
Abstract
Cypresses are characterized by their longevity and valuable timber. In Taiwan, two endemic cypress species, Chamaecyparis formosensis and C. obtusa var. formosana, are threatened by prevalent illegal logging. A DNA barcode system is urgently needed for reforestation and conservation of these two cypresses. In this study, both plastomes and 35S rDNAs from 16, 10, and 6 individuals of C. formosensis, C. obtusa var. formosana, and C. obtusa var. obtusa were sequenced, respectively. We show that the loss of plastid trnT-GGU readily distinguishes C. formosensis from its congeneric species. We demonstrate that entire sequences of plastomes or 35S rDNAs are capable of correctly identifying cypress species and varieties, suggesting that they are effective super-barcodes. We also discover three short hypervariable loci (i.e., 3′ETS, ITS1, and trnH-psbA) that are promising barcodes for identifying cypress species and varieties. Moreover, nine species-specific indels of > 100 bp were detected in the cypress plastomes. These indels, together with the three aforementioned short barcodes, constitute an alternative and powerful barcode system crucial for identifying specimens that are fragmentary or contain degraded/poor DNA. Our sequenced data and barcode systems not only enrich the genetic reference for cypresses, but also contribute to future reforestation, conservation, and forensic investigations.
Similar content being viewed by others
Introduction
Chamaecyparis, commonly named cypress or false cypress, is a small genus belonging to the Cupressaceae family in cupressophytes (also called conifers II). The cypress genus includes five to six species native to eastern Asia (Japan and Taiwan) and the western and eastern margins of North America1,2. Cypresses are characterized by their longevity and rot-resistant timbers, which are widely used in the construction of palaces, temples, and shrines as well as in furniture making. In Taiwan, two endemic cypress taxa, Chamaecyparis formosensis and C. obtusa var. formosana, are ecologically and economically important. They are montane conifers dominating cloud forests at 1500–2500 m above sea level3. Unfortunately, the IUCN red list regards them as vulnerable or endangered species4. It was estimated that ~ 60% of Taiwan’s cypress forests were logged during the early twentieth century5. Although logging of any primary forests is now prohibited in Taiwan, illegal logging is frequently reported and seriously damages not only the cypress populations but also the entire ecosystem of the cloud forest, where many stout and tall cypresses have survived for several thousand years.
Precise identification of seeds, seedlings, and timbers is vital for reforestation, and provides scientific support for prosecuting illegal logging crimes. Adult leaf tips can be used to distinguish C. formosensis from C. obtusa var. formosana, as the former’s are sharply pointed and the latter’s are abruptly acute (Fig. 1). In contrast, the winged seeds of both taxa are highly similar in size and morphology (Fig. 1), making it difficult to separate their seeds for managed reforestation. Timber identification based on wood anatomy relies on professional knowledge and practices6,7. Moreover, it is difficult to reliably identify species based on wood anatomical analyses8. In contrast, genetic approaches, such as DNA barcoding, offer an alternative and more straightforward line of evidence for timber identification9,10. Therefore, a comprehensive set of DNA references should be established prior to reliable identification11,12.

Photos of adult-leaves and winged seeds of C. formosensis (a) and C. obtusa var. formosana (b). The scale-bar unit is 1 mm.
DNA barcoding, which generally requires a small and recoverable DNA segment, has recently attracted much attention in the fields of systematics, ecology, bio-conservation, and forensic investigation13. For example, plastid rbcL and matK were designated as core barcodes of land plants14, while the plastid trnH-psbA and nuclear ITS loci were suggested to be supplementary to these core barcodes15,16. Molecular technology provides authentic genetic information for species identification without requiring morphological characters17. However, none of the above-mentioned loci has ever been used to identify cypress species.
With high-throughput NGS data and advances in genomic assembly and analytical tools, utilization of the entire plastome or 35S rDNA sequence as a super-barcode was previously proposed to be a cost-effective way to discriminate species and evaluate phylogenies18,19,20. This super-barcoding approach is also encouraged to avoid problems of primer specificity, PCR amplification rate, and loss or duplication of the corresponding loci21. In addition, deciphering plastomes allows researchers to exploit hypervariable loci and lineage-specific indels, which have been demonstrated to be very efficient at discriminating orchid22,23 and yew24 species, respectively.
Despite the ecological and economic importance of cypresses, their DNA barcodes and associated references have not been investigated or established. In this paper, we address two questions: (1) Can we use whole plastomes and 35S rDNA as effective super-barcodes for identifying cypress species and varieties? and (2) If yes, then do they contain hypervariable loci that can be used as promising barcodes? To address these questions, we sampled 16 C. formosensis individuals and 10 C. obtusa var. formosana individuals from natural populations scattered across the cloud forests of Taiwan. Six cultivated individuals of another C. obtusa variety, C. obtusa var. obtusa, were also sampled from two remote localities. Sampling of multiple individuals per species/variety allowed us to evaluate identification rates based on the degree of species/variety-level monophyly in tree methods. We reconstructed both plastomes and 35S rDNAs from shallowly sequenced total DNA of the sampled cypresses using an approach known as genome skimming25. Our results reveal that the entire sequences of plastomes or 35S rDNAs not only serve as an effective super-barcode, but also contain short hypervariable loci and long lineage-specific indels that together constitute an alternative and powerful barcode system for cypress identification at both interspecies and inter-variety levels.
Results
Characteristics of cypress plastomes and nuclear 35S rDNAs
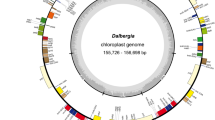
The plastomes of C. formosensis, C. obtusa var. formosana, and C. obtusa var. obtusa are GC-poor and mapped as circular molecules. Their lengths range from 126.8 to 127.8 kb (Table 1). Similar to C. hodginsii and C. lawsoniana, they lack the canonical repeat pair usually present in other seed plant plastomes. Notably, all cypress plastomes elucidated so far share a trnQ-containing repeat, which was previously called "trnQ-IR" in other cupressaceous species26,27,28. An approximately 20-kb region flanked by trnQ-IRs is inverted in C. hodginsii and C. lawsoniana compared to C. formosensis and C. obtusa varieties (Fig. 2). This suggests that the trnQ-IR remains active to trigger homologous recombination and generation of plastomic inversions in these cypresses. The plastid gene number is slightly variable among cypresses. For example, trnT-GGU is only missing in C. formosensis (Fig. 2). As a result, the absence of trnT-GGU readily distinguishes C. formosensis from other cypresses.

Circular maps of Chamaecyparis plastomes (above closed circle) and nuclear 35S rDNA (below open circle). Average inter-species and inter-variety pairwise substitution rates are depicted by histograms with gray-rose and steel-cyan colors, respectively. Repeat pairs are linked with lines. The region highlighted with blue is inverted in C. hodginsii and C. lawsoniana. A transfer RNA gene, trnT-GGU (bold), is uniquely missing in C. formosensis. Eight hypervariable loci are denoted by thick grey bars and Arabic numerals.
The nuclear 35S rDNA sequences of the sampled 32 cypresses were assembled into linear molecules encompassing seven known genetic loci in the following order: 5′ETS, 18S, ITS1, 5.8S, ITS2, 26S, and 3′ETS (Fig. 2). These 35S rDNAs are 8.9‒10.1 kb long, with GC contents over 50% (Table 1).
Plastomes and 35S rDNAs as effective super-barcodes
We evaluated the entire plastomes and 35S rDNA sequences to identify cypresses using maximum-likelihood tree approaches under the 50% majority-rule consensus. The plastome- and 35S rDNA-based trees share three monophyletic groups, each comprising individuals from the same species or varieties (Fig. 3). As a result, both plastome and 35S rDNA sequences achieved 100% identification rates at both inter-species and inter-variety levels.

ML trees inferred from the entire plastome (a) and 35S rDNA (b) sequences. The trees are condensed under the 50% majority-rule consensus. Bootstrap values estimated from 1000 pseudo-replicates are shown at the nodes. Taxa are indicated by their voucher numbers, and population localities are denoted by letters within parentheses.
We sampled one to two individuals from nine and six natural populations of C. formosensis and C. obtusa var. formosana, respectively (Supplementary Fig. 1). A 100% identification rate was not achieved at the inter-population level because individuals in the same population did not always form monophyletic groups in the plastome- (Fig. 3a) and 35S rDNA-based (Fig. 3b) trees. We did not estimate the inter-population identification rate of C. obtusa var. obtusa because the specimens were collected from reforestation areas or botanic gardens (Supplementary Table 1). Collectively, our results indicate that the entire sequences of both plastomes and 35S rDNAs are excellent super-barcodes capable of effectively identifying species and varieties, but not populations, of cypress.
Nucleotide substitution rates are heterogeneous across plastomes and 35S rDNAs
Our sliding window analyses across the plastomes show that plastid nucleotide substitution rates (NSRs) vary from 0 to 0.667 and 0 to 0.036 substitutions per site at the inter-species and inter-variety levels, respectively (Fig. 2; Supplementary file 1). Variations in inter-species and inter-variety NSRs in the 35S rDNAs are 0‒0.483 and 0‒0.278 substitutions per site, respectively (Fig. 2; Supplementary file 1). These data highlight the fact that cypress plastomes and 35S rDNAs contain heterogeneous NSRs at both inter-species and inter-variety levels. We noted that the degree of plastid NSR heterogeneity dropped drastically at the inter-variety level. As a consequence, the 35 rDNAs are 7.72 times higher than plastomes in terms of the loci containing the highest inter-variety NSRs. Therefore, in cypresses, 35S rDNAs are likely more vulnerable to mutations than plastomes at the inter-variety level.
Inter-species and inter-variety NSRs estimated from each sliding window were also compared to assess their relationships. We obtained 0.225 and 0.26 of Pearson’s correlation coefficients for the plastomes (Fig. 4a) and 35S rDNAs (Fig. 4b), respectively. This weak correlation suggests that the NSR evolution is not strongly linked between the inter-species and inter-variety levels, and that specific hypervariable loci may be required to identify cypresses at different taxonomic levels.

Estimated relationships between inter-species and inter-variety pairwise NSRs in the cypress plastomes (a) and 35S rDNAs (b).
ITS1 is a very promising barcode
Eight hypervariable loci were identified based on the estimated NSRs (Fig. 2). Seven of them (5′ETS1, 5′ETS2, 3′ETS, ITS1, accD-ycf1, trnE-psbD, and 16S-trnV) feature high inter-species NSRs, while the last one (5′rps12-clpP) has a relatively higher inter-variety NSR than other plastid loci. These loci are all noncoding regions that range from 524 to 1,835 bp in length (Table 2). We separated the 5′ETS region into 5′ETS1 and 5′ETS2 loci to exclude long repeats inside the loci (Fig. 2). We also included four loci that were previously reported to be promising barcodes. For example, the locus of trnH-psbA is a widely acceptable barcode in diverse plant lineages29,30,31. Two loci, matK and rbcL, constitute the core barcodes for land plants14. Meanwhile, the locus of accD was reported to be a promising barcode that can be used with the plastome super-barcode to identify yews24. Table 2 demonstrates that all 12 examined loci have a clear barcoding gap at the inter-species level because there is no overlap between inter-species and intra-species NSRs. In contrast, inter-variety barcoding gaps are observed only in the loci of 5′ETS1, ITS1, trnH-psbA, and rbcL.
Unrooted neighbor-joining trees inferred from each of the 12 loci were generated to identify the cypresses at both inter-species and inter-variety levels. Under the 50% majority-rule consensus, these 12 loci congruently suggest that all sampled C. formosensis individuals constitute a monophyletic group with strong bootstrap values ranging from 99 to 100% (Table 2). However, only the 3′ETS, ITS1, trnH-psbA, and accD loci successfully resolved the two C. obtusa varieties as separate monophyletic groups. When the bootstrap values, locus lengths, and existence of barcoding gaps are taken into account, ITS1 appears to be the most promising barcode capable of effectively identifying both cypress species and varieties.
Long indels for species discrimination
Lineage-specific indels are a straightforward tool for distinguishing species. We identified five and 287 plastid indels as variety- and species-specific, respectively (Supplementary Table 2). These indels occur mostly in intergenic spaces (76.7%), followed by coding regions (15.4%) and introns (7.9%). In addition, all variety-specific indels are located in the plastid intergenic spaces except for one, which is located in the ycf1 gene. Although the majority (74.4%) of the plastid indels are shorter than or equal to 10 bp, nine species-specific indels are longer than 100 bp (Fig. 5a). These long plastid indels are useful markers for separating C. formosensis from C. obtusa varieties because they are readily detectable using PCR gel electrophoresis.

Summary of species-specific (black bar) and variety-specific (grey bar) indels found in the cypress plastomes (a) and 35S rDNAs (b).
We detected 45 indels in 35S rDNAs. They are species-specific, shorter than 100 bp (Table S3; Fig. 5b; Supplementary Table 3), and located in the 3′ETS (51.1%), 5′ETS (40%), ITS1 (4.4%), and ITS2 (4.4%).
Discussion
We obtained 3.03‒3.76 Gb of NGS sequences for each of the 32 sampled cypresses (Supplementary Table 1). Our assemblies of the complete plastomes and full-length 35S rDNA sequences suggest that genome skimming is a cost-effective and robust strategy for assembling the two sequences in cypresses, whose genomes are huge (estimated to be 17.1‒18.6 pg/2C)32. We were not able to recover high-quality mitochondrial contigs from NGS reads, perhaps because (1) the copy number of mitochondrial DNAs is relatively low33,34 and (2) seed plant mitochondria frequently undergo inter- and intra-genomic recombination, generating a complicated set of subgenomes35,36,37. Nonetheless, our study clearly demonstrates that both the plastome and 35S rDNA are valuable resources for developing effective barcodes to identify cypress species and varieties.
Previous studies have used entire plastome sequences to identify various seed plant species, and argued that plastomes are effective super-barcodes20,23,24,38,39. Our analysis further confirms that entire plastomes are effective super-barcodes for identifying both cypress species and varieties (Fig. 3a). In addition, using 35S rDNAs as super-barcodes also yielded a 100% identification rate at both the inter-species and inter-variety levels (Fig. 3b). This finding further suggests that the 35S rDNAs are more cost-effective super-barcodes than plastomes since the former is approximately 13 times shorter than the latter in the cypresses (Table 1).
Plastomic rearrangements between congeneric species, albeit rare in seed plants, have been documented in some lineages, such as Pelargonium and Hypseocharis40, Podocarpus41, Juniperus26, Calocedrus28, and Halamphora42. Recently, plastomic rearrangements were found in conspecific individuals of yews, likely due to recurrent shifts between predominant and substoichiometric isomeric plastomes24. In this study, we detected a 23-kb plastomic inversion between cypress species (Fig. 2). This non-collinear genome organization impedes the performance of entire plastome alignments, thus decreasing the feasibility of utilizing plastomes as super-barcodes. In contrast, the component and organization of 35S rDNAs are highly uniform across the land plant kingdom43, thus making it a good candidate for DNA barcoding outside of cypresses too.
However, using plastomes and 35S rDNAs as super-barcodes has some limitations. First, the assembly of plastomes and 35S rDNAs from NGS reads is labor-intensive, including DNA extraction, library construction, sequencing, quality trimming, assembly, and annotation. Second, plastid DNA copies are highly variable among lineages, tissues, developmental stages, and even growth conditions44, making it tough to standardize the amount of NGS reads required to assemble plastomes. Similarly, 35S rDNA copy numbers vary greatly between species43. Third, NGS requires a larger amount of DNA than PCR-based methods, and extracting sufficient DNA from woody materials may be challenging when DNA is rare or degraded10.
Although concatenation of multiple loci is frequently adopted in plant DNA barcoding21, we demonstrate that four loci—3′ETS, ITS1, trnH-psbA, and accD—are individually capable of delimiting the cypress species and varieties (Table 2). In cupressophytes, however, accD is generally elongated and longer than 2 kb, with high lineage-specific repeat content45,46. A gene of this size requires at least four runs of Sanger sequencing to sequence the full gene. Moreover, an inverse relationship between amplicon lengths and recoverable rates was observed when PCR templates were the DNA extracted from wood10,47. As cypresses are threatened by illegal logging because of their valuable timber, accD may not be a suitable barcoding locus for forensic timber identification. Therefore, we screened nine species-specific indels > 100 bp long (Fig. 5; Supplementary Table 3). They can serve as supplementary markers that better discriminate among cypress species.
In conclusion, we successfully obtained entire plastomes and 35S rDNAs from a shallow sequencing of all sampled cypress species and varieties. Both plastomes and 35S rDNAs have a strong potential to be effective super-barcodes for identifying cypress species and varieties. We also identified three loci (3′ETS, ITS1, and trnH-psbA) with appropriate lengths, high NSRs, and 100% identification rates at both the inter-species and inter-variety levels. These loci, together with the nine supplementary indel markers, can be used in an alternative barcode system when the botanical specimen has poor or low DNA content. Our new sequence data and innovative barcode systems not only enrich the availability of genetic references for cypresses, but also contribute to their conservation, authentication, reforestation, and forensic timber identification to stop illegal logging and its associated trade.
Materials and methods
Sample collection
We sampled 26 endemic cypress individuals—16 C. formosensis and 10 C. obtusa var. formosana—from cloud forests in the Central Mountain Range of Taiwan (Supplementary Fig. 1), with 1‒2 individuals sampled in each population. In addition, six cultivated individuals of C. obtusa var. obtusa were also sampled from two remote localities (four and two from localities A and B, respectively; Supplementary Table 1). These closely related taxa were identified based on their morphologies (Fig. 1) and planting histories. All vouchers and the associated DNAs are deposited in the germplasm bank of the Ministry of Justice Investigation Bureau (MJIB), New Taipei City, Taiwan.
DNA extraction, sequencing, and assembling
Total genomic DNA was extracted from 2 g of fresh leaves using a modified CTAB method48 with 0.1% of polyvinylpyrrolidone (PVP-40, Sigma) incorporated into the extraction buffer. The extracted DNA was sheared into fragments 400‒600 bp long, and DNA libraries were constructed using Ovation Rapid Library Preparation kits (NuGEN). Each library was sequenced on an Illumina HiSeq 4000 platform in Tri-I Biotech Company (New Taipei City, Taiwan) to generate 16.7‒46.26 million 2 × 150 bp pair-end reads that amassed a total of 3.03‒7.36 Gb (Supplementary Table 1). After removing adapters and non-qualified bases using Trimmomatic 0.3849, these reads were de-novo assembled with kmer lengths = 21, 33, 55, 77, and 91 in SPAdes 3.14.050. Plastomic and 35S rDNA contigs were searched using BLAST + 2.10.051 with the plastome (NC034943) and ITS (AY211258) of C. formosensis as references. Gaps between plastomic contigs were closed using GapCloser v1.1252. We used SEQuel v1.0.253 to do base-scale corrections of the assembled plastomes and 35S rDNAs.
Plastome and 35S rDNA annotation
Plastome annotation was performed using GeSeq54 with the default settings for search identities. For 35S rDNA, the 18S, 5.8S, and 26S ribosomal RNAs were predicted on RNAmmer 1.2 Server55. The annotated genes were further adjusted manually based on alignments of their orthologous genes from other cupressaceous species. Plastomes and 35S rDNAs were visualized using Circos 0.6756.
Sequence alignment and phylogenetic tree construction
Plastomes and 35S rDNAs were aligned using MAFFT 7.07457 with the algorithm = auto, scoring matrix = 200 PAM/k = 2, gap open penalty = 1.53, and offset value = 0.123. Maximum likelihood (ML) trees inferred from these alignments were estimated under a GTRGAMMAI model and 1,000 bootstrap replicates in RAxML v8.258. Neighbor-joining (NJ) trees based on single loci were constructed with a P-distance model, uniform rates among sites, and 1000 bootstrap replicates in MEGA X 10.1.75959. All yielded trees were condensed under the 50% majority-rule consensus.
Exploration of plastomic inversions
We used progressiveMauve60 to estimate plastomic inversions among C. formosensis, C. hodginsii, C. lawsoniana, C. obtusa var. formosana, and C. obtusa var. obtusa.
Calculation of nucleotide substitution rates
The average number of nucleotide substitutions per site between species or varieties was calculated to explore the hypervariable loci using the non-overlapping sliding window approaches in DnaSP v661. The window length was set to be 200 and 50 bp for plastomes and 35S rDNAs, respectively. To investigate barcoding gaps, sequences of the examined loci were aligned using MUSCLE 3.8.3162, followed by an estimate of pairwise nucleotide substitution rates (NSRs) using MEGA X with a P-distance model and uniform rates among sites.
Detection of repeats and indels
NCBI blastn was employed to compare each plastome or 35S rDNA against itself with the default settings. We discarded the matched pairs with sequence identities less than 90%. The remaining pairs of repeats were further checked manually to remove redundancies. Indels between species or varieties were detected using DnaSP.
Data availability
All DNA sequences were deposited into DDBJ DataBank under accession numbers LC516824‒LC529365 (Supplementary Table 1).
References
Wang, W., Hwang, C., Lin, T. & Hwang, S. Y. Historical biogeography and phylogenetic relationships of the genus Chamaecyparis (Cupressaceae) inferred from chloroplast DNA polymorphism. Plant Syst. Evol. 241, 13–28. https://doi.org/10.1007/s00606-003-0031-0 (2003).
Liao, P. C., Lin, T. P. & Hwang, S. Y. Reexamination of the pattern of geographical disjunction of Chamaecyparis (Cupressaceae) in North America and East Asia. Bot. Stud. 51, 511–520 (2010).
Li, C. F. et al. Chamaecyparis montane cloud forest in Taiwan: ecology and vegetation classification. Ecol. Res. 30, 771–791. https://doi.org/10.1007/s11284-015-1284-0 (2015).
IUCN Red List of Threatened Species. https://www.iucnredlist.org/ (2013).
Horng, F. W., Ma, F. C., Yu, H. M., Hsui, Y. R. & Chang, H. M. An estimation of original Chamaecyparis forest area in Taiwan and its implication for conservation. Q. J. Chin. For. 17, 143–153 (2000).
Koch, G., Richter, H. G. & Schmitt, U. Design and application of CITESwoodID computer-aided identification and description of CITES-protected timbers. IAWA J. 32, 213–220. https://doi.org/10.1163/22941932-90000052 (2011).
Sarmiento, C. et al. Pl@ntwood: a computer-assisted identification tool for 110 species of Amazon trees based on wood anatomical features. IAWA J. 32, 221–232. https://doi.org/10.1163/22941932-90000053 (2011).
Gasson, P. How precise can wood identification be? Wood anatomy’s role in support of the legal timber trade, especially cites. IAWA J. 32, 137–154. https://doi.org/10.1163/22941932-90000049 (2011).
Jiao, L., Yin, Y., Cheng, Y. & Jiang, X. DNA barcoding for identification of the endangered species Aquilaria sinensis: comparison of data from heated or aged wood samples. Holzforschung 68, 487–494. https://doi.org/10.1515/hf-2013-0129 (2014).
Jiao, L. et al. DNA barcode authentication and library development for the wood of six commercial Pterocarpus species: the critical role of xylarium specimens. Sci. Rep. 8, 1945. https://doi.org/10.1038/s41598-018-20381-6 (2018).
Nithaniyal, S. et al. DNA barcode authentication of wood samples of threatened and commercial timber trees within the tropical dry evergreen forest of India. PLoS ONE 9, e107669. https://doi.org/10.1371/journal.pone.0107669 (2014).
Liu, J. et al. Integrating a comprehensive DNA barcode reference library with a global map of yews (Taxus L.) for forensic identification. Mol. Ecol. Resour. 18, 1115–1131. https://doi.org/10.1111/1755-0998.12903 (2018).
Kress, W. J. Plant DNA barcodes: applications today and in the future. J. Syst. Evol. 55, 291–307. https://doi.org/10.1111/jse.12254 (2017).
CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. U S A 106, 12794–12797. https://doi.org/10.1111/1755-0998.12194 (2009).
Hollingsworth, P. M., Graham, S. W. & Little, D. P. Choosing and using a plant DNA barcode. PLoS ONE 6, e19254. https://doi.org/10.1371/journal.pone.0019254 (2011).
Li, D. Z. et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 108(19641–19646), 2011. https://doi.org/10.1073/pnas.1104551108 (2011).
Purty, R. S. & Chatterjee, S. DNA Barcoding: an effective technique in molecular taxonomy. Austin J. Biotechnol. Bioeng. 3, 1059 (2016).
Kane, N. et al. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 99, 320–329. https://doi.org/10.3732/ajb.1100570 (2012).
Bock, D. G., Kane, N. C., Ebert, D. P. & Rieseberg, L. H. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phytol. 201, 1021–1030. https://doi.org/10.1111/nph.12560 (2014).
Ji, Y. et al. Testing and using complete plastomes and ribosomal DNA sequences as the next generation DNA barcodes in Panax (Araliaceae). Mol. Ecol. Resour. 19, 1333–1345. https://doi.org/10.1111/1755-0998.13050 (2019).
Li, X. et al. Plant DNA barcoding: from gene to genome. Biol. Rev. 90, 157–166. https://doi.org/10.1111/brv.12104 (2015).
Niu, Z. et al. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 7, 2073. https://doi.org/10.1038/s41598-017-02252-8 (2017).
Zhu, S. et al. Accurate authentication of Dendrobium officinale and its closely related species by comparative analysis of complete plastomes. Acta. Pharm. Sin. B. 8, 969–980. https://doi.org/10.1016/j.apsb.2018.05.009 (2018).
Fu, C. N. et al. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 9, 2773. https://doi.org/10.1038/s41598-019-39161-x (2019).
Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 20, 525–527. https://doi.org/10.1016/j.tplants.2015.06.012 (2015).
Guo, W. et al. Predominant and substoichiometric isomers of the plastid genome coexist within Juniperus plants and have shifted multiple times during cupressophyte evolution. Genome Biol. Evol. 6, 580–590. https://doi.org/10.1093/gbe/evu046 (2014).
Wu, C. S. & Chaw, S. M. Large-scale comparative analysis reveals the mechanisms driving plastomic compaction, reduction, and inversions in conifers II (cupressophytes). Genome Biol. Evol. 8, 3740–3750. https://doi.org/10.1093/gbe/evw278 (2016).
Qu, X. J., Wu, C. S., Chaw, S. M. & Yi, T. S. Insights into the existence of isomeric plastomes in Cupressoideae (Cupressaceae). Genome Biol. Evol. 9, 1110–1119. https://doi.org/10.1093/gbe/evx071 (2017).
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A. & Janzen, D. H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 102, 8369–8374. https://doi.org/10.1073/pnas.0503123102 (2005).
Pang, X. et al. Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: a meta-analysis. PLoS ONE 7, e48833. https://doi.org/10.1371/journal.pone.0048833 (2012).
Loera-Sánchez, M., Studer, B. & Kölliker, R. DNA barcode trnH-psbA is a promising candidate for efficient identification of forage legumes and grasses. BMC Res. Notes 13, 35. https://doi.org/10.1186/s13104-020-4897-5 (2020).
Zonneveld, B. J. M. Conifer genome sizes of 172 species, covering 64 of 67 genera, range from 8 to 72 picogram. Nord. J. Bot. 30, 490–502. https://doi.org/10.1111/j.1756-1051.2012.01516.x (2012).
Preuten, T. et al. Fewer genes than organelles: extremely low and variable gene copy numbers in mitochondria of somatic plant cells. Plant J. 64, 948–959. https://doi.org/10.1111/j.1365-313X.2010.04389.x (2010).
Shen, J., Zhang, Y., Havey, M. J. & Shou, W. Copy numbers of mitochondrial genes change during melon leaf development and are lower than the numbers of mitochondria. Hortic. Res. 6, 95. https://doi.org/10.1038/s41438-019-0177-8 (2019).
Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. https://doi.org/10.1111/nph.12395 (2013).
Gualberto, J. M. & Newton, K. J. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. https://doi.org/10.1146/annurev-arplant-043015-112232 (2017).
Kozik, A. et al. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLoS Genet. 15, e1008373. https://doi.org/10.1371/journal.pgen.1008373 (2019).
Ruhsam, M. et al. Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria?. Mol. Ecol. Resour. 15, 1067–1078. https://doi.org/10.1111/1755-0998.12375 (2015).
Chen, Q., Wu, X. & Zhang, D. Comparison of the abilities of universal, super, and specific DNA barcodes to discriminate among the original species of Fritillariae cirrhosae bulbus and its adulterants. PLoS ONE 15, e0229181. https://doi.org/10.1371/journal.pone.0229181 (2020).
Weng, M. L., Blazier, J. C., Govindu, M. & Jansen, R. K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. https://doi.org/10.1093/molbev/mst257 (2014).
Vieira Ldo, N. et al. The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 9, e90618. https://doi.org/10.1371/journal.pone.0090618 (2014).
Hamsher, S. E. et al. Extensive chloroplast genome rearrangement amongst three closely relatedHalamphora spp. (Bacillariophyceae), and evidence for rapid evolution as compared to land plants. PLoS ONE 14, e0217824. https://doi.org/10.1371/journal.pone.0217824 (2019).
Garcia, S., Kovařík, A., Leitch, A. R. & Garnatje, T. Cytogenetic features of rRNA genes across land plants: analysis of the Plant rDNA database. Plant J. 89, 1020–1030. https://doi.org/10.1111/tpj.13442 (2017).
Liere, K. & Börner, T. Development-dependent changes in the amount and structural organization of plastid DNA. In Plastid Development in Leaves during Growth and Senescence (ed. Biswal, B., K. Krupinska, K. & Biswal, U. C.) 215–237 (Amsterdam Springer, 2013).
Li, J., Su, Y. & Wang, T. The repeat sequences and elevated substitution rates of the chloroplast accD gene in cupressophytes. Front. Plant Sci. 9, 533. https://doi.org/10.3389/fpls.2018.00533 (2018).
Sudianto, E. & Chaw, S. M. Two independent plastid accD transfers to the nuclear genome of Gnetum and other insights on acetyl-coA carboxylase evolution in gymnosperms. Genome Biol. Evol. 11, 1691–1705. https://doi.org/10.1093/gbe/evz059 (2019).
Deguilloux, M. F., Pemonge, M. H. & Petit, R. J. Novel perspectives in wood certification and forensics: dry wood as a source of DNA. Proc. Biol. Sci. 269, 1039–1046. https://doi.org/10.1098/rspb.2002.1982 (2002).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bul. 19, 11–15 (1987).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421. https://doi.org/10.1186/1471-2105-10-421 (2009).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. https://doi.org/10.1186/2047-217X-1-18 (2012).
Ronen, R., Boucher, C., Chitsaz, H. & Pevzner, P. SEQuel: improving the accuracy of genome assemblies. Bioinformatics 28, i188-196. https://doi.org/10.1093/bioinformatics/bts219 (2012).
Tillich, M. et al. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. https://doi.org/10.1093/nar/gkx391 (2017).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. https://doi.org/10.1093/nar/gkm160 (2007).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. https://doi.org/10.1101/gr.092759.109 (2009).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. https://doi.org/10.1093/molbev/msy096 (2018).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147. https://doi.org/10.1371/journal.pone.0011147 (2010).
Rozas, J. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(1792–1797), 2004. https://doi.org/10.1093/nar/gkh340 (2004).
Acknowledgements
We thank the Forest Conservation and Management Administration for providing the seed specimens, and Forestry Bureau Council of Agriculture of Executive Yuan for providing plant materials. We also thank the three anonymous reviewers for their critical reading and helpful comments. This work was partially supported by the Grants Academia Sinica 23-23 and MOST 106-2311-B-001-005 to SMC, and Grant Ministry of Justice 108-1301-05-17-05 to C.T.C.
Author information
Authors and Affiliations
Contributions
C.T.C., S.M.C. conceived and designed the study. C.S.W., B.C.W., Y.M.H. performed the experiments. C.S.W. analyzed the data. C.S.W., C.J.H. contributed to the images in Fig. 1. C.S.W., E.S., C.T.C., S.M.C wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, CS., Sudianto, E., Hung, YM. et al. Genome skimming and exploration of DNA barcodes for Taiwan endemic cypresses. Sci Rep 10, 20650 (2020). https://doi.org/10.1038/s41598-020-77492-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77492-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
