
Abstract
The SiO2/TiO2/Nb2O5 material was set by the sol–gel method and was characterized by several techniques through thermogravimetric, spectroscopic, and textural analyzes. For the two synthesized materials, the specific surface area was 350.0 and 494.0 m2 g−1 (SiTiNb-A and SiTiNb-B, respectively). An enhance of the crystalline order with the temperature increase of the thermal treatment was observed. Through X-ray Photoelectron Spectroscopy analysis, the binding energy values for the Ti 2p and Nb 3d levels showed the insertion of Ti and Nb atoms in the silica matrix. The Electron Dispersive Spectroscopy analyses also confirmed the high dispersion of the metals presented on the materials surface. The Thermogravimetric Analysis showed weight loss for the of 37.6% (SiTiNb-A) and 29.7% (SiTiNb-B). The presence of the crystalline phases TiO2-anatase and monoclinic-Nb2O5 in the materials was confirmed through the data obtained by association of powder X-ray Diffraction and FT-Raman. Values obtained from optical band-gap aimed the dependence of the oxides concentration and the calcination temperature. Finally, the pyridine adsorption studies have indicated the presence of Lewis and Brønsted acid sites.
Similar content being viewed by others
Introduction
The sol–gel process is a synthesis methodology widely used to obtain inorganic materials, as it is based on the growth of a three-dimensional polymeric silica network, which may or may not be modified by the insertion of other metals in the network, adding new characteristics and properties and allowing applications of these materials in various areas. The sol–gel process has been widely applied in the synthesis of mixed oxides for being a simple and reproducible methodology. Thus, several research groups employ the sol–gel process to obtain oxide mixtures of the type SiO2/MxOy or SiO2/MxOy/NxOy, where MxOy or NxOy are metallic oxides dispersed in the silica matrix such as: Al2O3, TiO2, WO3, Nb2O5, MoO3, CeO2, CaO, Sb2O5 and others1,2,3,4,5,6,7,8,9,10.
The great importance of the sol–gel process is because of the different characteristics and properties obtained by the synthesis of mixed oxides which are unique. The characteristics of the sol–gel process are well known: the reactions operate at low temperatures obtaining materials with high thermal stability, high purity, controlled porosity and high uniformity for the distribution of the metal oxides in the silica matrix (MxOy or NxOy) and increasing the number of active sites on the surface. Besides this, these materials combine the mechanical properties of the silica matrix with the chemical properties of the metal oxides, exhibiting different chemical and physical properties from those observed in the oxide alone, especially because the presence of Brønsted and Lewis acid sites on the oxide mixture3,4,11,12,13,14. Hence, the oxides mixture are interesting materials for various applications3,4: as heterogeneous catalysis2,9,15,16, photoanodes for solar cells6, photocatalytic activity7, optical properties8, bioactive glass10, preconcentration systems of metallic ions11,17,18,19, dye remediation or adsorbent in aqueous solution5,20, adsorption of interest molecules21, development of new chemically modified electrodes22,23, among others.
Recently, Xu et al.24 exhibited the assembly of SiO2/Nb2O5/TiO2–SiO2 broadband antireflective coating by the sol–gel process. Firstly, there was the preparation of the Nb2O5 composite sol, and apart, the preparation of TiO2–SiO2 composite sol to get finally triple-layer broadband antireflective coatings. The methodology by Xu et al. is used to obtain mixed oxides in the shape of films; otherwise from obtaining mixed oxides in the form of micrometric materials with large surface area and porosity, as previously presented.
Differently from Xu et al., the article researching group has contributed to the development and applications of new micrometric ternary oxides obtained by sol–gel process. Among the applications, it can be highlighted that the development of new chemically modified electrodes22,23,25,26 and the preconcentration of metallic ions systems11,17,27,28,29. Based on that, the main purpose of this work is to obtain and characterize SiO2/TiO2/Nb2O5 ternary oxides obtained by the sol–gel process as well to study its properties, for further applications.
Materials and methods
Reagents and solutions
The tetraethylorthosilicate (TEOS, 98%), titanium (IV) butoxide (97%) and niobium (V) pentachloride (99%) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Ethanol (99.8%), HCl (37% v/v) and HNO3 (95% v/v) were purchased from Vetec (Duque de Caxias, RJ, Brazil). Ultra-pure Milli-Q water was used (resistivity > 18.2 MΩ cm−1, 25 °C, Millipore Milli-Q purification system, Billerica, MA, USA).
Synthesis of the SiO2/TiO2/Nb2O5 material
The SiO2/TiO2/Nb2O5 material (designed by SiTiNb) was prepared in two mass proportions, and the samples obtained were SiTiNb-A and SiTiNb-B. The choice theoretical mass percent of TiO2 and Nb2O5 on the materials was based on other studies of the group, to verify this dependence in the properties, such as acidic properties, specific surface area, optical properties, among others. The synthesis procedure for SiTiNb-A material was: 230.0 mL of a 50% (v/v) ethanol/TEOS solution and 7.0 mL of a 6.0 mol L−1 HCl solution were added to a 500.0 mL reactor. The mixture was stirred for 3 h at 353 K to initiate the pre-hydrolysis of the TEOS. After that, 22.0 mL of titanium (IV) butoxide dissolved in 50.0 mL of ethanol were added drop by drop to the mixture, then more 7.0 mL of a 6.0 mol L−1 HCl solution. The mixture was stirred for 1 h at 353 K. In a third stage, 35.0 g of niobium (V) pentachloride dissolved in 50.0 mL of ethanol were added drop by drop and the mixture was stirred for 30 min at 353 K. After, 15.0 mL of a 6.0 mol L−1 HCl solution were added, and the mixture was stirred at 353 K until the formation of the gel. The resulting mixture was transferred to a beaker glass and heated at 363 K until complete evaporation of the solvent and then heated for 4 h in an oven at 363 K to form the xerogel. The obtained product (xerogel) was precipitated and then dried under vacuum (10–5 Pa) at 363 K for 6 h. The resulting particles were washed with ethanol in a Soxhlet extractor for 24 h to remove any metallic oxide that was not incorporated into the silica matrix, precursors residues and possible soluble species. Then, SiTiNb-A material was washed with 100.0 mL of a 0.1 mol L−1 HNO3 solution, followed by some ethanol, ultra-pure water, and ethanol again. Finally, the ternary oxide was dried under vacuum (10–5 Pa) for 6 h at 363 K and stored.
The same synthesis procedure for SiTiNb-B material was adopted but using the following amounts of reagents: 230.0 mL of a 50% (v/v) ethanol/TEOS solution; 45.0 mL of titanium (IV) butoxide dissolved in 50.0 mL of ethanol and 25.0 g of niobium (V) pentachloride dissolved in 50.0 mL of ethanol.
Instrumentation and characterization
The amounts of TiO2 and Nb2O5 in the silica matrix were determined by using Wavelength Dispersive X-ray Fluorescence (WDXRF) on a Bruker AXS equipment (Tokyo, Japan), model S4 Pioneer, operating with Rh tube (E Kα1 = 20.2 keV).
The crystallinity of the thermally treated SiTiNb-A and SiTiNb-B materials was analyzed by powder X-ray Diffraction (XRD). A Bruker AXS D8 Advance instrument (Cu Kα1,α2 radiation, 40.0 kV and 40.0 mA) operating in a Bragg– Brentano θ/θ configuration. The diffraction patterns were collected in a flat geometry with steps of 0.02° and accumulation time of 3 s per step. Finally, the X-ray powder diffraction data were refined following the Rietveld Method with the TOPAS Academic version 5.0 software (Copyright 1992–2012 Alan A. Coelho. Where, for the activation of the program, DLL files are released by Alan).
The analysis of the Specific Surface Area (SBET) of the SiTiNb-A and SiTiNb-B materials was determined by using the BET (Brauner, Emmett and Teller) multipoint method on a Quantachrome Model Nova 1200e (Boynton Beach, USA) instrument. The samples were previously activated at 393 K in vacuum for 24 h and the samples with granulometry of 150.0 μm ≤ x ≤ 300.0 μm were used. The BJH (Barrett–Joyner–Halenda) method was used to obtain the average pore size and volume.
The Thermogravimetric Analysis was performed on a Shimadzu DTG-60 (Kyoto-Japan). The analyzes, in the range of 293 to 1173 K and the temperature scan rate of 5 K min−1, were performed with argon flow rate of 50.0 mL min−1.
Scanning Electron Microscopy (SEM) images were acquired using a JEOL model JSM 6460-LV (Tokyo, Japan) or FEI model Magellan 400 HXR scanning electron microscope at an acceleration voltage of 3.0 kV, 5.0 kV or 10.0 kV according to the analyzed ternary oxide, and 500 × or 1000 × of magnification. For Electron Dispersive Spectroscopy (EDS) analyses, the EDS Noran System Six model 200 (Waltham, USA) was linked to JSM 6460-LV equipment. For the SEM and EDS analyses, the sample was dispersed over double-sided conductive tape on a copper support and coated with gold before the experiment. For this analysis, the materials with a fraction of particle size less than granulometry of 90 μm were used.
The X-ray Photoelectron Spectroscopy (XPS) analysis was performed on the Esca Plus equipment from Omicron Nanotechnology instrument (Germany), in ultra-high vacuum (pressure: 10–9 mbar), using an Mg X-ray source (Κα = 1253.6 eV). The adjusted emission current was of 16 mA at a voltage of 12.5 kV. Survey spectra were obtained with 50.0 eV analyzer pass energy and 0.5 eV step size. The high-resolution spectra were obtained with 40.0 eV pass energy in the analyzer and 0.08 eV steps. The binding energies were referred to the carbon 1 s of high oriented pyrolytic graphite (HOPG) level, set as 284.6 eV. The peak fitting was performed using the CasaXPS Software Version 2.3.13 (License holder: INMETRO, 25250-020 Xerem Duque de Caxias RJ, Brazil).
The SiTiNb-A and SiTiNb-B materials samples were analyzed by powder XRD, FT-RAMAN scattering analyzes and Diffuse Reflectance Spectroscopy (DRS) in the UV–Visible region. For the analyses, the samples were thermally treated, separately, at temperatures of 473, 673, 873, 1073 and 1273 K for 8 h.
Solid-state FT-Raman spectra of the thermally treated SiTiNb-A and SiTiNb-B materials were recorded by a Bruker MultiRam spectrometer (Tokyo, Japan) at room temperature with a germanium detector, maintained at liquid nitrogen temperature. For the measurements, 1064 nm Nd–YAG laser line was used with a resolution of 2 cm−1 in the region of 1200–70 cm−1 and 256 scans at a laser power of 100.0 mW. The samples were measured in the hemispheric bore of an aluminum sample holder.
The value determination of optical band-gap of the thermally treated samples was performed through the Kubelka–Munk function (F(R)) of data interpretation from the DRS and using the best function applicable to each curve according to the software. The measurements were performed on a Cary 5000 Varian UV–Vis–NIR Spectrophotometer, with wavelengths between 190 and 950 nm and magnesium oxide as reference.
The acidic properties of the SiTiNb-A and SiTiNb-B materials were verified using pyridine as a probe molecule to detect Lewis and Brønsted acid sites in the samples. Drops of pyridine were added in the samples and they were dried under high vacuum at different temperatures (room temperature, 373 and 423 K). After dried, the samples were analyzed by Infrared Spectroscopy, obtained with pellets containing 12% of the material and 88% of KBr.
Results and discussions
Structural characterization
The XRF analysis was carried to access whether the synthesis procedure was efficient to obtain the oxide mixture4. The theoretical mass percent of TiO2 and Nb2O5 in the SiTiNb-A material are 10.0 and 30.0 wt.%; and for SiTiNb-B material are 20.0 and 20.0 wt.%. The results obtained for the TiO2 and Nb2O5 amounts in percentage by mass (wt.%) incorporated in the silica matrix, were 4.8 wt.% of TiO2 and 18.4 wt.% of Nb2O5 for SiTiNb-A material and 9.3 wt.% of TiO2 and 11.6 wt.% of Nb2O5 for SiTiNb-B material. Regarding the synthesis, the method showed a lower percentage of titanium oxide and niobium oxide after washing with ethanol in a Soxhlet apparatus with 0.1 mol L−1 HNO3 solution. These values are lower than expected probably due to the reaction medium pH and other factors, which were not favorable to the complete hydrolysis of titanium (IV) butoxide and niobium (V) pentachloride precursors.
As previously reported by Teixeira et al., it is extremely difficult to obtain SiO2/MxOy/NxOy type materials in the desired proportions considering the differences between the hydrolysis kinetics of the precursors in the same reaction media4. In addition to that, it can generate soluble species in the washing process, for example. However, the values obtained are satisfactory to employ the SiTiNb-A and SiTiNb-B materials, and the sol–gel process is a method extremely useful and reproductive for the SiO2/MxOy/NxOy type materials synthesis4.
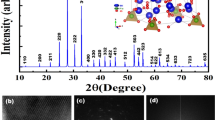
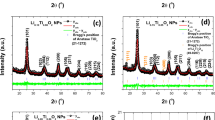
The Fig. 1 shows the diffractograms obtained after heat treatment at different temperatures for the SiTiNb-A (Fig. 1A) and SiTiNb-B (Fig. 1B) materials. It is observed that both materials, up to a temperature of 873 K do not present enough structural order to be observed peaks in the diffractograms and present low crystallinity, showing a peak for 2θ = 23° (around) due to the amorphous halo typical of glassy silica30. However, after 1073 K some peaks begin to appear without having a good definition. At a temperature of 1273 K, can be seen some peaks with greater definition, where the SiTiNb-A sample presents the highest crystallinity at this temperature. Through the values of 2θ of the peaks presented in the diffractograms, when comparing with the ICSD database, it can be identified that beyond the anatase phase of TiO2 ICSD#92,363 of space group I41/amd Z (141) (a = 3.7710(9) Å ; b = 3.7710(9) Å c = 9.430(2) Å; α = β = γ = 90°)31, there is the presence of monoclinic Nb2O5 ICSD#25,750 from space group C 12/m1 (12) (a = 28.51 Å; b = 3.83 Å; c = 17.48 Å; α = 90°; β = 120.8°; γ = 90°)32. A halo between 20° and 27° is also observed in 2θ indicating the presence of SiO2, as previously mentioned. As it will be seen under by analysis of Electron Dispersive Spectroscopy images (EDS), a high dispersion of metal oxides in the silica matrix is observed and it can be attributed to the strong interactions with siloxane groups forming strong covalent bonds. Also, there is a concentration below 10.0 wt.% of TiO2 in both materials. This high dispersion and low concentration of TiO2 in the silica matrix decreases the mobility TiO2 in the silica matrix, making it difficult in the formation of Anatase phase or Rutile at lower temperatures.

X-ray diffractograms for the SiTiNb-A (A) and SiTiNb-B (B) materials thermally treated in the range of 473–1273 K. The peaks marked with the letters A and M in the graph of samples heated to 1273 K indicate the indexed phases of TiO2 Anatase (A) and monoclinic Nb2O5 (M).
The Tables 1 and 2 summarize the crystallographic data obtained using the cell parameter values, crystallite size referring to TiO2 in the anatase phase and monoclinic-Nb2O5 phase obtained with the refinement by the Rietveld Method. In the case of SiTiNb samples, by virtue of their low crystallinity, it was only possible to perform the refinement for the diffractograms of the samples calcined from 1073 K. The Gof (Good of fitness) and Rwp (weighted profile R-factor) values were lower and better than those obtained in the refinement of samples containing niobium, showing a good approximation of the theoretical and experimental diffractograms.
It can be observed that, with the increase in the calcination temperature, there is an increment in the size of the crystallite, while the volume of the unit cell undergoes a small reduction, which is characteristic of the grow in the structural ordering of the sample. However, in the SiTiNb-B sample, there is a slight gain in the volume of the unit cell of the sample calcined at 1073 K in relation to that calcined at 1273 K, indicating a higher amount of anatase sites in relation to monoclinic niobium oxide may be the cause of this reduction (Tables 1, 2, 3).
According to refinement, the relative percentage between the anatase and monoclinic phases varies as specified by Table 3. It is possible to observe that the calcination temperature increase favors the anatase-TiO2 phase and decreases the percentage of the monoclinic-Nb2O5 phase. This behavior occurs in the same proportions in the two samples, demonstrating that it is only an effect of temperature and it is not related to the amount of metal oxide in the matrix of each material.
The BET analyses showed that the specific surface area was 494.0 m2 g−1 for SiTiNb-A and 350.0 m2 g−1 for SiTiNb-B. The BJH method revealed an average pore volume of 0.272 cm−3 g−1 for SiTiNb-A and 0.194 cm−3 g−1 for SiTiNb-B; and the average pore size of 11.0 Å for SiTiNb-A and 11.1 Å for SiTiNb-B, which indicates that these ternary oxides are microporous. These values are important, as it shows that the active sites are accessible in the SiTiNb-A and SiTiNb-B materials.
It can be seen by the adsorption and desorption curves (Fig. 2) for both materials, that the isotherms are type I(b)33, presenting a characteristic curve of microporous materials with a small external surface area34,35. In such cases, it is common for adsorption to occur on the accessible pore volume and not on the internal surface area. There is a small separation between the adsorption and desorption curves, pointing out a possible H4-type hysteresis33, which is normally associated with adsorption in micropores and characteristic of non-linear slits36. However, the BET analyses demonstrate a lower surface area and pore volume for SiTiNb-B, showing that the concentration of TiO2 and Nb2O5 oxides in the silica matrix had a greater influence on the parameters (surface area and pore volume) and probably hardens the structure due to the formation of a dense layer and/or the coalescence of pores7,8.

Nitrogen adsorption–desorption isotherm of SiTiNb-A (A) and SiTiNb-B (B) ternary oxides.
The thermal stability of SiTiNb-A and SiTiNb-B materials is evaluated by Thermogravimetric Analysis (TGA-DTA) (Fig. 3). For the SiTiNb-A material (Fig. 3A), in the temperature variation to approximately 383 K, an endothermic event (exo down) is observed with a mass loss of 21%, related to water desorption. From 383 to 528 K (exothermic event, exo up), it was found a mass loss of approximately 8%, due to the dehydroxylation of the groups –SiOH, –TiOH or –NbOH in virtue of structural water loss37,38. Above the temperature of 528 K, there was a change in the line referring to DTA, which may indicate an event where there is a change in energy generating a mass loss of approximately 4% (endothermic event, exo down). This event can also be seen in Raman's analysis which will be discussed later. In the range between 673 and 1093 K there is a mass loss also of approximately 4% (endothermic event, exo down) related to the loss of organic matter adhered to the sample38,39. Finally, there is a small loss (0.6%) ascribed, possibly, to organic matter aggregated or encapsulated in the pores of the sample40, totaling a mass loss of 37.6%.

Thermogravimetric analysis of SiTiNb-A (A) and SiTiNb-B (B). The graphs show the DTA data (blue line) and the relative weight loss (black line).
For SiTiNb-B material (Fig. 3B), the mass losses are similar, totaling 29.7%. Up to 408 K a mass loss of 15.4% is observed (endothermic event, exo down), related to water desorption37,38. Between 408 and 523 K the mass loss is approximately 5% (exothermic event, exo up) and in the range of 523 K to 698 K the mass loss also corresponds to 5% (endothermic event, exo down)38,39. At 698 K at the end of the process, the mass loss is 4%38,39,40.
As seen in Fig. 4, the Scanning Electron Microscopy (SEM) for SiTiNb-A (Fig. 4A) and SiTiNb-B (Fig. 4B) materials shows the morphology of the particles does not present a uniform size and it presents irregular shape and no-spherical particles. They have been characterized by a flat surface with a rough structure. This characteristic is quite usual for mixed oxides obtained by the sol–gel process3,4. Note that the particle size distribution is quite heterogeneous, which is characteristic for mixed oxides obtained by the sol–gel process; and the distribution cannot be estimated by SEM analysis. But there are particles in the vast majority between 10 and 90 µm. Some particles are even larger than 100 µm, but as rods form, they pass through the mesh of the sieve that was used for particles smaller than 90 µm.

Scanning Electron Microscopy (SEM) micrograph of SiTiNb-A (A) and SiTiNb-B (B) materials.
The Fig. 5 shows the scanning by EDS analysis of Si (Fig. 5B), Ti (Fig. 5C) and Nb (Fig. 5D) for SiTiNb-A material. The EDS shows the TiO2 or Nb2O5 particles are dispersed without phase segregation or formation of islands at the silica matrix, which was a desired feature. In addition, at the magnification level used (1000x), the SiTiNb-A material seems to be homogeneous with high uniformity on its surface. This dispersion is very important because increases the number of acidic sites on the surface or pores of SiTiNb-A material, and it can be attributed to the strong interactions with siloxane groups of the silica matrix forming strong covalent bonds as the –Si–O–Ti–OH, –Si–O–Nb–OH types, as observed by XRD analyzes.

Scanning Electron Microscopy (SEM) micrograph (A) and respective Electron Dispersive Spectroscopy images (EDS) of Si (B), Ti (C) and Nb (D) for SiTiNb-A material. Magnification of 1000x.
The percentage values on the surface of the SiTiNb-A material for the elements Si, Ti and Nb were determined from the 3-point analysis by EDS, which were homogeneous, corroborating with the XRF analysis. It was observed through the SEM and EDS analyzes that the SiTiNb-B material presented the same behavior, so the results were not presented.
The XPS analyses were performed to investigate the chemical states of the elements in the SiTiNb-A and SiTiNb-B materials. The survey spectra (Figs. 6A, 7A) show the presence of O, Ti, Nb and Si elements, which confirm the mixture of SiO2, TiO2 and Nb2O5 oxides on the samples surface, also observed in EDS analyses. It was also observed in spectra the presence of residual carbon, accordingly to Barr and Seal41 is common on XPS analysis due to the impurities on the air or the material of synthesis, such desorbed CO2. The high-resolution spectra in the Ti 2p region for the SiTiNb-A and SiTiNb-B samples (Figs. 6B, 7B), show the peaks at 464.5 and 458.4 eV correspond to the binding energy of components Ti 2p1/2 and Ti 2p3/2 related to Ti (IV) species in TiO242. Furthermore, the sample SiTiNb-A (Fig. 6A) present small peaks in 456.2 and 462.2 eV, according to literature it is related to Ti (III) species in Ti2O3 analogously to the works of Marino et al.43,44,45.

XPS analysis of SiTiNb-A material. Survey Spectrum (A) and the higher resolution spectra in the regions of Ti 2p (B), Nb 3d (C), Si 2p (D) e O 1 s (E).

XPS analysis of SiTiNb-B material. Survey Spectrum (A) and the higher resolution spectra in the regions of Ti 2p (B), Nb 3d (C), Si 2p (D) e O 1 s (E).
In the Nb 3d region for SiTiNb-A and SiTiNb-B samples (Figs. 6C, 7C) were observed peaks located at 211.8 and 208.9 eV corresponding to the components 3d3/2 and 3d5/2 respectively, referring to Nb2O5 and confirming the presence of Nb (V)46,47. The presence of Si (IV) on two samples was confirmed with a centralized peak in 104.7 eV Si 2p region, referring to the Si–O bond in SiO242,48 (Figs. 6D, 7D). The O 1 s region showed all oxides in the SiTiNb-A and SiTiNb-B samples composition, SiO2, TiO2 and Nb2O5 (Figs. 6E, 7E). The regions in 533.2 eV, 531.0 eV and 530.3 eV were related to the oxygen bonds of the Si(IV), Nb(V) and Ti(IV) species, respectively46.
Finally, when comparing the analysis of WDXRF and XPS, where in WDXRF the elements were also quantified in the bulk of the materials, it was observed that there was a higher concentration of Nb2O5 on the materials surface than in the bulk.
Spectroscopic characterization
According to the Raman spectra of thermally treated SiTiNb-A (Fig. 8A) and SiTiNb-B (Fig. 8B) materials, there was an appearance of crystalline phase at 1073 K, these bands being present in the sample without calcining and in those calcined at temperatures below 1073 K. With a higher temperature of calcination, this ordering increases and it can also be noticed in the medium and long distance as data observed in the XRD analyzes. According to the Raman spectra of thermally treated samples and the spectroscopic factor group analysis, the anatase phase presented six vibrational modes active in Raman (D4h19). Thus, for TiO2 in anatase form, there were six active Raman modes (3Eg + 2B1g + A1g), with them being centered at 144 (Eg), 197 (Eg), 399 (B1g), 513 (A1g), 519 (B1g) and 639 cm−1 (Eg)49. Graça and collaborators50 noticed that the Raman displacements for monoclinic Nb2O5 had better defined bands in samples treated from 1173 K, some of these bands being observed for the materials SiTiNb-A and SiTiNb-B at 1073 K. For both phases, there was an overlap of the bands referring to anatase and niobia in the region of 630–650 cm−1. Tables 4 and 5 summarize the Raman displacement values for the most defined peaks of the calcined samples.

Raman spectra of thermally treated for the SiTiNb-A (A) e SiTiNb-B (B) materials at different temperatures.
For the samples of SiTiNb-A, it was observed that up to 473 K there was no characteristic peak formation, except for the peak around 80–70 cm−1 that appeared in all samples. After 673 K the peaks referring to anatase in 513 (A1g), 519 (B1g) and 639 cm−1 (Eg)49, that appeared as broad bands, shifted to the left and lose definition, while the peaks referring to monoclinic Nb2O5 appeared with greater definition at 1273 K. The peaks were more evident in 998, 649, 270 and 115 cm−1, as described in the literature50. Finally, for the samples of SiTiNb-B, there was less definition of the bands in relation to sample A, however, the well-defined peak, at 144 cm−1, referring to the anatase, appears at 873 K. Note that in the 150–100 cm−1 region there was no overlapping of the bands referring to anatase and niobia.
The optical band-gap values were obtained using the Kubelka–Munk (F (R)) function in the Diffuse Reflectance data of the heat-treated samples (Fig. 9) and are shown in Table 6. Nb2O5 is n-type semiconductor with a band-gap of about 3.4 eV51, and that was dependent on oxygen stoichiometry in its structure, varying its band-gap from 3.2 to 4.0 eV. In general, Nb2O5 had a higher conduction band than TiO2 being indicated to obtain open-circuit voltage and efficiency in the conversion of photons. The most thermodynamically stable form of Nb2O5 is the monoclinic arrangement presented in the synthesized samples52. For the TiO2 anatase band-gap is observed in the 3.23 eV region53; and this transition occurring in anatase can be described as an absorption of the valence band (essentially 2p filled O2–) to the conduction band (essentially 3d voids of the Ti).

DRS (Kubelka–Munk function, F (R)) and extrapolation of the tangent line to obtain the optical band-gap of the SiTiNb-A (A) and SiTiNb-B (B) samples treated at different temperatures.
The samples which were not calcinated had a band-gap around 3.3 eV and when the appearance of the TiO2 anatase and Nb2O5 monoclinic phases begun, there was a slight increase in the band-gap followed by its reduction after temperatures of 1073 K. Whereas the monoclinic phase stabilizes, it is obtained a band-gap value of 3.1 eV in 1273 K. Thus, the transition from band 2p (full) of oxygen (O2–) to band 4d (empty) of Nb5+ should occur. It was observed in the spectrum of SiTiNb-A phase a tooth formation (electronic artifact) that masks the proper measurements, requiring deeper analysis to determine minor variations in the band-gap values.
Table 6 shows that the band-gap values did not vary significantly with the different percentages of the crystalline phases. However, in Tables 1 and 2 were shown a variation in the crystallites size of the crystalline phases and a smooth "red-shift" in the energy of band-gap, both in the heat-treated samples.
The acid properties of the SiTiNb-A (Fig. 10A) and SiTiNb-B (Fig. 10B) materials were studied using pyridine adsorbed as probe molecule, which is the most used in the literature54. The characteristic bands for Lewis and Brønsted acid sites can be observed in both materials: The Lewis acid sites are due to the titanium or niobium ions coordinatively unsaturated55,56,57,58, and the Brønsted acid sites are a contribution of Ti–OH or Nb-OH groups, for example11,59,60,61,62.

Infrared spectra of pyridine adsorbed on SiTiNb-A (A) and SiTiNb-B (B) at different temperatures under vacuum: (a) pure SiTiNb, (b) room temperature, (c) 373 K, (d) 423 K.
The weak band at 1575 cm−1 is due to the vibrational mode 8a of the pyridine physically adsorbed by weak forces such as van der Waals. Thus, the pyridine molecule exhibits low interaction with the material, and this band disappears whereas the temperature increases63,64. The bands at 1598 and 1446 or 1447 cm−1, certainly, are due to the 8a and 19b modes of pyridine bonded to the free silanol groups present at the surface of the materials. The –SiOH group of silica matrix by hydrogen bonds seen in the pure SiTiNb ternary oxide treated at room temperature disappears whereas the temperature increases65. The band at 1490 cm−1 possibly corresponds to the vibrational mode 19a. A band at 1544 cm−1 is attributed to the vibrational mode 19b of the pyridine molecule bound to the Brønsted acid sites (–TiOH or –NbOH groups), and this band is not observed for the pure SiO266. The 19b mode of pyridine bound to Lewis acid sites is observed as a small shoulder at approximately 1458 cm−1 that disappears after heat treatment at 423 K. Finally, the weak band detected at 1641 or 1639 cm−1 characterizes the presence of Brønsted acid sites, that decreases while increasing temperature67.
In comparison to the SiO2/TiO2/Sb2O5 (SiTiSb) material, already described by LaDANM Group4, the behavior is different in relation to the SiTiNb material, in this study. It can be reported that in the SiTiSb-A material the presence of the anatase phase is noticed in the sample treated at 473 K. However, in the system with niobium, SiTiNb material, it was only observed the presence of the anatase phase with the appearance of the crystalline oxide phase of monoclinic niobium at 1273 K. This indicates that the SiO2/Nb2O5 network binds more strongly to Ti than the SiO2/Sb2O5 network.
The different structural characteristics and properties showed for the SiTiNb materials with the presence of the Lewis and Brønsted acid sites on its surface made them interesting for use involving application studies, such as adsorbent for metal ions, in photocatalysis, etc. In addition, the band-gap values obtained for both SiTiNb-A and SiTiNb-B show they can be probably used as n-type semiconductors.
Conclusions
The SiTiNb materials obtained by the sol–gel method, showed a high dispersion of metal oxides at the silica matrix, showed no phase segregation, and presented significant Specific Surface Areas (SBET). The XPS analysis indicated the insertion of Ti and Nb atoms in the silica matrix and that antimony is in its higher oxidation state. These materials presented good thermal stability as observed by DRX and TGA‑DTA data. Crystalline phases in the ternary oxides were found only after heating at 1073 K and confirmed by the optical band-gap depend on the oxides concentration and the calcination temperature. The presence of the Lewis and Brønsted acid sites on its surface was confirmed by pyridine test. Due to the characteristics presented of these materials, they are (the most) promising for testing as electrochemical sensors, adsorbent for metal ions and other species in effluents, heterogeneous catalysts and photocatalysis, for instance.
References
Lima, L. F. D., Coelho, C. R., Gomes, G. H. M. & Mohallem, N. D. S. Nb2O5/SiO2 mesoporous monoliths synthetized by sol–gel process using ammonium niobate oxalate hydrate as porogenic agent. J. Sol-Gel Sci. Technhnol. 93, 168–174 (2020).
Vrbkova, E. et al. Contribution to MoO3–SiO2 and WO3–SiO2 utilization-active catalysts in jasmine aldehyde, 2-hexyl-1,3-dioxolane and methyllaurate synthesis. React. Kinet. Mech. Catal. 129, 645–658 (2020).
Silva, C. N. et al. Characterization of silicon-aluminum-zirconium oxide obtained by the sol-gel process. Quim. Nova 42, 513–521 (2019).
Fonseca, B. T. et al. Study of the characteristics, properties and characterization of new SiO2/TiO2/Sb2O5 ternary oxide obtained by the sol–gel process. J. Mater. Sci.: Mater. Electron. 29, 2159–2169 (2018).
Umpierres, C. S. et al. Mesoporous Nb2O5/SiO2 material obtained by sol–gel method and applied as adsorbent of crystal violet dye. Environ. Technol. 38, 566–578 (2017).
Laranjo, M. T. et al. Influence of ball milling on textural and morphological properties of TiO2 and TiO2/SiO2 xerogel powders applied in photoanodes for solar cells. J. Solid State Electrochem. 20, 1731–1741 (2016).
Roldan, M. V., Castro, Y., Pellegri, N. & Duran, A. Enhanced photocatalytic activity of mesoporous SiO2/TiO2 sol-gel coatings doped with Ag nanoparticles. J. Sol-Gel Sci. Technol. 76, 180–194 (2015).
Rocha, L. A., Schiavon, M. A., Ribeiro, S. J. L., Goncalves, R. R. & Ferrari, J. L. Eu3+-doped SiO2–Gd2O3 prepared by the sol-gel process: structural and optical properties. J. Sol-Gel Sci. Technol. 76, 260–270 (2015).
Stuyven, B. et al. Molybdenum–vanadium–antimony mixed oxide catalyst for isobutane partial oxidation synthesized using magneto hydrodynamic forces. Appl. Catal. A 474, 18–25 (2014).
Letaïef, N., Lucas-Girot, A., Oudadesse, H., Dorbez-Sridi, R. & Boullay, P. Investigation of the surfactant type effect on characteristics and bioactivity of new mesoporous bioactive glass in the ternary system SiO2–CaO–P2O5: Structural, textural and reactivity studies. Micropor. Mesopor. Mater. 195, 102–111 (2014).
Tarley, C. R. T. et al. Novel on-line sequential preconcentration system of Cr(III) and Cr(VI) hyphenated with flame atomic absorption spectrometry exploiting sorbents based on chemically modified silica. Talanta 100, 71–79 (2012).
Graham, T. On the properties of silicic acid and other analogous colloidal substances. J. Chem. Soc. 17, 318–327 (1864).
Hench, L. L. & West, J. K. The sol-gel process. Chem. Rev. 90, 33–72 (1990).
Brinker, C. J. & Scherer, G. W. Sol-gel Science—The Physics and Chemistry of Sol-Gel Processing (Academic Press, San Diego, 2001).
Sultana, S. S. P. et al. Promoting effects of thoria on the nickel-manganese mixed oxide catalysts for the aerobic oxidation of benzyl alcohol. Arab. J. Chem. 10, 448–457 (2017).
Serio, M. et al. Valuation of Nb2O5–SiO2 catalysts in soybean oil epoxidation. Catal. Today 192, 112–116 (2012).
Oliveira, L. L. G. et al. Development of new analytical method for preconcentration/speciation of inorganic antimony in bottled mineral water using FIA-HG AAS system and SiO2/Al2O3/SnO2 ternary oxide. Food Chem. 294, 405–413 (2019).
Wutke, N. B. et al. Preconcentration of nickel(II) by a mini-flow system with a novel ternary oxide solid phase and flame atomic absorption spectrometry. Anal. Lett. 49, 723–736 (2016).
Vieira, E. G. et al. Preconcentration and determination of metal ions from fuel ethanol with a new 2,2′-dipyridylamine bonded silica. J. Colloid Interface Sci. 391, 116–124 (2013).
Wawrzkiewicz, M., Wiśniewska, M., Gun’ko, V. M. & Zarko, V. I. Adsorptive removal of acid, reactive and direct dyes from aqueous solutions and wastewater using mixed silica–alumina oxide. Powder Technol. 278, 306–315 (2015).
Wiśniewska, M., Nowicki, P., Bogatyrov, V. M., Nosal-Wiercińska, A. & Pietrzak, R. Comparison of adsorption properties of MgxOy–SiO2 and ZnxOy–SiO2 in the mixed oxide-poly(vinyl alcohol) system. Colloids Surf. A 492, 12–18 (2016).
Giarola, J. F. et al. Development and application of graphite-SiO2/Al2O3/Nb2O5-methylene blue (GRP-SiAlNb-MB) composite for electrochemical determination of dopamine. Arab. J. Chem. 10, 430–438 (2017).
Ribeiro, M. S. et al. On the applicability of SiO2/Al2O3/Nb2O5 and SiO2/Al2O3/TiO2 as a biocompatible platform for chloroperoxidase. Anal. Methods 6, 521–528 (2014).
Xu, S. et al. Low-temperature preparation of SiO2/Nb2O5/TiO2–SiO2 broadband antireflective coating for the visible via acid-catalyzed sol–gel method. Coatings 10, 737 (2020).
Marco, J. P., Borges, K. B., Tarley, C. R. T., Ribeiro, E. S. & Pereira, A. C. Electrochemical study of oxidation process of promethazine using sensor based on carbon nanotubes paste containing immobilized DNA on inorganic matrix. Semin Ciênc Exatas Tecnol 35, 215–230 (2014).
Marco, J. P., Borges, K. B., Tarley, C. R. T., Ribeiro, E. S. & Pereira, A. C. Development of a simple, rapid and validated square wave voltametric method for determination of promethazine in raw material and pharmaceutical formulation using DNA modified multiwall carbon nanotube paste electrode. Sens. Actuator B Chem. 177, 251–259 (2013).
Diniz, K. M. et al. Preparation of SiO2/Nb2O5/ZnO mixed oxide by sol-gel method and its application for adsorption studies and on-line preconcentration of cobalt ions from aqueous medium. Chem. Eng. J. 239, 233–241 (2014).
Lima, G. F. et al. Flow injection on-line minicolumn preconcentration and determination of trace copper ions using an alumina/titanium oxide grafted silica matrix and FAAS. Microchim. Acta 178, 61–70 (2012).
Costa, L. M. et al. Adsorption studies of Cd(II) onto Al2O3/Nb2O5 mixed oxide dispersed on silica matrix and its on-line preconcentration and determination by flame atomic absorption spectrometry. Spectrochim. Acta B 66, 329–337 (2011).
Reed, C., Lee, Y. K. & Oyama, S. T. Structure and oxidation state of silica-supported manganese oxide catalysts and reactivity for acetone oxidation with ozone. J. Phys. Chem. B 110, 4207–4216 (2006).
Weirich, T. E., Winterer, M., Seifried, S., Hahn, H. & Fuess, H. Rietveld analysis of electron powder diffraction data from nanocrystalline anatase, TiO2. Ultramicroscopy 81, 263–270 (2000).
Andersson, S. The crystal structure of N-Nb2O5, prepared in the presence of small amounts of LiF. Z. Anorg. Allg. Chem. 351, 106–112 (1967).
Thommes, M. et al. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 87, 1051–1069 (2015).
Gregg, S. J. & Sing, K. S. W. Adsorption, Surface Area and Porosity (Academic Press Inc, New York, 1982).
Sing, K. S. W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 57, 603–619 (1985).
Li, T. et al. Effect of pore structure on shale oil accumulation in the lower third member of the Shahejie formation, Zhanhua Sag, eastern China: evidence from gas adsorption and nuclear magnetic resonance. Mar. Petrol. Geol. 88, 932–949 (2017).
Llano, B., Hidalgo, M. C., Rios, L. A. & Navío, J. A. Effect of the type of acid used in the synthesis of titania-silica mixed oxides on their photocatalytic properties. Appl. Catal. B 150–151, 389–395 (2014).
Montoya, I. A., Viveros, T., Domínguez, J. M., Canales, L. A. & Schifter, I. On the effects of the sol-gel synthesis parameters on textural and structural characteristics of TiO2. Catal. Lett. 15, 207–217 (1992).
Aronne, A. et al. Sol-gel synthesis and structural characterization of niobium-silicon mixed-oxide nanocomposites. J. Sol-Gel Sci. Technol. 43, 193–204 (2007).
Pabón, E., Retuert, J., Quijada, R. & Zarate, ,. TiO2–SiO2 mixed oxides prepared by a combined sol-gel and polymer inclusion method. Micropor. Mesopor. Mater, 67, 195–203 (2004).
Barr, T. L. & Seal, S. Nature of the use of adventitious carbon as a binding energy standard. J. Vac. Sci. Technol. A 13, 1239–1246 (1995).
Zhang, L. et al. Catalytic reduction of SO2 by CO over CeO2–TiO2 mixed oxides. Trans. Nonferr. Met. Soc. 26, 2960–2965 (2016).
dos Santos, A. et al. A study of the physical, chemical and biological properties of TiO2 coatings produced by micro-arc oxidation in a Ca–P-based electrolyte. J. Mater. Sci. Mater. Med. 25, 1769–1780 (2014).
Marino, C. E. B., Nascente, P. A. P., Biaggio, S. R., Rocha-Filho, R. C. & Bocchi, N. XPS characterization of anodic titanium oxide films grown in phosphate buffer solutions. Thin Solid Films 468, 109–112 (2004).
Bharti, B., Kumar, S., Lee, H.-N. & Kumar, R. Formation of oxygen vacancies and Ti3+ state in TiO2 thin film and enhanced optical properties by air plasma treatment. Sci. Rep. 6, 32355 (2016).
Campos, E. A., Gushikem, Y., Gonçalves, M. C. & Castro, S. C. Preparation and characterization of niobium oxide coated cellulose fiber. J. Colloid Interface Sci. 180, 453–459 (1996).
Denofre, S., Gushikem, Y., Castro, S. C. & Kawano, Y. Stability and surface acidity of niobium(V) oxide grafted on a silica gel surface. J. Chem. Soc. Faraday Trans. 89, 1057–1061 (1993).
Alfonsetti, R., Lozzi, L., Passacantando, M., Picozzi, P. & Santucci, S. XPS studies on SiOx thin films. Appl. Surf. Sci. 70–71, 222–225 (1993).
Choi, H. C., Jung, Y. M. & Kim, S. B. Size effects in the raman spectra of TiO2 nanoparticles. Vib. Spectrosc. 37, 33–38 (2005).
Graça, M. P. F., Meireles, A., Nico, C. & Valente, M. A. Nb2O5 nanosize powders prepared by sol–gel-structure, morphology and dielectric properties. J. Alloys Compd. 553, 177–182 (2013).
Raba, A. M., Bautista-Ruíz, J. & Joya, M. R. Synthesis and structural properties of niobium pentoxide powders: a comparative study of the growth process. Mater. Res. 19, 1381–1387 (2016).
Le Viet, A., Jose, R., Reddy, M. V., Chowdari, B. V. R. & Ramakrishna, S. Nb2O5 photoelectrodes for dye-sensitized solar cells: choice of the polymorph. J. Phys. Chem. C 114, 21795–21800 (2010).
Litter, M. I. Heterogeneous photocatalysis transition metal ions in photocatalytic systems. Appl. Catal. B 23, 89–114 (1999).
Emeis, C. A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. J. Catal. 141, 347–354 (1993).
Corma, A. & García, H. Lewis acids as catalysts in oxidation reactions: from homogeneous to heterogeneous systems. Chem. Rev. 102, 3837–3892 (2002).
Imamura, S., Nakai, T., Kanai, H. & Ito, T. Effect of tetrahedral Ti in titania–silica mixed oxides on epoxidation activity and Lewis acidity. J. Chem. Soc. Faraday Trans. 91, 1261–1266 (1995).
Imamura, S., Nakai, T., Kanai, T. & Ito, T. Titanium sites of titania silica mixed oxides for epoxidation activity and Lewis acidity. Catal. Lett. 28, 277–282 (1994).
McAteer, J. C. Acidic and basic properties of mixed tin–antimony oxides. J. Chem. Soc. Faraday Trans. 1(75), 2762–2767 (1979).
Camposeco, R., Castillo, S., Mejía-Centeno, I., Navarrete, J. & Nava, N. Boosted surface acidity in TiO2 and Al2O3–TiO2 nanotubes as catalytic supports. Appl. Surf. Sci. 356, 115–123 (2015).
Damyanova, S., Petrov, L., Centeno, M. A. & Grange, P. Characterization of molybdenum hydrodesulfurization catalysts supported on ZrO2-Al2O3 and ZrO2–SiO2 carriers. Appl. Catal. A 224, 271–284 (2002).
ZakiMI, H. M. A., Al-Sagheer, F. A. & Pasupulety, L. In situ FTIR spectra of pyridine adsorbed on SiO2-Al2O3, TiO2, ZrO2 and CeO2: general considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf. A 190, 261–274 (2001).
Damyanova, S., Grange, P. & Delmon, B. Surface characterization of zirconia-coated alumina and silica carriers. J. Catal. 68, 421–430 (1997).
Morterra, C. & Cerrato, G. On the use of pyridine adsorption as an analytical tool in surface chemistry. Langmuir 6, 1810–1812 (1990).
Busca, G., Centi, G., Marchetti, L. & Trifiro, F. Chemical and spectroscopic study of the nature of a vanadium oxide monolayer supported on a high-surface-area TiO2 anatase. Langmuir 2, 568–577 (1986).
Benvenutti, E. V., Gushikem, Y., Davanzo, C. U., de Castro, S. C. & Torriani, I. L. Antimony (V) oxide grafted onto a silica gel surface: acidic properties and thermal stability. J. Chem. Soc. Faraday Trans. 88, 3193–3196 (1992).
Lefrancois, M. & Malbois, G. The nature of the acidic sites on mordenite: Characterization of adsorbed pyridine and water by infrared study. J. Catal. 20, 350–358 (1971).
Elassal, Z. et al. Synthesis and FT–IR study of the acido–basic properties of the V2O5 catalysts supported on zirconia. Arab. J. Chem. 4, 313–319 (2011).
Acknowledgements
The authors thank to Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support. The authors wish to acknowledge Fernando Celso Ribeiro Soares Junior for checking the English and Priscila Pio of LACCO-INT for the XRF analysis.
Author information
Authors and Affiliations
Contributions
All authors wrote the main manuscript text and prepared figures. Finally, all authors reviewed the manuscript. The article was written in an equitable manner.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Fonseca, B.T., D’Elia, E., Siqueira Júnior, J.M. et al. Study of the characteristics and properties of the SiO2/TiO2/Nb2O5 material obtained by the sol–gel process. Sci Rep 11, 1106 (2021). https://doi.org/10.1038/s41598-020-80310-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80310-4
This article is cited by
-
Disposable electrochemical sensor based on carbon black and ternary oxide (SiO2/TiO2/Nb2O5) for determination of triclosan in mouthwash and water samples
Journal of Solid State Electrochemistry (2023)
-
Curing treatments of silica gel for fire-retardant glass by intumescence
SN Applied Sciences (2023)
-
Effect of carbonate additive on the microstructure and corrosion resistance of plasma electrolytic oxidation coating on Mg-9Li-3Al alloy
International Journal of Minerals, Metallurgy and Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
