executed and analyzed the experiments and drafted the manuscript, C.R. performed the sequence data clean up and mapping and published the finished genome. J.B. created the customized E.D.G.A.R. project and ran the COG database comparison. K.R. isolated and provided the KRP1 strain. J.K. supervised the sequencing work. M.A.R. conceived of the study, designed and interpreted the experiments and edited the manuscript. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Berger, C., Rückert, C., Blom, J. et al. Estimation of pathogenic potential of an environmental Pseudomonas aeruginosa isolate using comparative genomics. Sci Rep 11, 1370 (2021). https://doi.org/10.1038/s41598-020-80592-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80592-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
