
Abstract
Enzalutamide (ENZ) is an important drug used to treat castration-resistant prostate cancer (CRPC), which inhibits androgen receptor (AR) signaling. Previous study showed that 3,3′-diindolylmethane (DIM) is an AR antagonist that also inhibits Wnt signaling and epithelial-mesenchymal transition (EMT). To investigate whether combined treatment with ENZ and DIM can overcome ENZ resistance by regulating Wnt signaling to inhibit AR signaling and EMT in ENZ-resistant prostate cancer cells, 22Rv1 cells were cultured in normal medium and treated with ENZ, DIM, and DIM with ENZ. Exposure of ENZ-resistant cells to both DIM and ENZ significantly inhibited cell proliferation without cytotoxicity and invasion in comparison with the control. DIM significantly increased the E-cadherin expression and inhibited the expressions of Vimentin and Fibronectin, subsequently inhibiting EMT. Co-treatment with ENZ and DIM significantly increased the expressions of GSK3β and APC and decreased the β-catenin protein expression, causing inhibition of Wnt signaling and AR expression, it also significantly decreased the AR-v7 expression and down-regulated AR signaling. Via suppression of Wnt and AR signaling, co-treatment increased the E-cadherin and decreased the Vimentin and Fibronectin RNA and protein expressions, then inhibited EMT. Co-treatment with DIM and ENZ regulated Wnt signaling to reduce not only the AR expression, but also the AR-v7 expression, indicating suppression of EMT that inhibits cancer cell proliferation, invasion and migration to ameliorate ENZ resistance.
Similar content being viewed by others
Introduction
Prostate cancer (PCa) is not only the second most common cancer in men; it is also the fifth leading cause of cancer mortality among men worldwide1. In Taiwan, the prevalence and mortality of PCa are increasing, and around 50% of patients are diagnosed with PCa at the late stage2. At the late stage, androgen deprivation therapy (ADT) is considered the standard therapy3; however, after the beginning of ADT, the duration of response to castration is short and, in almost all patients, is followed by the emergence of a castration-resistant phenotype4. Recurrence of PCa is regarded as castration-resistant prostate cancer (CRPC). It has been confirmed that CRPC is still an androgen-dependent disease that relies on androgen receptor (AR) signaling5. AR signaling can be activated through various mechanisms, such as AR overexpression, binding of alternate AR ligands, upregulation of AR coactivators and downregulation of AR corepressors, steroidogenesis, inflammation pathways, growth-factor signaling, and Wnt/β-catenin signaling6. Therefore, second-generation anti-androgen drugs, such as abiraterone and enzalutamide (ENZ), were developed as new therapies for CRPC7.
ENZ, also named MDV3100, is an AR antagonist that competitively antagonizes androgen binding to AR and inhibits nuclear translocation of AR, recruitment of AR co-factors, and AR binding to DNA8. ENZ has been proven to increase the overall survival duration and improve quality of life of PCa patients. Owing to its safety characteristics, it has become a critical drug for use at different stages of PCa9. Although ENZ has many benefits, 20–40% of patients have primary resistance, and nearly all treated CRPC patients develop ENZ resistance eventually10. Recent evidence has shown that several mechanisms are involved in ENZ resistance, including AR mutation, the existence of AR splice variants (AR-Vs), AR and glucocorticoid receptor (GR) overexpression, autophagy, intracrine androgen biosynthesis, and epithelial-mesenchymal transition (EMT)11,12,13.
Wnt proteins are cysteine-rich, secreted lipoglycoproteins that play critical roles in normal embryonic development, and are involved in a variety of biological processes, but aberrant activation of the Wnt signaling pathway has been noted in carcinogenesis14. Our previous studies have shown that dysregulation of Wnt signaling is associated with hepatoma15, ovarian cancer16,17, and cervical cancer18, while other studies have demonstrated its involvement in prostate cancer19 and colorectal cancer20 in terms of affecting cell proliferation, invasion or migration. The two distinct pathways of Wnt signals are a canonical or Wnt/β‐catenin pathway and a noncanonical pathway or pathways that are β-catenin-independent21. The stabilization and nuclear translocation of β-catenin play crucial roles while Wnt signaling is engaged. β-catenin no longer binds to some proteins, such as adenomatous polyposis coli (APC) protein and glycogen synthase kinase‑3 (GSK3), but does bind to T-cell factor (TCF) family transcription factors, whereupon it activates target genes determining carcinogenesis, including MYC, MMP7, VEGF and AR14,19.
Relatively high gene expression levels of both β-catenin and AR have been observed in CRPC22. Of several mechanisms involved in ENZ-resistant CRPC, aberrant AR signaling is a crucial one. In general, AR signaling is activated while AR is bound to androgen, and then translocates to the nucleus to bind with androgen-responsive elements (ARE) in the promoter regions of target genes6. AR-Vs have been found to translocate into the nucleus without androgen, and can form homodimers or heterodimers with androgen-bound AR23. AR-Vs may be associated with the initiation and progression of PCa. Evidence has shown that AR-V expressions are involved in EMT, which plays important roles in cancer progression and drug resistance23,24,25. Among approximately 20 different variants, increased AR-v7 expression has been identified in CRPC patients, and is related to ENZ resistance26,27. Moreover, interaction of AR with Wnt signaling has been suggested, and both could activate the process of EMT24,28,29. Therefore, a new adjuvant therapy might be helpful in improving the treatment of ENZ-resistant CRPC.
According to epidemic evidence, high intakes of cruciferous vegetables reduce the risk of PCa, especially in the early stages30,31,32, and dietary and plant-based drugs have been suggested as possible alternative strategies in the treatment of cancer. The phytochemical indole-3-carbinol (I3C) and its metabolite 3,3′-diindolylmethane (DIM) are present in high levels in cruciferous vegetables, and have been shown to exert anticancer effects, but DIM has a greater bioactivity and is safer than I3C33,34. Previous studies have shown that DIM induces cell-cycle arrest in AR positive, p53 wild-type LNCaP and AR negative, p53 mutant DU145 human prostate cancer cell lines35. It also has inhibitory effects on AR signaling, Wnt signaling and the process of EMT30,33,34,36. Moreover, in clinical experiments, a formulated DIM (Bio-Response 3,3′-diindolylmethane, BR-DIM) was used as an adjuvant therapy in the early stages of PCa, and a good response was observed29,30,34. However, no research has been performed to examine the effects of DIM through Wnt/β-catenin signaling in the late stages of ENZ-resistant PCa. Therefore, in the current study, we focused on AR, AR-v7 and Wnt signaling to investigate whether combined treatment with ENZ and DIM could overcome ENZ resistance by regulating Wnt signaling to inhibit AR signaling and EMT in ENZ-resistant PCa cells. We found that co-treatment with DIM and ENZ inhibited prostate cancer cell proliferation, invasion and migration through regulating Wnt signaling to reduce the expressions of AR and AR-v7.
Results
DIM and ENZ treatment suppress cell proliferation and colony formation of PCa cells
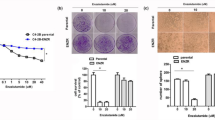
Exposure to a low dose of ENZ for 48 and 72 h had no suppressive effect on 22Rv1 cells (data not shown). It was confirmed that 22Rv1 cells exhibited ENZ resistance. Treatment of cells with DIM alone for 48 and 72 h caused significant dose- and time-dependent inhibition of cell proliferation, with 25% suppression following 72 h of exposure to 30 μM DIM. Therefore, in order to investigate the adjuvant effects of DIM on ENZ resistance, 22Rv1 cells were treated with DIM and ENZ simultaneously, and the results showed that treatment of 22Rv1 cells with 30 μM DIM alone for 48 and 72 h significantly decreased the cell viability (Fig. 1; p < 0.05 vs. control), and the effect of co-treatment with DIM and 10 μM ENZ was similar to that of treatment with 40 μM ENZ. Co-exposure to DIM and ENZ resulted in significant inhibition of cell proliferation (Fig. 1; p < 0.05 vs. DIM or ENZ alone).

Inhibition of cell proliferation by DIM (30 μM) and ENZ (10 or 40 μM), alone and in combination, in 22Rv1 cells at 48 h (A) and 72 h (B) post-exposure (n = 6). DIM 3,3′-diindolylmethane; ENZ enzalutamide. Values are presented as means ± SD. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
In 22Rv1 cells, treatment with DIM alone significantly inhibited the clonogenic ability (Fig. 2; p < 0.05 vs. control), while there was no suppression when cells were treated with 10 μM ENZ. In cells treated with a high concentration of ENZ, colony formation was significantly inhibited (Fig. 2; p < 0.05 vs. control), and the inhibition was of the same magnitude as that caused by treatment with 30 μM DIM. Combined DIM and ENZ treatment significantly increased the inhibition of colony formation (Fig. 2; p < 0.05 vs. control and 10 μM ENZ). The results of the present study indicated that ENZ and DIM may have a synergistic effect in suppressing cell proliferation and colony formation in PCa cells.

DIM and ENZ treatment suppress cell colony formation of PCa cells (n = 3). DIM 3,3′-diindolylmethane; ENZ enzalutamide. Representative images (A), number of colonies (B), colony formation normalized to control (C). Values are presented as means ± SD. a,b,cBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
DIM potentiates the inhibition effect of ENZ on PCa cell invasion
In order to examine whether combined treatment with DIM and ENZ resulted in stronger inhibition of invasion in PCa cells, 22Rv1 cells were treated with DIM and ENZ alone and together for 24 h. The results illustrated that both DIM and ENZ significantly inhibited cell invasion (Fig. 3B; p < 0.05 vs. control), but DIM had a better inhibition effect (Fig. 3B; p < 0.05 vs. 10 and 40 μM ENZ). In addition, exposure to DIM and ENZ in combination significantly increased the efficacy of ENZ in suppressing cell invasion (Fig. 3B; p < 0.05 vs. 10 and 40 μM ENZ). The results of the present study indicated that DIM may potentiate the efficacy of ENZ with regards to invasion of 22Rv1 cells.


DIM potentiates the inhibition effect of ENZ on PCa cell invasion (n = 3). DIM 3,3′-diindolylmethane; ENZ enzalutamide. Results of cell migration (A) and invasion (B) assays. Values are presented as means ± SD. a,b,cBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
DIM and ENZ co-treatment augments the suppression of PCa cell migration
To investigate whether combined treatment with DIM and ENZ inhibited PCa cell migration, a wound-healing assay and a Transwell migration assay were employed. As shown in Fig. 4A,C, treatment with 10 μM ENZ did not inhibit cancer cell migration; however, exposure to DIM significantly suppressed cell migration (p < 0.05 vs. control and 10 μM ENZ). Treatment with ENZ (10 and 40 μM) and DIM combined resulted in a stronger inhibition effect (p < 0.05 vs. control and 10 μM ENZ). Furthermore, as shown in Fig. 3A, treatment of 22Rv1 cells with ENZ alone did not repress cell migration, but treatment with DIM or combined treatment with DIM and ENZ significantly inhibited cancer cell migration (p < 0.05 vs. control and ENZ alone). The results showed that combined treatment with DIM and ENZ resulted in better suppression of migration in PCa cells after 24 h. Therefore, a wound-healing assay was employed to detect cell migration at 48 and 72 h. Exposure to DIM and ENZ alone did not suppress migration of 22Rv1 cells (Fig. 4B,D), but combined treatment with DIM and ENZ significantly suppressed migration of 22Rv1 cells (Fig. 4B,D; p < 0.05 vs. control, DIM and ENZ alone). The results of the present study indicated that co-treatment with DIM and ENZ resulted in better suppression of migration of PCa cells.

DIM and ENZ co-treatment augments the suppression of PCa cell migration (n = 4). Representative images at 12 and 16 h (A), 48 and 72 h (B); wound-healing rate (C). DIM 3,3′-diindolylmethane; ENZ enzalutamide. Values are presented as means ± SD. a,b,cBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
DIM and ENZ co-treatment inhibits Wnt signaling in PCa cells
Wnt signaling is an important mechanism in CRPC. In order to elucidate whether co-treatment with DIM and ENZ inhibits Wnt signaling, western blot analysis was employed to detect the expressions of β-catenin, APC, and GSK3β, which play important roles in Wnt signaling. After 48 h of treatment, exposure to DIM and ENZ in combination significantly increased the expressions of APC and GSK3β, and downregulated the expression of β-catenin (Fig. 5; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly upregulated the level of GSK3β and decreased the expression of β-catenin (Fig. 5; p < 0.05 vs. 10 μM ENZ).

Inhibition of Wnt signaling by DIM and ENZ co-treatment in 22Rv1 cells at 48 h. Representative western blot image (A); quantitative results for β-catenin (B), APC (C), and GSK3β (D). DIM 3,3′-diindolylmethane; ENZ enzalutamide. Values are presented as means ± SD. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
After 72 h, combined treatment with DIM and ENZ significantly increased the expressions of APC and GSK3β (Fig. 6A–C; p < 0.05 vs. control), but there was no significant difference in the expression of β-catenin (Fig. 6A,D). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly upregulated the level of GSK3β and decreased the expression of β-catenin (Fig. 6; p < 0.05 vs. 10 μM ENZ). The results demonstrated that combined treatment with DIM and ENZ may inhibit Wnt signaling.

Inhibition of Wnt signaling by DIM and ENZ co-treatment in 22Rv1 cells at 72 h. Representative western blot image (A); quantitative results for β-catenin (B), APC (C), and GSK3β (D). DIM 3,3′-diindolylmethane; ENZ enzalutamide. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
DIM and ENZ downregulate AR and AR-v7 expressions
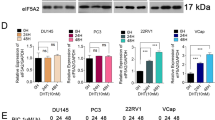
Aberrant AR signaling is considered a critical mechanism in CRPC. Moreover, a mechanism of cross-talk between Wnt and AR signaling has been identified. Therefore, to elucidate whether AR signaling is inhibited when Wnt signaling is inhibited, western blot analysis was employed to detect the expression of AR. After 48 h of treatment, 40 μM ENZ significantly increased the expression of AR (Fig. 7A; p < 0.05 vs. control); however, combined treatment with DIM and 40 μM ENZ significantly decreased the level of AR (Fig. 7A; p < 0.05 vs. 40 μM ENZ). After 72 h of treatment, exposure to DIM and 40 μM ENZ significantly downregulated the AR expression (Fig. 7B; p < 0.05 vs. control).

DIM and ENZ downregulate AR and AR-v7 protein expressions. AR expression at 48 (A) and 72 h (B); AR-v7 expression at 48 (C) and 72 h (D). DIM 3,3′-diindolylmethane; ENZ enzalutamide. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
Besides AR overexpression, a high AR-v7 expression has been found in ENZ-resistant PCa, and AR-v7 may also activate AR signaling. Therefore, to examine the effect of DIM on AR-v7 expression, western blot analysis was used to detect the protein expression of AR-v7 in 22Rv1 cells. After 48 h of treatment, exposure to ENZ alone or in combination with DIM significantly decreased the AR-v7 expression (Fig. 7C; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly downregulated the level of AR-v7 (Fig. 7C; p < 0.05 vs. 10 μM ENZ). After 72 h of treatment, 40 μM ENZ and combined treatment with DIM and ENZ significantly decreased the AR-v7 expression (Fig. 7D; p < 0.05 vs. control). The results indicated that co-treatment with DIM and ENZ may inhibit AR signaling by downregulating the expressions of AR and AR-v7.
DIM and ENZ treatment alter EMT-related mRNA and protein expressions in PCa cells
The process of EMT plays a pivotal role in CRPC and is related to drug resistance. Moreover, Wnt signaling and AR signaling activate EMT in CRPC. To investigate the role of EMT in CRPC, qPCR was performed to examine the mRNA expressions of an epithelial marker (E-cadherin, CDH1) and mesenchymal markers (Fibronectin and Vimentin, FN1 and VIM). After treatment for 48 h, exposure to DIM or ENZ alone, and combined treatment with DIM and ENZ, significantly upregulated the expression of CDH1 (Fig. 8A; p < 0.05 vs. control). On the other hand, cells treated with DIM alone and co-treated with DIM and ENZ exhibited significantly decreased expressions of FN1 and VIM, but cells treated with 10 μM ENZ alone exhibited a significantly increased expression of FN1 (Fig. 8A; p < 0.05 vs. control). Exposure to 40 μM ENZ significantly downregulated the level of FN1 but increased the expression of VIM (Fig. 8A; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly upregulated the level of CDH1 and downregulated the levels of FN1 and VIM (Fig. 8A; p < 0.05 vs. 10 μM ENZ).

DIM and ENZ treatment alter EMT-related gene expressions in 22Rv1 cells at 48 (A) and 72 (B) h (n = 3). DIM 3,3′-diindolylmethane; ENZ enzalutamide; VIM Vimentin; FN1 Fibronectin; CDH1 E-cadherin. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
After treatment for 72 h, exposure to DIM alone and combined treatment with DIM and ENZ significantly upregulated the expression of CDH1, but cells treated with ENZ alone exhibited a significantly decreased level of CDH1 (Fig. 8B; p < 0.05 vs. control). Exposure to DIM or ENZ alone, and co-treatment with DIM and ENZ, significantly inhibited the FN1 expression, but only ENZ treatment and combined treatment decreased the VIM expression (Fig. 8B; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly upregulated the level of CDH1 and downregulated the levels of FN1 and VIM (Fig. 8B; p < 0.05 vs. 10 μM ENZ).
Furthermore, western blot analysis was employed to detect the protein expressions of E-cadherin (E-cad), Fibronectin (FIN) and Vimentin (VIM). After treatment for 48 h (Fig. 9A), exposure to DIM or 10 μM ENZ alone, and combined treatment with DIM and 40 μM ENZ, significantly upregulated the expression of E-cad (Fig. 9C; p < 0.05 vs. control). In contrast, cells treated with DIM alone and co-treated with DIM and 40 μM ENZ had significantly decreased expressions of FIN and VIM (Fig. 9B,D; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with DIM and ENZ significantly downregulated the expression of FIN (Fig. 9B; p < 0.05 vs. 10 μM ENZ).

DIM and ENZ treatment alter EMT-related protein expressions in 22Rv1 cells at 48 h. Representative western blot image (A); quantitative results for FIN (B), E-cad (C), and VIM (D). DIM 3,3′-diindolylmethane; ENZ enzalutamide; VIM Vimentin; FIN Fibronectin; E-cad E-cadherin. Values are presented as means ± SD. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
After treatment for 72 h (Fig. 10A), exposure to DIM and 10 μM ENZ alone and combined treatment with DIM and ENZ significantly increased the expression of E-cad (Fig. 10C; p < 0.05 vs. control). Exposure to DIM and 40 μM ENZ alone, and co-treatment with DIM and ENZ, significantly inhibited the FIN expression, but only combined treatment decreased the VIM expression (Fig. 10B,D; p < 0.05 vs. control). Compared with treatment of 22Rv1 cells with 10 μM ENZ alone, co-treatment with ENZ and DIM significantly decreased the expressions of FIN and VIM (Fig. 10B,D; p < 0.05 vs. 10 μM ENZ). The results revealed that co-treatment with DIM and ENZ possibly inhibits EMT and improves drug resistance.

DIM and ENZ treatment alter EMT-related protein expressions in 22Rv1 cells at 72 h. Representative western blot image (A); quantitative results for FIN (B), E-cad (C), and VIM (D). DIM 3,3′-diindolylmethane; ENZ enzalutamide; VIM Vimentin; FIN Fibronectin; E-cad E-cadherin. Values are presented as means ± SD. a,b,cBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
DIM and ENZ treatment do not promote cell cytotoxicity in PCa cells
To ascertain whether combined treatment with DIM and ENZ had a cytotoxic effect on PCa cells, 22Rv1 cells were treated with DIM and ENZ alone and together for 48 and 72 h. The results showed that treatment with DIM for 48 and 72 h significantly inhibited PARP and c-Caspase 3, respectively, and treatment of 22Rv1 cells with DIM and ENZ together for 48 and 72 h significantly decreased the levels of PARP, c-PARP, Caspase 3 and c-Caspase 3 (Figs. 11 and 12; p < 0.05 vs. control). The results showed that ENZ and DIM treatment may not enhance cell cytotoxicity.

No evident cell cytotoxicity in 22Rv1 cells treated with DIM and ENZ for 48 h. Representative western blot image (A); quantitative results for PARP (B), C-PARP (C), Caspase 3 (D) and C-Caspase 3 (E). DIM 3,3′-diindolylmethane; ENZ enzalutamide; PARP poly (ADP-ribose) polymerase; C-PARP cleaved poly (ADP-ribose) polymerase; C-Caspase 3 cleaved caspase 3. Values are presented as means ± SD. a,b,cBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).

No evident cell cytotoxicity in 22Rv1 cells treated with DIM and ENZ for 72 h. Representative western blot image (A); quantitative results for PARP (B), C-PARP (C), Caspase 3 (D) and C-Caspase 3 (E). DIM 3,3′-diindolylmethane; ENZ enzalutamide; PARP poly (ADP-ribose) polymerase; C-PARP cleaved poly (ADP-ribose) polymerase; C-Caspase 3 cleaved caspase 3. Values are presented as means ± SD. a,b,c,dBars without the same letters at the top indicate statistically-significant differences between treatments when compared with each other (one-way ANOVA, Duncan’s new multiple range test, p < 0.05).
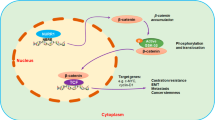
According to these results, we attempted to outline a possible model to elucidate the role of DIM in an ENZ-resistant prostate cancer cell line (Fig. 13). The cell treatment with 10 μM ENZ is as a model of ENZ resistant control group. Our results showed that co-treatment with DIM and ENZ resulted in the strongest inhibition of cell proliferation, colony formation, invasion and migration. Furthermore, co-treatment with DIM and ENZ significantly increased the AR and AR-v7 protein expressions to inhibit AR signaling (vs. control and 10 μM ENZ). In addition, treatment with DIM and ENZ for 72 h significantly increased the expressions of APC and GSK3β, and decreased the expression of β-catenin (vs. control), which indicated that co-treatment suppressed Wnt signaling. Both signaling pathways are related to the progression of EMT, and the results showed that co-treatment significantly increased the expression of E-cadherin and decreased the expressions of Fibronectin and Vimentin to suppress EMT (vs. control and 10 μM ENZ).

Co-treatment with DIM and ENZ regulates Wnt and AR signaling. AR androgen receptor, ARE androgen receptor element, DHT dihydrotestosterone, APC adenomatous polyposis coli protein, GSK3 glycogen synthase kinase‑3, TCF/LEF T-cell factor/lymphoid enhancer factor transcription factors.
Discussion
The 22Rv1 cell line is a CRPC cell line that expresses AR and has the highest expression of AR-v7 as compared with other CRPC cell lines37,38. In comparison with these other cell lines, 22Rv1 cells have higher levels of expression of AR and AR-v7, the other cells barely expressing AR-v7 at all. In the present study, we aimed to investigate whether inhibition of AR and AR-v7 expressions by DIM is a main cause of enzalutamide resistance. Therefore, we employed 22Rv1 cells as the main model in this study.
The results of previous studies and the present study showed that treatment of 22Rv1 cells with ENZ (0–40 μM) did not significantly inhibit cell proliferation, proving that 22Rv1 is ENZ-resistant39,40. Therefore, in this study, we used 22Rv1 cells to ascertain whether DIM treatment could improve ENZ resistance. According to previous studies, DIM suppresses cell proliferation in various cancers, including prostate cancer, breast cancer and ovarian cancer41,42. Exposure of LNCaP, DU145, PC-3, C4-2B and 22Rv1 PCa cell lines to DIM or BR-DIM was found to reduce the cell viability43,44,45,46. Moreover, BR-DIM inhibited PCa cell invasion and proliferation by decreasing platelet-derived growth factor expression and activity47. In addition, Admad et al.48 showed that treatment of PC-3 cells with 25 μM BR-DIM downregulated uPA by decreasing the expressions of VEGF and MMP-9 to inhibit cell proliferation, invasion and migration. The results of this study showed that DIM had significant anticancer effects on 22Rv1 cells, and combined treatment with DIM and ENZ had stronger effects in terms of inhibiting cancer cell proliferation, colony formation, invasion and migration. DIM re-sensitizes ENZ-resistant cells to ENZ treatment, proving that it may be able to be developed as an adjuvant therapy for PCa.
In the clinical treatment of PCa, repressing AR signaling is a critical strategy6,7. ENZ is one of the newer and potentially more effective AR-targeting agents49; however, the response to ENZ is not permanent, and almost all patients eventually develop resistance to ENZ, demonstrating that targeting AR signaling alone is not sufficient for CRPC therapy6,10. Lee et al.50 revealed that Wnt signaling might be active in PCa cells after ADT, partly due to enforcement of interaction between β-catenin and TCF. In androgen-independent PCa cell lines, the combination of a GSK3β inhibitor or APC knockdown with ENZ increases growth inhibition through repressing both AR signaling and Wnt signaling50. In addition, overexpressions of AR and CTNNB1 (β-catenin) were found in ENZ-resistant PCa cells, and the combination of β-catenin inhibitor ICG001 with ENZ improved ENZ resistance22. A previous study indicated that exposure of LNCaP and C4-2B PCa cells to 50 μM BR-DIM decreased the phosphorylation of GSK3β, and then inhibited nucleus translocation of β-catenin; in addition, it caused FOXO3a to bind to the p27kip1 promoter rather than the AR promoter51. Leem et al.52 showed that treatment of colon cancer cells with DIM downregulated the expressions of β-catenin, Myc and FOS, which are related to cancer prevention and prognosis. In gastric cancer cells, treatment with different concentrations of DIM produces contrary results. A high level of DIM (30 μM) resulted in inhibition of cell proliferation, but a low level (1 and 10 μM) activated Wnt4 signaling, which enhanced the progression of gastric cancer53. In the present study, we decided to use a high level of DIM to avoid any adverse influence, and our results showed that DIM did not significantly inhibit Wnt signaling, but combined treatment with DIM and ENZ significantly inhibited Wnt signaling through upregulating the expressions of APC and GSK3β and downregulating the β-catenin expression.
In addition, there is a connection between AR signaling and Wnt signaling. In normal prostate epithelial cells, androgen modulates the activation of AR signaling19,28. However, in PCa, the increased level of β-catenin leads to the formation of β-catenin–AR complexes, and the formation of β-catenin–TCF complexes is preferred under the condition of absence of androgen, which express target genes, including AR, c-myc, MMP7, and VEGF19,28. DIM has been proven to be a strong androgen antagonist that is a competitive inhibitor of DHT binding to AR54. DIM binds to AR directly, and then inhibits nucleus translocation of AR and transcription activation54. However, Palomera-Sanchez et al.55 treated LNCaP cells with DIM for 48 h, and the results showed that DIM inhibited the protein expression of AR, but increased the mRNA expression of AR. Although the mRNA expression of AR increased, chromatin modifications at its promoter region had no significant effects55. In our study, DIM downregulated the protein expression of AR, but upregulated its mRNA expression (data not shown), consistent with previous studies. DIM treatment might cause AR turnover by inducing AR-protein instability. Several studies have revealed that AR overexpression and increased expressions of AR-Vs alleviate the efficacy of ENZ and induce progression of PCa11,23,27,56,57. Of the variants, AR-v7 is the most common in patients with ENZ resistance, and activates ligand-independent AR signaling58,59,60. Ong et al.61 showed that exposure of PCa cells to BR-DIM for 96 h significantly downregulated the AR-v7 mRNA expression. However, DIM did not significantly decrease the level of AR-v7 protein expression in this study. Although the effects of DIM on AR-v7 expression were not significant, combined DIM and ENZ treatment significantly inhibited the AR-v7 expression.
Recent studies have indicated that EMT is involved in PCa progression, migration and therapy resistance, but the effects of ADT on EMT are still unclear62. There are several mechanisms, including AR signaling and Wnt signaling, that activate the process of EMT62. Numerous molecular processes mediate the process of EMT, which is associated with loss of epithelial factors (e.g., E-cadherin and zona occludens protein-1) and gain of mesenchymal factors (e.g., N-cadherin, Fibronectin and Vimentin)24. In normal prostate tissue, canonical AR signaling suppresses EMT24; however, a previous study showed that androgen deprivation increased the expressions of AR and AR-Vs, and upregulated EMT markers (ZEB1, Snail, Twist, N-cadherin and Vimentin), which caused therapy resistance63. Li et al.63 reported that BR-DIM increased the E-cadherin expression and decreased the vimentin expression by inhibiting miR-92a expression, and then suppressed the process of EMT. In addition, miR-34a, miR-27b, miR-320m, miR-200 and let-7 were re-expressed by BR-DIM, which then repressed the expressions of AR, AR-Vs and EMT marker45,61,64. In the present study, DIM increased the E-cadherin expression and decreased the expressions of Fibronectin and Vimentin, then inhibited the process of EMT, which was consistent with the results of previous studies. Compared with treatment with ENZ alone, combined treatment with DIM and EMT resulted in stronger inhibition of EMT, which confirmed that DIM may mediate ENZ resistance by inhibiting EMT.
Although our results demonstrated that DIM could inhibit both Wnt signaling and AR signaling, the inhibition effects were not as obvious as those reported in previous studies, which might be because most of those studies used BR-DIM, rather than DIM. DIM is stable under acidic conditions, but the administration of DIM to female Sprague–Dawley rats by the i.p. route has better effects than the oral route65. Therefore, in order to enhance bioavailability, a formulation of DIM (BR-DIM) was developed to contain small particles of DIM within a water-soluble matrix, containing α-tocopherol polyethylene glycol succinate and phosphatidyl choline, by solubility-enhancing microencapsulation technology66. Anderton et al. administered DIM and BR-DIM to mice and then analyzed the concentration of DIM in the plasma and liver, kidney and lung tissues. The results showed that BR-DIM exhibited an approximate 50% higher bioavailability than DIM66. Moreover, in clinical studies, adjuvant intervention in the early stage of PCa has been performed using BR-DIM, and was proven to be an effective method by which to improve the therapeutic outcome34.
In our study, DIM was effective in ameliorating carcinogenesis through modulating AR signaling, Wnt signaling and EMT in ENZ-resistant cells. The findings may have several implications for the use of DIM as a dietary supplement or a therapeutic agent. The results only relate to patients who have already exhibited ENZ resistance, and in future study, cells could be treated with ENZ for a long duration in order to build a model of long-term ENZ resistance to solve the clinical problem. Furthermore, transcriptional activation of ARE and TCF directly influences AR and Wnt signaling, and can therefore be analyzed to confirm the results in future study.
Conclusion
Combined treatment with DIM and ENZ significantly inhibited the proliferation, invasion and migration of 22Rv1 cells. DIM could regulate Wnt signaling to inhibit the recurrence of AR signaling by reducing the expressions of AR and AR-v7. In addition, inhibition of these two signaling pathways is related to suppression of the EMT process. Our results demonstrated that treatment with DIM, or a combination of DIM and ENZ, could potentially represent other ADTs.
Methods
Cell line and reagents
22Rv1 (ATCC CRL-2505) is an androgen-independent PCa cell line that expresses AR and AR-v7, and is often used as a CRPC model. Cells were cultured in RPMI 1640 (Life Technologies/Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Waltham, MA, USA), sodium pyruvate and 0.2% penicillin/streptomycin (Biological Industries, Cromwell, CT, USA) in a humidified incubator containing 5% CO2 at 37 °C. DIM (Sigma-Aldrich, St. Louis, MO, USA) and ENZ (Toronto Research Chemicals, North York, ON, Canada) were dissolved in dimethyl sulfoxide (DMSO) and diluted in medium before use.
Cell proliferation assay (MTS assay)
Cells were seeded at 5 × 104 cells per well in 96-well plates and grown for 24 h, following which different concentrations of DIM and ENZ were added and incubated for 48 and 72 h. Viable concentrations were tested using a CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Durham, NC, USA). Cell viability was analyzed from absorbance readings at 490 nm.
Colony formation
The anchorage independent growth (AIG) assay of colony formation is performed in soft agar, and detects malignant transformation of cells. 7 × 103 cells were mixed with 0.7% melting agar (Invitrogen, Carlsbad, CA, USA), and the mixtures were then placed on a solidified layer of 0.5% agar made from 1% melting agar, 2X RPMI and 20% FBS in a 6-well plate. The plate was incubated for 24 h, and 2 ml RPMI with different concentrations of DIM and ENZ were added. Fresh medium was added every 7 days during the different treatments. After incubation for 28 days, colonies were stained with crystal violet for 1 h, then captured and counted using Cell3iMager Neo (SCREEN Holdings Co., Rolling Meadows, IL, USA).
Wound-healing
A wound-healing assay was employed to evaluate cell migration using a culture insert (ibidi GmbH, Munich, Germany). First, the culture insert was placed on a 24-well plate, and 106 cells were seeded onto the insert. After the cells had grown in the medium until approximate confluence, the culture insert was removed and washed with PBS. Under varying conditions, different treatments were added to the well, and microscopy was employed to capture images at different time points. Images were analyzed using Image J (National Institutes of Health, Bethesda, MD, USA).
Cell migration and invasion assays
Cell migration and invasion assays were performed using Transwell chambers with an 8.0-µm-pore PET membrane (Becton Dickinson Biosciences, San Jose, CA, USA USA). Transwell chambers were coated with 15 μL Matrigel (Becton Dickinson Labware, Bedford, MA, USA) for the invasion assay, while uncoated chambers were used for the migration assay. Cells at densities of 2 × 105 and 105 for the invasion and migration assays, respectively, were resuspended in 300 μL serum-free medium with different treatments and then added to the upper chamber. Then, 800 μL medium containing 10% FBS were added to the lower chamber. After 24 h of incubation, cells were fixed with 1% paraformaldehyde at 4 °C for 24 h and then stained with Giemsa (Merck, Darmstadt, Germany) for 2 min. Images were captured using a microscope, and invaded cells were counted using Image J (National Institutes of Health)16,67,68.
Quantitative polymerase chain reaction (qPCR)
Cells were treated with different concentrations of DIM and ENZ for 48 and 72 h, and then total RNA was extracted using a RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany). cDNA was generated from 2 μg of total RNA using SuperScript III Reverse Transcriptase (Invitrogen, Karlsruhe, Germany). qPCR analysis was performed using 2X SYBR Green I master on a LightCycler 480 system (Roche, Penzberg, Germany). The data were analyzed using the 2−ΔΔCt method. The gene expression levels were normalized to GAPDH, and the results are presented as the relative fold change compared with the control. The sequences of the primers used are presented in Supplementary Table 116,67.
Western blot analysis
After treatment with different concentrations of DIM and ENZ for 48 and 72 h, cells were washed with PBS three times and then lysed in RIPA buffer (Thermo Scientific, Waltham, MA, USA), protease inhibitor and phosphatase inhibitor. The lysates were cleared by centrifugation at 4 °C for 30 min at 14,000 rpm, and the concentrations of proteins were measured using a BCA protein assay kit (PIERCE Biotechnology, Rockford, IL, USA). Equal amounts of the proteins were loaded per well, then separated using 8% SDS-PAGE gels and transferred onto polyvinyl difluoride (PVDF) membranes (Millipore, Burlington, MA, USA). The PVDF membranes were blocked with 5% non-fat milk at room temperature for 1 h and then incubated with primary antibodies (Supplementary Table 2) overnight at 4 °C. Following three washes with 0.1% tris-buffered saline with Tween 20 (TBST) buffer (BioMan, Taiwan), the membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature, and detection was performed using enhanced chemiluminescence (ECL, Bio-Rad, Hercules, CA, USA). Images were captured with SPOT Xplorer and quantitated using Image J (National Institutes of Health)16,67,69.
Statistical analysis
The results are presented as the mean ± SD of at least three experiments. The data were analyzed using one-way analysis of variance (ANOVA) with Duncan’s multiple range test to determine significant differences between groups. These analyses were performed using SAS 9.4 software (SAS Institute Inc., Cary, NC, USA). p < 0.05 was considered to indicate a statistically-significant difference16,67.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Cancer Registry Annual Report, 2015 (Taiwan). (Health Promotion Administration Ministry of Health and Welfare Taiwan, 2017).
Assikis, V. J. & Simons, J. W. Novel therapeutic strategies for androgen-independent prostate cancer: An update. Semin. Oncol. 31, 26–32 (2004).
Harris, W. P., Mostaghel, E. A., Nelson, P. S. & Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Practice Urol. 6, 76–85 (2009).
Longo, D. L. New therapies for castration-resistant prostate cancer. N. Engl. J. Med. 363, 479–481 (2010).
Katzenwadel, A. & Wolf, P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer Lett. 367, 12–17 (2015).
Crawford, E. D., Petrylak, D. & Sartor, O. Navigating the evolving therapeutic landscape in advanced prostate cancer. Urol. Oncol. 35s, S1–s13 (2017).
Beer, T. M. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 371, 424–433 (2014).
Golshayan, A. R. & Antonarakis, E. S. Enzalutamide: An evidence-based review of its use in the treatment of prostate cancer. Core Evidence 8, 27–35 (2013).
Boudadi, K. & Antonarakis, E. S. Resistance to novel antiandrogen therapies in metastatic castration-resistant prostate cancer. Clin. Med. Insights Oncol. 10, 1–9 (2016).
Narayanan, S., Srinivas, S. & Feldman, D. Androgen–glucocorticoid interactions in the era of novel prostate cancer therapy. Nat. Rev. Urol. 13, 47–60 (2016).
Prekovic, S. et al. Molecular underpinnings of enzalutamide resistance. Endocr. Relat. Cancer 25, R545-r557 (2018).
Li, P., Yang, R. & Gao, W. Q. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol. Cancer 13, 55 (2014).
Murillo-Garzon, V. & Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 14, 683–696 (2017).
Shih, Y. L. et al. SFRP1 suppressed hepatoma cells growth through Wnt canonical signaling pathway. Int J Cancer 121, 1028–1035 (2007).
Yen, H. Y. et al. Regulation of carcinogenesis and modulation through Wnt/beta-catenin signaling by curcumin in an ovarian cancer cell line. Sci. Rep. 9, 17267 (2019).
Su, H. Y. et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int. J. Cancer 127, 555–567 (2010).
Chung, M. T. et al. SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecol. Oncol. 112, 646–653 (2009).
Kypta, R. M. & Waxman, J. Wnt/β-catenin signalling in prostate cancer. Nat. Rev. Urol. 9, 418 (2012).
Malcomson, F. C., Willis, N. D. & Mathers, J. C. Is resistant starch protective against colorectal cancer via modulation of the WNT signalling pathway?. Proc. Nutr. Soc. 74, 282–291 (2015).
Reya, T. & Clevers, H. Wnt signalling in stem cells and cancer. Nature 434, 843–850 (2005).
Zhang, Z. et al. Inhibition of the Wnt/beta-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 78, 3147–3162 (2018).
Lu, J., Van der Steen, T. & Tindall, D. J. Are androgen receptor variants a substitute for the full-length receptor?. Nat. Rev. Urol. 12, 137–144 (2015).
Das, R., Gregory, P. A., Hollier, B. G., Tilley, W. D. & Selth, L. A. Epithelial plasticity in prostate cancer: Principles and clinical perspectives. Trends Mol. Med. 20, 643–651 (2014).
Tucci, M. et al. Enzalutamide-resistant castration-resistant prostate cancer: Challenges and solutions. OncoTargets Ther. 11, 7353–7368 (2018).
Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 371, 1028–1038 (2014).
Cao, S., Zhan, Y. & Dong, Y. Emerging data on androgen receptor splice variants in prostate cancer. Endocr. Relat. Cancer 23, T199-t210 (2016).
Pakula, H., Xiang, D. & Li, Z. A tale of two signals: AR and WNT in development and tumorigenesis of prostate and mammary gland. Cancers 9, 14 (2017).
Bitting, R. L., Schaeffer, D., Somarelli, J. A., Garcia-Blanco, M. A. & Armstrong, A. J. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 33, 441–468 (2014).
Higdon, J. V., Delage, B., Williams, D. E. & Dashwood, R. H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 55, 224–236 (2007).
Gerhauser, C. Epigenetic impact of dietary isothiocyanates in cancer chemoprevention. Curr. Opinion Clin. Nutr. Metabolic Care 16, 405–410 (2013).
Giovannucci, E., Rimm, E. B., Liu, Y., Stampfer, M. J. & Willett, W. C. A prospective study of cruciferous vegetables and prostate cancer. Cancer Epidemiol. Biomark. Prevent. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prevent. Oncol. 12, 1403–1409 (2003).
Maruthanila, V. L., Poornima, J. & Mirunalini, S. Attenuation of carcinogenesis and the mechanism underlying by the influence of indole-3-carbinol and its metabolite 3,3’-diindolylmethane: A therapeutic marvel. Adv. Pharmacol. Sci. 2014, 832161 (2014).
Banerjee, S. et al. Attenuation of multi-targeted proliferation-linked signaling by 3,3’-diindolylmethane (DIM): From bench to clinic. Mutat. Res. 728, 47–66 (2011).
Vivar, O. I., Lin, C. L., Firestone, G. L. & Bjeldanes, L. F. 3,3’-Diindolylmethane induces a G(1) arrest in human prostate cancer cells irrespective of androgen receptor and p53 status. Biochem. Pharmacol. 78, 469–476 (2009).
Li, Y. & Sarkar, F. H. Role of BioResponse 3,3’-diindolylmethane in the treatment of human prostate cancer: Clinical experience. Med. Principles Practice Int. J. Kuwait Univ. Health Sci. Centre 25(Suppl 2), 11–17 (2016).
Cunningham, D. & You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2, e17 (2015).
Ma, Y. et al. Droplet digital PCR based androgen receptor variant 7 (AR-V7) detection from prostate cancer patient blood biopsies. Int. J. Mol. Sci. 17, 1264 (2016).
Bai, Y. et al. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide-resistance in castration-resistance prostate cancer. J. Biol. Chem. 294, 9911–9923 (2019).
Khurana, N. et al. Multimodal actions of the phytochemical sulforaphane suppress both AR and AR-V7 in 22Rv1 cells: Advocating a potent pharmaceutical combination against castration-resistant prostate cancer. Oncol. Rep. 38, 2774–2786 (2017).
Bradlow, H. L. Review. Indole-3-carbinol as a chemoprotective agent in breast and prostate cancer. In vivo (Athens, Greece) 22, 441–445 (2008).
Kandala, P. K. & Srivastava, S. K. DIMming ovarian cancer growth. Curr. Drug Targets 13, 1869–1875 (2012).
Garikapaty, V. P., Ashok, B. T., Tadi, K., Mittelman, A. & Tiwari, R. K. 3,3’-Diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem. Biophys. Res. Commun. 340, 718–725 (2006).
Singh-Gupta, V. et al. B-DIM impairs radiation-induced survival pathways independently of androgen receptor expression and augments radiation efficacy in prostate cancer. Cancer Lett. 318, 86–92 (2012).
Kong, D. et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS ONE 7, e33729 (2012).
Chinnakannu, K. et al. Cell cycle-dependent effects of 3,3’-diindolylmethane on proliferation and apoptosis of prostate cancer cells. J. Cell. Physiol. 219, 94–99 (2009).
Kong, D. et al. Mammalian target of rapamycin repression by 3,3’-diindolylmethane inhibits invasion and angiogenesis in platelet-derived growth factor-D-overexpressing PC3 cells. Cancer Res. 68, 1927–1934 (2008).
Ahmad, A. et al. Inactivation of uPA and its receptor uPAR by 3,3’-diindolylmethane (DIM) leads to the inhibition of prostate cancer cell growth and migration. J. Cell. Biochem. 107, 516–527 (2009).
Lorente, D., Mateo, J., Perez-Lopez, R., de Bono, J. S. & Attard, G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol. 16, e279-292 (2015).
Lee, E., Ha, S. & Logan, S. K. Divergent androgen receptor and beta-catenin signaling in prostate cancer cells. PLoS ONE 10, e0141589 (2015).
Li, Y. et al. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3’-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J. Biol. Chem. 282, 21542–21550 (2007).
Leem, S. H., Li, X. J., Park, M. H., Park, B. H. & Kim, S. M. Genome-wide transcriptome analysis reveals inactivation of Wnt/beta-catenin by 3,3’-diindolylmethane inhibiting proliferation of colon cancer cells. Int. J. Oncol. 47, 918–926 (2015).
Zhu, Y. et al. Anti-cancer drug 3,3’-diindolylmethane activates Wnt4 signaling to enhance gastric cancer cell stemness and tumorigenesis. Oncotarget 7, 16311–16324 (2016).
Le, H. T., Schaldach, C. M., Firestone, G. L. & Bjeldanes, L. F. Plant-derived 3,3’-Diindolylmethane is a strong androgen antagonist in human prostate cancer cells. J. Biol. Chem. 278, 21136–21145 (2003).
Palomera-Sanchez, Z. et al. The phytochemical 3,3’-diindolylmethane decreases expression of AR-controlled DNA damage repair genes through repressive chromatin modifications and is associated with DNA damage in prostate cancer cells. J. Nutr. Biochem. 47, 113–119 (2017).
Buttigliero, C. et al. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 41, 884–892 (2015).
Yamamoto, Y. et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin. Cancer Res Off. J. Am. Assoc. Cancer Res. 21, 1675–1687 (2015).
Li, Y. et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73, 483–489 (2013).
Yu, Z. et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 20, 1590–1600 (2014).
Hu, R. et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 72, 3457–3462 (2012).
Kong, D. et al. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate 75, 161–174 (2015).
Montanari, M. et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 8, 35376–35389 (2017).
Sun, Y. et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Res. 72, 527–536 (2012).
Li, Y., Kong, D., Ahmad, A., Bao, B. & Sarkar, F. H. Targeting bone remodeling by isoflavone and 3,3’-diindolylmethane in the context of prostate cancer bone metastasis. PLoS ONE 7, e33011 (2012).
Jellinck, P. H. et al. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem. Pharmacol. 45, 1129–1136 (1993).
Anderton, M. J. et al. Physiological modeling of formulated and crystalline 3,3’-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab. Disposit. Biol. Fate Chem. 32, 632–638 (2004).
Liu, C. Y. et al. Characterization of LMX-1A as a metastasis suppressor in cervical cancer. J. Pathol. 219, 222–231 (2009).
Li, D. Q. et al. Effects of mifepristone on invasive and metastatic potential of human gastric adenocarcinoma cell line MKN-45 in vitro and in vivo. World J. Gastroenterol. 10, 1726–1729 (2004).
You, L. et al. Inhibition of Wnt-1 signaling induces apoptosis in beta-catenin-deficient mesothelioma cells. Cancer Res. 64, 3474–3478 (2004).
Acknowledgements
Our project was partially funded through research Grant TSGH-D-109070 from Tri-Service General Hospital, Taiwan.
Author information
Authors and Affiliations
Contributions
C.-Y.L.: planned, revised, and supervised the writing and concept of the article. C.-W.T.: wrote, edited and revised the article. J.-S.L.: analyzed data and wrote the article. Y.-W.L.: reviewed and edited the article with comments. S.-T.W. and T.-L.C.: reviewed and edited the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsao, CW., Li, JS., Lin, YW. et al. Regulation of carcinogenesis and mediation through Wnt/β-catenin signaling by 3,3′-diindolylmethane in an enzalutamide-resistant prostate cancer cell line. Sci Rep 11, 1239 (2021). https://doi.org/10.1038/s41598-020-80519-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80519-3
This article is cited by
-
Wnt/β-catenin-driven EMT regulation in human cancers
Cellular and Molecular Life Sciences (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
