
Abstract
The plant-available pools of calcium, magnesium and potassium are assumed to be stored in the soil as exchangeable cations adsorbed on the cation exchange complex. In numerous forest ecosystems, despite very low plant-available pools, elevated forest productivities are sustained. We hypothesize that trees access nutrient sources in the soil that are currently unaccounted by conventional soil analysis methods. We carried out an isotopic dilution assay to quantify the plant-available pools of calcium, magnesium and potassium and trace the soil phases that support these pools in 143 individual soil samples covering 3 climatic zones and 5 different soil types. For 81%, 87% and 90% of the soil samples (respectively for Ca, Mg and K), the plant-available pools measured by isotopic dilution were greater than the conventional exchangeable pool. This additional pool is most likely supported by secondary non-crystalline mineral phases in interaction with soil organic matter and represents in many cases (respectively 43%, 27% and 47% of the soil samples) a substantial amount of plant-available nutrient cations (50% greater than the conventional exchangeable pools) that is likely to play an essential role in the biogeochemical functioning of forest ecosystems, in particular when the resources of Ca, Mg and K are low.
Similar content being viewed by others


Introduction
Magnesium (Mg), calcium (Ca) and potassium (K) are three major and essential nutrients for plants1. In forest ecosystems, their plant-available pools are assumed to be stored in the soil as dissolved cations in solution and as exchangeable cations adsorbed on the cation exchange complex2,3. This exchangeable pool is commonly measured in soil samples from an extraction with a concentrated salt reactant (e.g. NH4Cl, BaCl2, etc.). Because, in most cases, fertilization and liming are not common practices in forestry, plant-available nutrient cations are, on the long term, mainly supplied by atmospheric deposition and mineral weathering and vary as a function of the net nutrient losses from the ecosystems: mainly harvested biomass exportation and nutrient leaching below the rooting zone.
Forest ecosystems are often developed on poorly fertile soils where the plant-available pools of nutrient cations are frequently very low4. In the context of global change, the sustainability of forest ecosystems and their chemical fertility is highly at risk due to increasing nutritional, sivilcultural and/or climatic pressures. Despite the decreasing acidity of atmospheric inputs since the 1980s in Europe and North America, many forest soils are still suffering from on-going acidification5,6,7,8. In many cases, atmospheric inputs of nutrient cations have decreased over the past decades7,9,10,11,12. Finally, nutrient outputs from the ecosystem are often increasing due to the intensification of silvicultural practices (biomass export, etc.) to meet the increasing demand for wood biomass. These practices may severely impact soil fertility13, especially in forest ecosystems where the nutrient level is low14. As a result, over the last decade degradation of forest soil fertility have been reported worldwide and are expected to increase6,15,16,17.
It is however not yet fully understood how trees cope with very low nutrient resources in the soil and sustain long term forest productivity18. Numerous studies have reported significant discrepancies between modelled and empirically measured changes in plant-available pools of nutrient cations in the soil: mass balance and dynamic models tend to exaggerate nutrient cation depletion from the soil19 compared to measured changes in soil available pool between two dates20. In addition, many studies have reported discrepancies between chemical fertility (exchangeable pools, nutrient fluxes), tree nutrition (foliar nutrient concentrations) and forest productivity indicators21,22,23,24,25.
To explain these discrepancies, it has been hypothesized that the plant-available pool of nutrient cations may be larger than the exchangeable pool20,26,27,28. Aluminium and iron (hydr)oxides as well as amorphous aluminosilicate structures, which are abundant in acidic soils, may develop a cationic exchange capacity and adsorb cations29. The amorphous nature and the dynamic dissolution/precipitation of these structures may cause the adsorbed cations to become temporally occluded30,31. In a highly weathered tropical soil, Hall and Huang32 showed that a significant amount of Ca, Mg, and K was sequestered in iron (hydr)oxide secondary mineral phases. Moreover, potassium stored in part as “non-exchangeable” or fixed potassium33 (held between adjacent tetrahedral phyllosilicate layers of micas and 2:1 clay minerals such as vermiculite or illite) may be available to plant uptake. The intensity of “non-exchangeable” K release has been related to root absorption and root activity34 and may be a quite significant source of K for plant nutrition35.
Quantifying plant-available pools in the soil through soil extraction methods is challenging because it is difficult to define a chemical reagent with the same nutrient extraction potential as plant roots and their associated microorganisms. Nevertheless, the isotopically exchangeable pool (noted EK, ECa and EMg) quantified by the isotopic dilution method has been shown to be the most adequate approach to quantify the plant-available pools36,37. Using radio-isotopes, previous studies found that the K and Ca isotopically exchangeable pool was larger than the exchangeable pool (over 45 agricultural soil studied)38,39,40,41,42,43. Two recent studies44,45, focused on soil samples from the Breuil-Chenue experimental forest and using a stable isotopic dilution approach, (i) showed that soil pools of Mg, Ca and K in addition to the exchangeable pool significantly contributed to chemical equilibrium reactions between the liquid and solid phases of the soil, and (ii) suggested that these pools were supported by secondary non-crystalline mineral phases such as Al and Fe (hydr)oxides, and amorphous aluminosilicates.
The objectives of this study are twofold: (i) to test the hypothesis that nutrient cation pools other than the conventional exchangeable pool contribute to solid solution equilibrium in a variety of forest soils covering a wide range of climatic, edaphic, chemical fertility and tree species cover conditions; (ii) to characterize the variability of the isotopically exchangeable pools in relation to the physical and chemical properties of the soil in order to better describe the different soil phases that may act as a support for this additional plant-available pool.
Results
Isotopically exchangeable pools
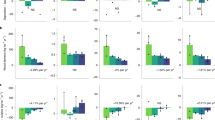
The ratio of the isotopically exchangeable over the conventional exchangeable pool (Ex:Exchx) ranged from 0.28 to 31.76 for Ca, from 0.67 to 2.79 for Mg and from 0.47 to 3.99 for K (Fig. 1, for individual results Table S3 supplementary data) but ECa, EMg and EK were larger than their respective exchangeable pool for the great majority of the soil samples: 81%, 87% and 90%, respectively, of soil samples had Ex:Exchx ratios above 1. The median ratios were respectively 1.30, 1.23 and 1.47 for Ca, Mg and K. The ECa:ExchCa ratio was greater than 2 for one third of the samples and greater than 3 for 17%. For Mg and K, the EX:ExchX ratio was greater than 2 for fewer samples compared to Ca: 10% and 24% respectively. The EX:ExchX was higher for the samples with low ExchX pools (Fig. 2). Over the entire dataset, the difference between the EX and ExchX pools was statistically significant (paired Wilcoxon-Mann–Whitney test) for all three elements (p-value < 0.01 for K and Mg and p-value < 0.05 for Ca), although homoscedasticity was not verified for Mg.

Histogram and boxplot of the distribution of the Ex:Exchx ratios for Ca, Mg and K. Ex represents the isotopically exchangeable pool and Exchx the conventional exchangeable pool. The number of validated samples (n) is given in brackets.

Relationship between the Ex:Exchx ratio and the conventional exchangeable (Exchx) pools for Ca, Mg and K.
The repeatability of EX measurements, estimated as the relative standard deviation was 16%, 14% and 8% for Ca, Mg and K respectively. Even when taking this analytical uncertainty into account, 58% (Ca), 62% (Mg) and 88% (K) of the samples had an Ex-pool significantly greater than the Exchx pool (data not shown). A small proportion of samples had Ex-pools lower than the Exchx pools (Ex:Exchx < 1): 19%, 13% and 10% for Ca, Mg and K respectively.
Strong linear relationships were found between isotopically exchangeable pools and conventional exchangeable pools (Fig. 3). Correlation coefficients were respectively 0.92, 0.92 and 0.88 for Ca, Mg and K (p-values < 0.001). Linear regressions slopes were greater than the 1:1 slope for Ca and Mg whereas for K, linear regression slope was similar to the 1:1 slope but intercept was different. No other significant relationships were found between Ex pools of Ca, Mg and K and experimental data or soil physico-chemical properties over the entire dataset.

Relationship between isotopically exchangeable (Ex) and conventional exchangeable (Exchx) pools for Ca, Mg and K. Linear regression parameters are given in each figure. Solid and dotted lines represent, respectively, y:x = 1 and linear regression.
Tracer recovery in the different soil pool
For all three elements, most of the tracer amounts recovered in the soil pools were found in the NH4 extracted pools (Fig. 4, for individual results Table S4 supplementary): the median relative contribution for NH4#1 and NH4#2 was 68% and 17% for Ca, 90% and 5% for Mg and 85% and 7% for K, respectively. For Ca and Mg, the median relative contribution in the three pools was as follows: NH4#1 > NH4#2 > HNO3. Potassium was similar to Ca and Mg but the median relative contribution in the NH4#2 pool was close to that in the HNO3 pool (~ 7%). The relative contribution in the NH4#1 pool showed higher variability for Ca and Mg compared to the other soil pools, the interquartile was 37% and 71%, respectively, whereas only 7% for K.

Relative contribution (%) of each extracted soil pool (NH4#1, NH4#2 and HNO3) to the isotopically exchangeable pool (Ex) for Ca, Mg and K. The number of validated samples (n) is given in brackets.
Analytically significant amounts of isotope tracers were found in the HNO3-extracted pool for almost all samples for Ca and K: 95% and 96% respectively whereas only 19% for Mg (Fig. 4). For all three elements, the relative contribution of the HNO3 extracted pool was lower than in the NH4 extracted pools: the median relative contribution was 10%, 0% and 7% for Ca, Mg and K, respectively, and the maximum value across soil samples reached 45% for Ca, 74% for Mg and 34% for K.
Significant relationships were found between the Ca-tracer recovery in the HNO3-extracted and the total carbon content in the soil sample, as well as with HNO3-extracted iron content (Fig. 5a,b). Correlation coefficients were 0.65 and 0.42 respectively (p-value < 0.001). For Breuil samples, significant relationships were also found between the Ca-tracer recovery in the HNO3-extracted pool with Tamura and Pyrophosphate extractible Fe (Fig. 5c,d). Correlation coefficients were 0.48 and 0.62 respectively (p-value < 0.001). To a lesser extent, the relative contribution of Ca HNO3-extracted pool to ECa was significantly correlated to the HNO3-extracted pool of Al (correlation coefficient of 0.17 and p-value < 0.001) (Fig. 5e).

Relationship between the isotopic recovery of Ca in the HNO3-extrated pool and (a) total carbon, (b) Fe HNO3-extracted pool, (c) Tamura and (d) pyrophosphate extractible Fe. (e) shows the relationship between the relative contribution of the Ca HNO3-extracted pool and the Al HNO3-extracted pool. The linear regressions are represented by the dotted lines.
A significant relationship was found between the isotopically exchangeable fraction of the HNO3-extracted pool of K and the clay content in soil samples (r-squared = 0.47, p-value < 0.001) (Fig. 6), but the relationship was not significant for Ca and Mg.

Relationship between the pourcentage of clay (texture class < 2 µm) in soil samples and the isotopically exchangeable fraction of the HNO3-extracted K pool. The linear regression is represented by the dotted line.
Discussion
Isotopically exchangeable pools
Our results show over the whole dataset, for the majority of these samples, that the isotopically exchangeable pools (EX) of Ca, Mg and K are larger than the conventional exchangeable pool (ExchX). The majority of samples showed an EX:ExchX ratio greater than 1 for Ca, Mg and K. For a substantial proportion of the dataset (43%, 27% and 47% respectively for Ca, Mg and K), Ex pools were more than 50% larger than the ExchX pools. This trend was confirmed by a Wilcoxon–Mann–Whitney comparison test that showed that isotopically exchangeable pools were significantly larger than their respective exchangeable pool, though homoscedasticity between the compared pools was not met for Mg. Our results are consistent with previous studies for Ca and K. Blum and Smith41 showed, in an isotopic dilution assay using the 45Ca radiogenic isotope over 16 different type of soils, that ECa was significantly greater than ExchCa for 6 soil samples, lower for 2 samples and not significantly different for 8 samples. Reeve et al.43 showed that 5 soil samples out of 7 showed an ECa pool greater than the ExchCa pool. For K, apart from the study by Graham and Fox46 in which 4 out of the 11 studied soil samples showed an EK pool lower than the ExchK pool, all other studies showed EK pools systematically greater than the ExchK pools38,39,40.
For a limited proportion of the dataset, the Ex pool was smaller than the ExchX pool. This result was unexpected because the conventional exchangeable pool is assumed to be in rapid equilibrium with soil solution2,3. The isotopically exchangeable pool was thus expected to be at least equal to the exchangeable pool. However, for the majority of the samples of this study, as reported in previous assays44,45, only a fraction of the exchangeable pool contributed to the isotopic equilibrium (the isotopically exchangeable proportion of the NH4#1 and NH4#2 extracted pools were (median): 68% and 54% for Ca, 81% and 28% for Mg, and 88% and 53% for K). That less than 100% of the exchangeable pool reached isotopic equilibrium with the isotopically labelled solution is most likely due to the fact that certain sites that are extractible with a highly concentrated reagent may not be exchangeable with a dilute solution47. The isotopically exchangeable proportion of the NH4#1 and NH4#2 extracted pools of Ca, Mg and K was non linearly and positively correlated to their respective conventional exchangeable pool. This result agrees with the mechanisms of ion exchange by which the likelihood of a given dissolved cation to undergo exchange with a cation adsorbed on the cationic exchange complex is proportional to the abundance of the adsorbed cation on the cationic exchange complex48.
Despite these results, for all samples showing Ex lower or similar in size to Exchx, analytically significant amounts of isotope tracer were recovered in the HNO3-extracted soil pool (Table S3 supplementary). Hence, for these samples as well, the results of this study show that soil phases other than the conventional exchangeable pool directly contribute to the geochemical equilibrium processes for Ca, Mg and K. These geochemically reactive pools of Ca, Mg and K unaccounted for by conventional soil analysis methods are likely to play a very important role in the biogeochemically functioning of forest ecosystems, most particularly in soils where the conventional exchangeable pools are low (Fig. 2).
Storage forms
Unsurprisingly, the isotopically exchangeable pools of Ca, Mg and K were mainly composed of NH4-exchangeable pools of Ca, Mg and K and their variability was mainly explained by the Ca, Mg and K exchangeable pool variability (Fig. 3). Nevertheless, for the great majority of the dataset, isotope tracers were also recovered in the HNO3-extracted pool. The amounts of isotope tracer recovered in this pool were analytically significant and, for numerous samples, substantial (Fig. 4). It is unlikely that this recovery may be explained by residuals amounts of isotope tracer after the two AcONH4 extractions (NH4#1 and NH4#2) because an intermediary rinsing extraction was performed. Concentrations of nutrient cations measured in this rinsing extraction were generally low and often too low (for 41%, 51% and 91% of samples for Ca, Mg and K, respectively) for isotopic analysis.
Calcium
The HNO3-extracted pool of Ca accounted for a significant proportion of the measured ECa pool (Fig. 4), and was larger for Ca compared to Mg and K. The Ca-tracer recovery in the HNO3-extracted pool was strongly correlated to the soil carbon content (R2 = 0.65, p-value < 0.001) (Fig. 5a) thus suggesting that significant amounts of Ca adsorbed or chelated to organic compounds in the soil contribute directly to geochemical equilibrium reactions but are not extracted by conventional exchangeable cation extractions. This may be explained by the much higher affinity of organic compounds for Ca compared to most other cations such as NH4+49,50,51.
A strong and significant correlation was also found between the Ca-tracer recovery in the HNO3-extracted pool and the HNO3-extracted pool of Fe (R2 = 0.42, p-value < 0.001) (Fig. 5b). For the Breuil samples, Tamura-extracted and pyrophosphate-extracted Fe (Fig. 5c,d) were also correlated to Ca-tracer recovery in the HNO3-extracted pool (r2 0.48 and 0.62 respectively for Tamura and pyrophosphate. p-values < 0.01). The Ca HNO3-extracted pool relative contribution was also significantly correlated to the HNO3-extracted pool of Al (R2 = 0.17, p-value < 0.001) (Fig. 5e). Because these different soil extractions mainly dissolve Fe and Al (hydr)oxides, it is likely that these amorphous secondary minerals act as a support to a pool of isotopically exchangeable Ca. In agreement with these results, the greatest ECa:ExchCa ratios were found in podzol and alumic cambisol soil samples where the soil organic matter and Fe and Al (hydr)oxides are abundant and likely to play an important role in calcium biogeochemistry.
Iron and Aluminium (hydr)oxides are common in soils52 and occur as amorphous minerals ranging from short-range-ordered to increasingly crystalline phases. The point of zero charge (pzc) of synthetic Fe and Al (hydr)oxides is generally above 653. Yet in natural environment, Fe and Al (hydro)oxides pzc are lower54 so that negative charges could be developed at pHwater < 6 and direct adsorption of Ca on such minerals has been demonstrated for the range of soil pH in our soil samples (pHH2O range 3.4–6.2, median 4.6)29,55,56,57. However direct adsorption is unlikely to be the main storage mechanism for the isotopically exchangeable Ca in the HNO3-extracted pool. Instead, the results of our study strongly suggest that geochemically reactive Ca is retained on the surface of Al and Fe (hydr)oxide minerals through an anion-bridge such as sulfate or organic acids as suggested by previous studies32,45,58.
Van der Heijden et al.44,45 in a similar isotopic dilution experiment carried out on an alumic cambisol soil, showed a non-negligible contribution of Ca, Mg and K pools extracted with a Tamm reagent and with a HNO3 reagent (1 mol L−1) to the isotopically exchangeable pools. They suggested that the main storage form of Ca and Mg in the Tamm and HNO3-extracted pools was cations indirectly adsorbed on Al and Fe oxides and hydroxides through (i) P or organic acid-mediated bridging or (ii) occluded within Fe and Al phases or their organic co-precipitates. Additionally, Hall and Huang32 showed the role of occluded cations in sustaining plant nutrition through Fe-(hydr)oxide reduction, whereby these occluded cations may act as a bank to replenish exchangeable pools on timescales of hours to months. Iron and aluminium (hydr)oxides are thus likely to be the support of a geochemically reactive HNO3 pool of Ca which is not extracted by conventional exchangeable cation methods. This may be explained by possible temporary occlusion of Ca in these soil phases: occluded in supramolecular aggregates where various large organic molecules are held together by van der Waals forces, hydrogen bonds and metal bridging involving Ca and Fe-hydr(oxides)59,60,61. These amorphous minerals are known to very dynamically precipitate and dissolve over time as a result of changes in the physical and chemical properties of the soil solution31 and under the influence of microbial and plant activity32.
Potassium
Similar to Ca, K tracer was recovered in the HNO3-extracted pool for 97% of all validated samples and represented on average 8% of the isotopically exchangeable pool. A strong linear and positive correlation was found between the clay content and the isotopically exchangeable fraction of the HNO3 extracted K pool (R2 = 0.52; p < 0.001) (Fig. 6). This relation is most likely explained by the presence of “fixed” potassium (K-specific exchange sites) in phyllosilicates33, held between adjacent tetrahedral phyllosilicate layers of micas and 2:1 clay minerals such as vermiculite or illite, but not extractible with concentrated salt extractions such as NH4+62,63. K specific ion-exchange reactions between the soil solution and the clay-interlayer potassium pools may have occurred during the isotopic equilibrium stage of the experiment. The HNO3 reagent (1 mol L−1) is likely to have caused a sufficient weathering of the phyllosilicates and a subsequent release of interlayer potassium. However, quantitative phyllosilicate mineralogy would be necessary to better characterize the role of clay minerals and K-specific exchange sites because (1) not all phyllosilicates contain pools of interlayer potassium and (2) the pools of interlayer K may respond very differently depending on the phyllosilicate. For instance, at the Breuil site where quantitative mineralogy was available64, a positive correlation was found between the vermiculite content with K tracer recovery, the relative contribution of the HNO3-extracted K pool to EK and the EK:ExchK ratio. By contrast, no correlations were found with the kaolinite (1:1 clay mineral) or illite (2:1 clay mineral) contents (data not shown).
The highest relative contribution of the HNO3-extracted pool of K and EK:ExchK ratios were however found for the andosol and podzol soil types where the clay content is low. It is thus likely that other forms of storage in addition to interlayer K contribute to supporting the isotopically exchangeable pools of K. Relationships in our dataset suggest that potassium may also be adsorbed through ion-exchange processes on the surface of amorphous silica gels and aluminosilicates. For the Breuil-Chenue site, a positive correlation was found between, on the one hand, the difference between EK and ExchK and, on the other hand, the difference between the Tamm-extracted K pool and ExchK (R2 = 0.36, p < 0.001) (data not shown). The Tamm reagent, although non-selective, primarily dissolves (acid dissolution and chelation) poorly crystallized Al and Fe (hydr)oxides and amorphous aluminosilicates. These adsorption sites may be K-specific or occluded and thus not accounted for by conventional exchangeable cation pool extractions. A previous isotopic dilution assay also suggested that Tamm labile pools of K were mainly linked to amorphous aluminosilicate phases45.
Magnesium
In contrast with Ca and K, analytically significant amounts of Mg tracer were only found in the HNO3-extracted pool in 19% of the dataset. However, it is likely that this difference with Ca and K is not or not solely the result of a very contrasting behaviour of Mg in the geochemical equilibrium processes. Indeed, a previous study using a similar isotopic dilution approach showed that soil phases extracted with Tamm and HNO3 reagents significantly contributed (up to 11%) to the Mg geochemical equilibrium between the solution and the soil44 in the Breuil-Chenue soil profile. It is most likely that the behaviour of Mg observed in the current isotopic dilution assay is due to the experimental design: the quantity of isotopically enriched Mg was probably too small to efficiently isotopically label soil phases other than the exchangeable pool. Compared to Ca and K, in the majority of samples, large amounts of Mg were extracted from the soil during the HNO3-extraction step most likely due to the dissolution of Mg-bearing soil minerals (biotite, muscovite, vermiculite, chlorite, etc.) making isotope tracer recovery difficult to analytically resolve (high isotopic dilution). Indeed, the ratio of the amount of isotope tracer applied over the HNO3-extracted pool size was much lower for Mg (0.074), than for Ca (1.33) or K (0.246). This demonstrates the importance of the experimental parameters when setting up and the limits of isotopic dilution assays: the concentration of the tracing solution should be as close as possible as in situ concentrations and in the same time, the amount of applied tracer should be sufficiently high to ensure the isotopic labelling of the different studied soil phases.
For the limited number of samples for which Mg isotope tracer was recovered in the HNO3 extracted Mg pool, no significant relations were found with the other variables of the dataset. However, the highest EMg:ExchMg ratios were found in andosol and podzol soil types similarly to both Ca and K. In a previous isotopic dilution assay45, geochemically reactive Mg was shown to be stored in Tamm and HNO3 extracted soil phases in forms close to those previously discussed for Ca: adsorbed to Al and Fe (hydr)oxide secondary minerals through anion-bridges.
Implication at the soil profile scale
Nearly all sites had Ex soil profile stocks (kg ha−1) greater than the Exchx stocks (respectively 82%, 94% and 95% of samples for Ca, Mg and K) (Table S5 supplementary). For calcium, ECa stocks were two-fold greater than ExchCa stocks for nearly half of the sites (47%), three-fold greater for 24% of the sites. Comparatively, less sites had EMg and Ek stocks which were at least two-fold greater than ExchMg and ExchK (6% and 32%, respectively). The relative differences between EX and ExchX stocks were greatest for the sites with low exchangeable pools of Ca, Mg and K. Differences represented in median + 73 kg ha−1 for Ca (range from − 37 to + 960 kg ha−1), + 12 kg ha−1 (from − 9 to + 927 kg ha−1) for Mg and + 121 kg ha−1 for K (from − 3 to + 441 kg ha−1) (Fig. 7). These results highlight that at the soil profile scale, the conventional exchangeable pools may greatly underestimate the pool of cations that contribute to geochemical equilibrium between the soil and the solution and thus to the plant-available pools.

Boxplot distribution of the differences between Ex and Exchx stocks at the soil profile scale for Ca, Mg and K. The number of soil profiles (n) is given in brackets.
In the European beech (Fagus sylvatica L.) plot of the Breuil-Chenue experimental site, nutrient input–output budgets predicted a depletion of the exchangeable pools of Ca (3.1 kg ha−1 year−1) and Mg (0.8 kg ha−1 year−1) in the soil profile. Evidence from an isotopic tracing experiment28 concurred with the predicted Mg depletion but showed that the exchangeable pools of Ca had not decreased. The present study shows that isotopically exchangeable pool of Ca (0–70 cm) was much greater (138 kg ha−1) than the exchangeable pool (73 kg ha−1) whereas the isotopically exchangeable and exchangeable pools of Mg were similar (35 and 33 kg ha−1, respectively). It is most likely that the unaccounted pool of isotopically exchangeable Ca has contributed to buffering the depletion of the conventional exchangeable pools over time.
Plant driven processes in specific soil zones can also increase the stocks of plant-available nutrient cations. The consequences of biogeochemical processes on nutrient release within the rhizosphere of plant roots are well documented65. A recent study at the Itatinga experimental site (Brazil) sampled in our study suggested that root-induced weathering of K-bearing minerals, partly related to enhanced rhizosphere acidification could explain the observed increase in exchangeable K concentration within the rhizosphere of Eucalyptus grandis trees66.
Conclusion
This study validated, for a wide variety of forest soils, the hypothesis that pools of Ca, Mg and K in the soil in addition to the exchangeable pools contribute directly and on short time scales to the geochemical equilibrium processes between the soil and solution. Although the isotopically exchangeable pools of Ca, Mg and K vary widely between the different soil samples, these pools are in many cases substantially greater compared to their respective conventional exchangeable pool. The differences between the EX and ExchX pools were most remarkable for Ca and K, and lesser for Mg. These previously unaccounted pools of Mg, Ca and K in the soil fertility diagnostic are most likely to play an essential role in the biogeochemical functioning of forest ecosystems, in particular in ecosystems where the resources of Ca, Mg and K are low, by providing a supplementary buffer capacity to the depletion of cations. These groundbreaking results enable to reframe the conceptual model of plant available pool by integrating this additional pool of available nutrient cations (Fig. 8).

Conceptual model of Ca, Mg and K plant-availability. Dot-ended lines symbolize a support function. Black boxes and arrows symbolize the pools and fluxes of nutrient cations that are taken in the current conceptual model. Red boxes and arrows represent the pools and fluxes evidenced by the present study.
Soil phases extracted with weak to strong acid dissolution extraction methods are likely to support source and sink pools of Ca, K and possibly Mg. Hypotheses of the nature of these soil phases may be formulated. The isotopically exchangeable pool of Ca appears to be associated with amorphous and poorly crystalized secondary minerals through interactions with soil organic matter, whereas the isotopically exchangeable pool of K is likely associated with K-specific sites of phyllosilicates and amorphous aluminosilicates. These soil phases in addition to being a support for plant-available cations over short time scales may also significantly contribute to the long-term plant-availability as a source of cations released by their mineral weathering.
This study shows that the use of stable isotopic tracers to quantify the plant-available pools of Ca, Mg, and K on short time scales (source and sink pools) is both adequate and relevant in order to better understand biogeochemical cycling and tree nutrition in forest ecosystems. Although the precise identification and characterization of the soil phases that support the geochemically reactive pools and their interactions is a challenge, it is a vital step to better understanding and quantifying their role in soil geochemistry and stable isotope approaches are a powerful tool to achieve this goal.
Material and methods
Study sites
Twenty-six sites from 4 countries were selected amongst long-term forest monitoring networks (French ICP-forests level II sites and Swedish ICP-Integrated Monitoring sites), ANAEE/IN-SYLVA experimental sites as well as the Luquillo Critical Zone Observatory in Puerto Rico (Table S1 supplementary) in order to cover a range of acidic (pHwater < 6) and non-hydromorphic soils from a wide variety of climatic, edaphic, chemical fertility and tree species cover conditions. The scope of this study focuses on acidic soils as they are representative of a large proportion of forest ecosystems and are particularly sensitive to disturbances. When several replicates of soil profiles were sampled in each plot, a composite soil profile was established from the archived soil samples. Depending on the number of sampled soil layers, a maximum of 5 soil layers covering the entire available profile were selected for each plot. Each plot was covered by one dominant tree species. All sites, except the Swedish IM sites which are located in natural reserves, followed conventional and local forest management. The dataset contained no fertilized plots apart from Itatinga (Brazil), where background fertilization was added in all plots for every rotation similar to commercial plantations (300 kg ha−1 NPK—10:20:10). The global dataset was composed of 143 individual samples and encompasses 5 different types of soil, 11 tree species and 3 climatic zones.
The dataset was compiled from different databases encompassing soil physical and chemical properties measured in different laboratories following different protocols (Table S2 supplementary). The soil physical and chemical property dataset included particle size content distribution (i.e. clay, silt, sand), bulk density, total carbon and total nitrogen measured by wet combustion (Kjeldhal method for N; Walkey and Black or Anne method for C) or dry combustion and exchangeable cations (NH4Cl, KCl, BaCl2, NH4OAc or cobaltihexamine), noted ExchCa, ExchMg and ExchK. The sum of exchangeable base cations (S) and exchange acidity (EA) were respectively defined as the sum of exchangeable Ca, Mg and K and as the sum of exchangeable Al and protons. In addition, specific extractions of soil phases were included, in particular Tamm, Tamura, Mehra-Jackson and pyrophosphate extractions.
Isotopically exchangeable pools of Mg, Ca and K
The stable isotopic dilution technique was used to quantify the pools of Ca, Mg and K stored in the soil that may exchange rapidly with ions of the same element in the soil solution, so as to replace these ions in solution as they become lost from the system through plant uptake, leaching, or other output fluxes (isotopically exchangeable pool noted ECa, EMg and EK).
A 44Ca, 26Mg and 41K labelled solution (concentration: 800, 200 and 500 μg L−1 for Ca, Mg and K respectively) was made up from dissolved 44CaCO3 (96.45 atom% 44Ca), 26MgO (99.25 at% 26Mg) and 41KCl (97 at% 41K). The pHwater of the labelled solution was adjusted with purified nitric acid to the soil pHH2O of each sample. For each soil sample, 2.50 g of 2 mm-sieved dry soil were placed in a 50 mL polypropylene tube and 50 mL of the labelled solution were introduced. Tubes were immediately caped and placed in a continuous shaker. After 1 h, 6 h, 24 h and 48 h, the tubes were centrifuged (3000 rot min−1) during 20 min to sample a 12.5 mL aliquot of the supernatant solution for chemical and isotopic analyses. Tubes were then vortexed and replaced in the continuous shaker until the following time step. The isotopic variation in the solution enables to quantify the isotopically exchangeable pools of Ca, Mg and K at different stages of the equilibrium process.
After the 48 h time step, a four-stage sequential soil extraction protocol was conducted:
-
1.
50 mL of 1 mol L−1 ammonium acetate (unbuffered) continuously shaken during 1 h. Hereafter referred as NH4#1.
-
2.
50 mL of 1 mol L−1 ammonium acetate during (unbuffered) continuously shaken during 24 h. Hereafter referred as NH4#2.
-
3.
50 mL of 0.1 mol L−1 ammonium acetate at pH 3 (nitric acid) continuously shaken during 3 h.
-
4.
50 mL of 1 mol L−1 nitric acid continuously shaken during 20 h. Hereafter referred as HNO3.
Ammonium acetate extracts the pools of cations stored in an ion-exchangeable form. The 0.1 mol L−1 ammonium acetate stage (at pH 3) was included as a rinsing step between the ammonium acetate and HNO3 extractions and ensured that all exchangeable cations were extracted before moving on to the 1 mol L−1 nitric acid stage which is a strong non-selective extraction capable of dissolving many secondary mineral phases such allophane, iron and aluminium organometallic complexes, some part of hydrated iron and aluminium oxides, clay minerals and readily weathered primary minerals67.
Sample analysis
Major element concentrations were measured by ICP-AES (AGILENT 7500 series). 44/40Ca, 26/24Mg and 41/39K isotope ratios were measured by ICP-MS (Analytik Jena 820MS). Instrument optimization and methods are detailed in van der Heijden et al.45. All samples were diluted or evaporated to the same concentration: 100 µg L−1 Mg, 100 µg L−1 Ca, and 200 µg L−1 K. Ammonium acetate was digested prior to isotopic analysis with 5 mL hydrogen peroxide (H2O2 30%) after complete sample evaporation. Instrumental mass discrimination was corrected using the standard bracketing technique by measuring standards of known isotopic composition every 12 samples: the bracketing standards were selected to ensure that enrichment of each sample fell between two standards. The tracer detection limit was set at 10‰ for 26Mg and 44Ca and 20‰ for 41K45. Tracer recovery was also considered below the detection limit when the sample elemental concentrations were too low to conduct isotope ratio analysis.
Calculation methodology
Calculation methodology
The isotopically exchangeable pool is in isotopic equilibrium with the soil solution. The quantification of this pool is thus based on the assumption that its isotopic composition is equal to that measured in the solution using (shown for Mg):
where \({E}_{Mg}\) is the isotopically exchangeable pool (µg g dry soil−1), \({Mg}_{label}\) is the amount of isotopically enriched Mg added into the system (µg g dry soil−1) and \({\alpha }_{solution}^{Mg}\) is the fraction of Mg in the solution originating from the initial tracing solution, calculated as:
where \((\%^{26} Mg)_{solution}\) is the 26Mg atomic abundance in the solution, \((\%^{26} Mg)_{nat}\) is the natural 26Mg atomic abundance, and \((\%^{26} Mg)_{label}\) is the 26Mg atomic abundance of the initial tracing solution.
The relative distribution of the stable isotope tracers in the different extracted pools of the soil was calculated as the amount of Ca, Mg and K originating from the tracing solution (tracer recovery) in each pool divided by the sum of tracer recovered in all three extractions, and quantifies the relative contribution of each extracted soil pool (i.e. NH4#1, NH4#2 and HNO3) to the isotopically exchangeable pool of Ca, Mg and K.
The isotopically exchangeable fraction of each extracted soil pool (the proportion of each pool that is isotopically exchangeable) was calculated by assuming that its isotopic composition is equal to that of the solution:
where \((\%^{26} Mg)_{pool}\) is the 26Mg atomic abundance in the extracted pool and \((\%^{26} Mg)_{solution}\) the fraction of Mg in the solution originating from the initial enriched tracing solution at the last time step of the isotopic dilution experiment (i.e. 48 h).
Sample validation
Because isotopically enriched samples are sensitive to contamination; the isotopic dilution results for each soil sample were verified and samples were excluded from the dataset according to the following criteria.
-
ECa, EMg and EK data. Samples displaying missing or contaminated data for one or more time steps during the isotopic dilution experiment were excluded.
-
Soil extraction data. Samples were excluded if one or more of the 3 extractions (i.e. NH4#1, NH4#2 and HNO3) was missing or if the cumulated tracer recovery in the whole system (soil + solution) was above 120%.
After data validation, the ECa, EMg and EK dataset was composed of 83 samples for Ca (22 sites), 90 samples for Mg (24 sites), and 114 samples for K (24 sites) over a global dataset of 143 samples. The soil extraction dataset was composed of 119 samples for Ca (26 sites), 93 samples for Mg (23 sites), and 122 samples for K (25 sites).
Statistical methods
Statistical analyses were computed using R version 3.6.168 (https://www.r-project.org/).
The difference between Ex and Exchx were tested over the entire dataset with a non-parametric Wilcoxon–Mann–Whitney signed rank test for Ca, Mg and K. Ex and Exchx normal distribution was tested with a Shapiro–Wilk test and homoscedasticity was tested with a Levene test. Homoscedasticity was met for Ca and K but not for Mg (p-value < 0.05).
The repeatability of Ex measurements was estimated by double replicates for 55 samples and the EX:ExchX ratio was calculated for each replicate. The mean relative error for this replicated dataset was 16%, 14% and 8% for Ca, Mg and K, respectively. A sample was defined as significantly greater than Exchx if Ex:Exchx ratio was greater than the respective mean relative error (i.e. if Ex:Exchx greater than 1.16 for Ca, 1.14 for Mg and 1.08 for K).
Correlations between the different variables of the dataset were tested with a Spearman correlation test. Only statistically significant correlations are presented.
Change history
16 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-81982-2
References
Marschner, H. Mineral Nutrition of Higher Plants (Academic Press, Cambridge, 1995).
Bormann, F. & Likens, G. Nutrient cycling. Science 155, 424–429 (1967).
Ranger, J. & Turpault, M. P. Input–output nutrient budgets as a diagnostic tool for sustainable forest management. For. Ecol. Manage. 122, 139–154 (1999).
Badeau, V., Dambrine, E. & Walter, C. Propriétés des sols forestiers français: Résultats du premier inventaire systématique. Étude Gest. des Sols 6, 165 (1999).
van der Heijden, G. et al. Long-term sustainability of forest ecosystems on sandstone in the Vosges Mountains (France) facing atmospheric deposition and silvicultural change. For. Ecol. Manage. 261, 730–740 (2011).
Johnson, J. et al. The response of soil solution chemistry in European forests to decreasing acid deposition. Glob. Change Biol. 24, 3603–3619 (2018).
Jonard, M. et al. Deterioration of Norway spruce vitality despite a sharp decline in acid deposition: A long-term integrated perspective. Glob. Change Biol. 18, 711–725 (2012).
Bailey, S. W., Horsley, S. B. & Long, R. P. Thirty years of change in forest soils of the Allegheny Plateau, Pennsylvania. Soil Sci. Soc. Am. J. 69, 681–690 (2005).
Hedin, L. O. et al. Steep declines in atmospheric base cations in regions of Europe and North America. Nature 367, 351–354 (1994).
Hedin, L. O. & Likens, G. E. Atmospheric dust and acid rain. Sci. Am. 275, 88–92 (1996).
Likens, G. E. et al. The biogeochemistry of calcium at Hubbard Brook. Biogeochemistry 41, 89–173 (1998).
Lövblad, G., Persson, C., & Roos, E. Deposition of Base Cations in Sweden. Swedish Environmental Protection Agency, Report 5119, ISBN 91-620-5119-9, ISSN 0282-7298. 60 (Stockholm, Sweden, 2000). https://www.naturvardsverket.se/Documents/publikationer/620-6145-3.pdf?pid=2834. Accessed 11 Aug 2020.
Achat, D. L. et al. Quantifying consequences of removing harvesting residues on forest soils and tree growth—A meta-analysis. For. Ecol. Manage. 348, 124–141 (2015).
Thiffault, E. et al. Effects of forest biomass harvesting on soil productivity in boreal and temperate forests—A review. Environ. Rev. 19, 278–309 (2011).
Talkner, U. et al. (2019) Nutritional status of major forest tree species in Germany. In Status and Dynamics of Forests in Germany: Results of the National Forest Monitoring (eds Wellbrock, N. & Bolte, A.) 261–293 (Springer, New York, 2019).
Jonard, M. et al. Tree mineral nutrition is deteriorating in Europe. Glob. Change Biol. 21, 418–430 (2015).
De Oliveira Garcia, W., Amann, T. & Hartmann, J. Increasing biomass demand enlarges negative forest nutrient budget areas in wood export regions. Sci. Rep. 8, 1–7 (2018).
Legout, A., Hansson, K., van der Heijden, G., Augusto, L. & Ranger, J. Chemical fertility of forest soils: Basic concepts. Rev. For. Française 66, 21–32 (2014).
Löfgren, S., Ågren, A., Gustafsson, J. P., Olsson, B. A. & Zetterberg, T. Impact of whole-tree harvest on soil and stream water acidity in southern Sweden based on HD-MINTEQ simulations and pH-sensitivity. For. Ecol. Manage. 383, 49–60 (2017).
Casetou-Gustafson, S. et al. Current, steady-state and historical weathering rates of base cations at two forest sites in northern and southern Sweden: A comparison of three methods. Biogeosciences 17, 281–304 (2020).
van der Heijden, G. et al. Tracing and modeling preferential flow in a forest soil—Potential impact on nutrient leaching. Geoderma 195–196, 12–22 (2013).
van Sundert, K. et al. Towards comparable assessment of the soil nutrient status across scales—Review and development of nutrient metrics. Glob. Change Biol. 26, 392–409 (2020).
Hansson, K. et al. Chemical fertility of forest ecosystems. Part 1: Common soil chemical analyses were poor predictors of stand productivity across a wide range of acidic forest soils. For. Ecol. Manage. 461, 117843 (2020).
Legout, A. et al. Chemical fertility of forest ecosystems. Part 2: Towards redefining the concept by untangling the role of the different components of biogeochemical cycling. For. Ecol. Manage. 461, 117844 (2020).
Lucash, M. S., Yanai, R. D., Blum, J. D. & Park, B. B. Foliar nutrient concentrations related to soil sources across a range of sites in the northeastern United States citation details. Soil Sci. Soc. Am. J. 76, 674–683 (2012).
Rosenstock, N. P. et al. Base cations in the soil bank: Non-exchangeable pools may sustain centuries of net loss to forestry and leaching. Soil 5, 351–366 (2019).
Richardson, J. B., Petrenko, C. L. & Friedland, A. J. Base cations and micronutrients in forest soils along three clear-cut chronosequences in the northeastern United States. Nutr. Cycl. Agroecosyst. 109, 161–179 (2017).
van der Heijden, G., Legout, A., Pollier, B., Ranger, J. & Dambrine, E. The dynamics of calcium and magnesium inputs by throughfall in a forest ecosystem on base poor soil are very slow and conservative: Evidence from an isotopic tracing experiment (26Mg and 44Ca). Biogeochemistry 118, 413–442 (2014).
Smeck, N. E., Saif, H. T. & Bigham, J. M. Formation of a transient magnesium-aluminum double hydroxide in soils of southeastern Ohio. Soil Sci. Soc. Am. J. 58, 470–476 (1994).
van Reeuwijk, L. P. & de Villiers, J. M. Potassium fixation by amorphous aluminosilica gels. Soil Sci. Soc. Am. J. 32, 238–240 (1968).
Collignon, C., Ranger, J. & Turpault, M. P. Seasonal dynamics of Al- and Fe-bearing secondary minerals in an acid forest soil: Influence of Norway spruce roots (Picea abies (L.) Karst.). Eur. J. Soil Sci. 63, 592–602 (2012).
Hall, S. J. & Huang, W. Iron reduction: A mechanism for dynamic cycling of occluded cations in tropical forest soils?. Biogeochemistry 136, 91–102 (2017).
Sparks, D. L. Potassium dynamics in soils. In Advances in Soil Science (ed. Stewart, B. A.) 1–63 (Springer, New York, 1987).
Hinsinger, P. & Jaillard, B. Root-induced release of interlayer potassium and vermiculitization of phlogopite as related to potassium depletion in the rhizosphere of ryegrass. J. Soil Sci. 44, 525–534 (1993).
Falk Øgaard, A. & Krogstad, T. Release of interlayer potassium in Norwegian grassland soils. J. Plant Nutr. Soil Sci. 168, 80–88 (2005).
Hamon, R. E., Bertrand, I. & McLaughlin, M. J. Use and abuse of isotopic exchange data in soil chemistry. Aust. J. Soil Res. 40, 1371–1381 (2002).
Ebelhar, S. A. Labile pool. In Encyclopedia of Earth Sciences Series (ed. Chesworth, W.) 425–426 (Springer, Dordrecht, 2008).
Tendille, C., de Ruere, J. G. & Barbier, G. Echanges isotopiques du potassium peu mobile des sols. C.R Acad. Sci. 243, 87–89 (1956).
Masozera, C. & Bouyer, S. Potassium et calicum labiles dans quelques types de sols tropicaux. in Sur l’emploi des radioisotopes et des rayonnments dans la recherche sur les relations sol-plante, vol. 12 (1971).
Fardeau, J. C., Hétier, J. M. & Jappe, J. Potassium assimilable du sol: Identification au comportement des ions isotopiquement diluables. C.R Acad. Sci. 288, 1039–1042 (1979).
Blume, J. M. & Smith, D. Detrmination of exchangeable calcium and cation-exchange capacity by equilibration with Ca-45. Soil Sci. 77, 9–18 (1954).
Newbould, P. & Russell, R. S. Isotopic equilibration of calcium-45 with labile soil calcium. Plant Soil 18, 239–257 (1963).
Reeve, N. G. & Sumner, M. E. Determination of exchangeable calcium in soils by isotopie dilution. Agrochemophysica 1, 13–18 (1969).
van der Heijden, G., Legout, A., Mareschal, L., Ranger, J. & Dambrine, E. Filling the gap in Ca input-output budgets in base-poor forest ecosystems: The contribution of non-crystalline phases evidenced by stable isotopic dilution. Geochim. Cosmochim. Acta 209, 135–148 (2017).
van der Heijden, G. et al. Measuring plant-available Mg, Ca, and K pools in the soil—An isotopic dilution assay. ACS Earth Sp. Chem. 2, 292–313 (2018).
Graham, E. R. & Fox, R. L. Tropical soil potassium as related to labile pool and calcium exchange equilibria calcium soil analysis. Soil Sci. 3, 318–322 (1971).
Ross, D. S., Matschonat, G. & Skyllberg, U. Cation exchange in forest soils: The need for a new perspective. Eur. J. Soil Sci. 59, 1141–1159 (2008).
Reuss, J. O. & Johnson, D. W. Soil-solution interactions. In Acid Deposition and the Acidification of Soils and Waters (eds Reuss, J. O. & Johnson, D. W.) 33–54 (Springer, New York, 1986).
Salmon, R. C. Cation exchange reactions. J. Soil Sci. 15, 273–283 (1964).
André, J. P. & Pijarowski, L. Cation exchange properties of Sphagnumpeat: Exchange between two cations and protons. J. Soil Sci. 28, 573–584 (1977).
Ponette, Q. Downward movement of dolomite, kieserite or a mixture of CaCO3 and kieserite through the upper layers of an acid forest soil. Water. Air. Soil Pollut. 95, 353–379 (1997).
Sparks, D. L. Inorganic soil components. In Environmental Soil Chemistry (ed. Sparks, D. L.) 43–73 (Academic Press, Cambridge, 2003).
Kosmulski, M. Compilation of PZC and IEP of sparingly soluble metal oxides and hydroxides from literature. Adv. Colloid Interface Sci. 152, 14–25 (2009).
Schwertmann, U. & Fechter, H. The point of zero charge of natural and synthetic ferrihydrites and its relation to adsorbed silicate. Clay Miner. 17, 471–476 (1982).
Grove, J. H., Sumner, M. E. & Syers, J. K. Effect of lime on exchangeable magnesium in variable surface charge soils. Soil Sci. Soc. Am. J. 45, 497–500 (1981).
Kinniburgh, D. G., Jackson, M. L. & Syers, J. K. Adsorption of alkaline earth, transition, and heavy metal cations by hydrous oxide gels of iron and aluminum. Soil Sci. Soc. Am. J. 40, 796–799 (1976).
Myers, J. A., McLean, E. O. & Bigham, J. M. Reductions in exchangeable magnesium with liming of acid Ohio soils. Soil Sci. Soc. Am. J. 52, 131–136 (1988).
Rowley, M. C., Grand, S. & Verrecchia, ÉP. Calcium-mediated stabilisation of soil organic carbon. Biogeochemistry 137, 27–49 (2018).
Simpson, A. J. et al. Molecular structures and associations of humic substances in the terrestrial environment. Naturwissenschaften 89, 84–88 (2002).
Clarholm, M., Skyllberg, U. & Rosling, A. Organic acid induced release of nutrients from metal-stabilized soil organic matter—The unbutton model. Soil Biol. Biochem. 84, 168–176 (2015).
Sowers, T. D., Stuckey, J. W. & Sparks, D. L. The synergistic effect of calcium on organic carbon sequestration to ferrihydrite. Geochem. Trans. 19, 4 (2018).
Meyer, D. & Jungk, A. A new approach to quantify the utilization of non-exchangeable soil potassium by plants. Plant Soil 149, 235–243 (1993).
Moritsuka, N., Yanai, J. & Kosaki, T. Possible processes releasing nonexchangeable potassium from the rhizosphere of maize. Plant Soil 258, 261–268 (2004).
Mareschal, L. Effet des substitutions d’essences forestières sur l’évolution des sols et de leur minéralogie: Bilan après 28 ans dans le site expérimental de Breuil (Morvan) (Henri Poincaré, Nancy, 2008).
York, L. M., Carminati, A., Mooney, S. J., Ritz, K. & Bennett, M. M. The holistic rhizosphere: Integrating zones, processes, and semantics in the soil influenced by roots. J. Exp. Bot. 67, 3629–3643 (2016).
Pradier, C. et al. Rainfall reduction impacts rhizosphere biogeochemistry in eucalypts grown in a deep Ferralsol in Brazil. Plant Soil 414, 339–354 (2017).
Nezat, C. A., Blum, J. D., Yanai, R. D. & Hamburg, S. P. A sequential extraction to determine the distribution of apatite in granitoid soil mineral pools with application to weathering at the Hubbard Brook Experimental Forest, NH, USA. Appl. Geochem. 22, 2406–2421 (2007).
R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2019). https://www.r-project.org/. Accessed 17 Mar 2019.
Acknowledgements
This study was financed by the Agence de l'environnement et de la maîtrise de l'énergie (ADEME), the Région Lorraine and the Office National des Forêts (ONF). We also kindly thank the ONF for providing data and archived soil samples from the Renecofor ICP Forests network. The UR-1138 INRAE—Biogéochimie des Ecosystèmes Forestiers is supported by a grant overseen by the French National Research Agency (ANR) as part of the "Investissements d'Avenir" program (ANR-11-LABX-0002-01, Lab of Excellence ARBRE). We thank all who contributed with data to this study: J. C. R. Almeida, E. Dambrine, A. El Gh’Mari, A. Ezzaïm, J. Fichter, J.L.M. Gonçalves, V. Maquere, R. Marques, L. Mareschal, D. Mohamed Ahamed, J. Ranger, M.-P. Turpault, A. Versini and others. We kindly thank Benoit Pollier for his support conducting experimental analysis. We also thank the 3 anonymous reviewers who helped improve this manuscript. The funding was also provided by Conseil Régional de Lorraine.
Author information
Authors and Affiliations
Contributions
J.B. contributed to the experimental design, carried out the experimental work, analyzed the data and wrote the manuscript. G.H., A.L., L.S.-A. contributed to the experimental design and edited the manuscript, S.L., S.J.H. and J.-P.L. provided soil samples, corresponding databases and additionnal informations, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bel, J., Legout, A., Saint-André, L. et al. Conventional analysis methods underestimate the plant-available pools of calcium, magnesium and potassium in forest soils. Sci Rep 10, 15703 (2020). https://doi.org/10.1038/s41598-020-72741-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72741-w
This article is cited by
-
Magnesium addition increases microbial metabolic efficiency during decomposition of Patagonian leaf litter
Plant and Soil (2024)
-
Effect of freeze–thaw manipulation on phytostabilization of industrially contaminated soil with halloysite nanotubes
Scientific Reports (2023)
-
Nitrogen-bedrock interactions regulate multi-element nutrient limitation and sustainability in forests
Biogeochemistry (2023)
-
Bio-fertilizer Affects Structural Dynamics, Function, and Network Patterns of the Sugarcane Rhizospheric Microbiota
Microbial Ecology (2022)
-
Forest biomass accumulation is an important source of acidity to forest soils: Data from Swedish inventories of forests and soils 1955 to 2010
Ambio (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
