
Abstract
Ready-to-eat (RTE) foods have been considered to be reservoirs of antibiotic resistance bacteria, which constitute direct threat to human health, but the potential microbiological risks of RTE foods remain largely unexplored. In this study, the metagenomic approach was employed to characterize the comprehensive profiles of bacterial community and antibiotic resistance gene (ARG) in 18 RTE food samples (8 RTE meat, 7 RTE vegetables and 3 RTE fruit) in southern China. In total, the most abundant phyla in RTE foods were Proteobacteria, Firmicutes, Cyanobacteria, Bacteroidetes and Actinobacteria. 204 ARG subtypes belonging to 18 ARG types were detected with an abundance range between 2.81 × 10−5 and 7.7 × 10−1 copy of ARG per copy of 16S rRNA gene. Multidrug-resistant genes were the most predominant ARG type in the RTE foods. Chloramphenicol, macrolide-lincosamide-streptogramin, multidrug resistance, aminoglycoside, bacitracin, tetracycline and β-lactam resistance genes were dominant, which were also associated with antibiotics used extensively in human medicine or veterinary medicine/promoters. Variation partitioning analysis indicated that the join effect of bacterial community and mobile genetic elements (MGEs) played an important role in the resistome alteration. This study further deepens the comprehensive understanding of antibiotic resistome and the correlations among the antibiotic resistome, microbiota, and MGEs in the RTE foods.
Similar content being viewed by others


Introduction
Due to the overuse and misuse of antibiotics in agricultural, livestock breeding and human medical environments, the problem of antibiotic resistant (AR) bacteria is growing, and constitutes a real threat to public health1,2. Many environments, such as soil, sludge, surface water and animal waste, have been proven to be important reservoirs for antibiotic resistance genes (ARGs) because many ARGs have been detected in these environments3,4. Recent studies have found that food (meat, vegetables and fruit) not only serve as a reservoir of ARGs and AR bacteria but also as a mediator to transfer ARGs and AR bacteria between the environment and humans by direct contact or indirectly through the consumption of contaminated foods5,6. Humans consume foods containing AR bacteria, and the exchange of ARGs between bacteria from food and human intestinal microorganisms may lead to the accumulation of ARGs in humans, which may affect the efficacy of antibiotics7,8. Consequently, in-depth investigations of the diversity and abundance of bacterial community and ARGs in food are central to establishing the overall picture and essential for management decision frameworks aimed at controlling antibiotic resistance.
Food contains a wide range of microorganisms, including AR bacteria5,9. Ready-to-eat (RTE) foods are not intended to undergo a serve heating step before consumption; hence, RTE foods have been recognized as potential vehicles of microbial food-borne bacteria10. ARGs present in RTE foods can be transmitted to bacteria in the human gut by horizontal gene transfer through mobile genetic elements (MGEs) such as plasmids, transposons and integrons11. This constitutes a direct threat to human health and necessitates more public concern worldwide. Current studies have mainly focused on the prevalence, antimicrobial resistance and genetic diversity of the food-borne pathogens, such as Escherichia coli12,13, Salmonella14, and Staphylococcus aureus15. Guo et al. revealed that out of 99 Escherichia coli isolates, 24.2% (24/99) were resistant to at least one antimicrobial agent in RTE food in Singapore12. Yang et al. observed that the prevalence of Salmonella was 9% among the 221 RTE food samples from Shanghai and twenty isolates were resistant to trimethoprim/sulfamethoxazole and sulfisoxazole14. Yang et al. reported that 75.8% (47/62) of the methicillin-susceptible Staphylococcus aureus (MRSA) were isolated from retail RTE foods in China and all of the MRSA isolates were resistant to three or more antibiotics15.
However, due to the limitations of traditional methods of microbiological isolation culture, a large number of microbiota cannot be isolated16,17. Thus, we cannot fully understand the distribution of AR bacteria in food using such methods. Although many molecular technologies, such as polymerase chain reaction (PCR)18, quantitative PCR (qPCR)19,20 and droplet digital PCR (ddPCR)21 have been reported to determine the distribution and occurrence of ARGs in food-borne bacteria, these methods also suffer from several limitations, including a limited number of validated primers for targeted genes, low-throughput and amplification bias. Fortunately, high-throughput sequencing (HTS)-based metagenomic analysis can overcome these problems and comprehensively provide information about the antibiotic resistomes in microorganisms from different samples22. For example, Ma et al. used a metagenomic approach and network analysis to establish a catalogue of antibiotic resistomes and perform host tracking in drinking water in 25 cities23. HTS-based metagenomic analysis has also been applied to activated sludge4, soil, and sediment samples24. Nevertheless, there are no reports on metagenomic analyses of RTE foods.
In this study, we investigated the distribution patterns of bacterial community and ARGs in 18 RTE foods (including RTE meat, RTE vegetable and RTE fruit) using the HTS-based metagenomic approaches. We then characterized the correlations between bacterial community and antibiotic resistome, and finally revealed the roles of bacterial community and MGEs in the resistome alteration in RTE foods. The data provide new insight into our standing of the distribution of bacterial community and ARGs and could be helpful to knowledge the complicated correlations among bacterial community, MGEs and ARGs in the RTE foods.
Results
Bacterial community structures in the RTE foods
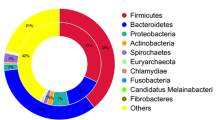
First, 16S rRNA genes were extracted by sequence alignment against the SILVA_128_SSURef_Nr99 database. The results showed that the bacteria in RTE meat, vegetables and fruit were annotated to 41, 61 and 41 phyla, respectively (Supplementary Table S2). Figure 1a showed that the dominate phyla among RTE food samples was similar. The most abundant phyla in RTE foods were Proteobacteria (average = 54.6%), Firmicutes (average = 28.7%), Cyanobacteria (average = 11.9%), Bacteroidetes (average = 2.9%) and Actinobacteria (average = 1.4%) (Relative abundance ≥ 1%; Supplementary Table S2), reaching up to 90%. To study the similarity of different samples, a clustering tree of samples based on the phyla level was constructed using the unweighted Pair-group Method with Arithmetic Means (UPGMA), and the results showed that except for RTE vegetables.1, the bacterial community in the 17 samples could be then clustered into three groups: RTE meat, RTE vegetables and RTE fruit (Fig. 1b). These results were in good accordance with the sample types.

Distributions of bacterial community structures at different taxonomic levels (phylum, family and genus) in the 18 ready-to-eat samples. (a) The top 10 abundant bacterial community structures at the phylum level and the rest were set to “others”. (b) Sample classification using the unweighted Pair-group Method with Arithmetic Means based on the phyla level. (c) The top 10 abundant bacterial community structures at the family level and the rest were set to “others”. (d) The top 10 abundant bacterial community structures at the genus level and the rest were set to “others”.
Figure 1c,d showed that the relative abundance of family and genus differed greatly among the RTE food samples. Vibrionaceae (average = 17.8%) and Leuconostocaceae (average = 11.5%) were the most abundant families in RTE meat samples, Leuconostocaceae (average = 20.5%) and Moraxellaceae (average = 18.2%) were the most abundant families in RTE vegetables samples, Mitochondria (average = 50.4%) and Enterobacteriaceae (average = 22.1%) were the most abundant families in RTE fruit samples (relative abundance ≥ 10%; Fig. 1c; Supplementary Table S3). Weissella (average = 10.7%) was the most abundant genus in RTE meat samples, Acinetobacter (average = 15.9%) and Weissella (average = 11.4%) were the most abundant genus in RTE vegetables samples, unidentified_Mitochondria (average = 24.1%) were the most abundant genus in RTE fruit samples (relative abundance ≥ 10%; Fig. 1d; Supplementary Table S4).
Phylogenetic origins of antibiotic resistance genes in the RTE foods
In order to compare the microbial origin of ARGs with the total microbial genes, the ARGs and the total microbial genes were assigned to different taxa with the assistance of the resistance gene identifier (RGI) and DIAMOND software. In RTE meat samples, the distribution of ARGs and total microbial genes at the phylum level was 61% and 25% for Proteobacteria, 14% and 4% for Firmicutes, respectively (Fig. 2a). In RTE vegetable samples, the assignment of ARGs and total microbial genes at the phylum level was 62% and 39% for Proteobacteria, 17% and 31% for Firmicutes (Fig. 2b). In RTE fruit samples, the distribution of ARGs and total microbial genes at the phylum level was 70% and 21% for Proteobacteria and 9% and 4% for Firmicutes (Fig. 2c). The results suggested that compared with the other genes, the ARGs were more prone to exist in Proteobacteria in RTE foods samples.

Comparison of the distribution of the antibiotic resistance genes (inner cycle) and the total microbial gene set (outer cycle) at the bacterial phylum level. (a) in ready-to-eat meat; (b) in ready-to-eat vegetables; (c) in ready-to-eat fruit. The ratios of genes (> 1%) assigned to each phylum are shown in the pie charts.
Broad-spectrum profiles of ARG abundance in the RTE foods
In total, 204 ARG subtypes belonging to 18 ARG types were detected in at least one of the 18 samples (Fig. 3a and Supplementary Table S5). The ARG diversity differed greatly among the 18 samples, and the number of ARG subtypes was in the range from 32 (RTE fruit.2) to 117 (RTE vegetables.5). The abundance of different ARG types in the samples varied greatly, from a ratio of 2.81 × 10−5 (Methicillin resistance genes in “RTE fruit.3”) to 7.7 × 10−1 (chloramphenicol resistance genes in “RTE meat.1”; Supplementary Table S6). Figure 3b showed the ARG-type composition in the 18 samples. In general, resistance genes associated with chloramphenicol, multidrug resistance, macrolide-lincosamide-streptogramin (MLS), aminoglycoside, bacitracin, tetracycline and β-lactam were more abundant and commonly distributed than other ARG types in these samples. Multidrug-ARGs, which encode resistance to multiple antimicrobial drugs, were found to be the most dominant in the RTE foods, with an abundance of 3.7 × 10−2–5.8 × 10−1 ratios (Supplementary Table S6). Additionally, the core-pan rarefaction curve indicated that as increase in the amount of sequencing data and sample size would not yield more new genes, illustrating that the current depth and the sample size was sufficient for microbial diversity investigation (Fig. 1S).

Characterization of antibiotic resistance gene (ARG) and mobile genetic elements (MGEs) in the 18 ready-to-eat (RTE) samples. (a) Broad-spectrum quantitative profile of ARG types (copy of ARG per copy of 16S rRNA gene) in the 18 samples. (b) Composition of ARG types in the 18 samples. (c) Comparison of the relative abundance of the antibiotic resistance genes in RTE meat, RTE vegetables and RTE fruit. Relative abundance refers to the proportion of the antibiotic resistance genes in the total genes in each type of RTE food samples. The line and the square in the diamond boxes denote the median and mean, respectively. The samples are shown as dots on the left. RTE meat: n = 8; RTE vegetables: n = 7; RTE fruit: n = 3. (d) The relative abundance of MGEs including integrons, plasmids and transposons in the 18 samples.
In order to compare the abundance of ARGs in the RTE meat, RTE vegetables and RTE fruit, we computed the relative enrichment of each of the genes based on sequencing coverage by using the original Illumina GA short reads. Figure 3c showed that the ARGs accounted for 0.86%, 0.30% and 0.45% of the total microbial genes in the RTE meat, RTE vegetables and RTE fruit, respectively. However, the RTE meat did not display the statistical difference with RTE vegetables and RTE fruit in AGRs abundance (Mann–Whitney test, P = 0.52445 and 0.35833, respectively). In addition, the relative abundance of MGEs differed greatly among the RTE food samples (Fig. 3d).
Representative ARG types in the RTE foods
There were 204 ARG subtypes detected in the 18 samples, and we considered the top 10 most abundant ARG subtypes in each sample type to be representative ARGs (Table 1). For RTE meat, catA1 was the most abundant ARG, but it was not among the top 10 abundant gene types in RTE vegetables or fruit (Table 1). The bacitracin resistance gene bacA was the second, first, and third most abundant ARG type in RTE meat, vegetables and fruit, respectively. The tetracycline resistance genes tetM (average = 2.22E−04), tetL (average = 2.20E−04) and tetA (average = 1.57E−04) were much more abundant in RTE meat than other types of RTE food. The MLS resistance gene ermB and the aminoglycoside resistance gene aph(3`)-IIIa were the fifth and seventh most abundant gene types in RTE meat, respectively. The multidrug resistance genes acrB and emrD were very common in all RTE foods (Supplementary Table S7), and both were in the top 10 most abundant gene types in RTE vegetables and fruit.
For RTE vegetables and fruit, representative ARG subtypes mainly belonged to multidrug resistance (mexF, acrB, mexB, mexW, emrD, tolC, acrA, mdtH and mdfA), bacitracin (bacA), tetracycline (tet39), aminoglycoside (ksgA) and macrolide (macB) resistance genes (Table 1). acrB and bacA were found in 100% of RTE food samples (Supplementary Table S7).
Shared ARGs among RTE meat, vegetable and fruit samples
Venn diagram was further applied to analyse the composition of shared ARGs among the RTE foods. We identified 82 AGRs belonging to 13 types shared by RTE meat, vegetables and fruit (Fig. 4a). The shared ARGs accounted for 56.94%, 44.09% and 83.67% of the total number of ARGs detected in RTE meat, vegetables and fruit, respectively (Supplementary Table S8). Among the shared ARGs, resistance genes related to multidrug resistance, bacitracin, aminoglycosides and tetracycline were the most abundant in RTE foods, accounting for > 60% of the total shared ARGs in each type of RTE food (Fig. 4b). Figure 4c shows a comparison of ARGs and their abundance among RTE meat, vegetable and fruit samples. The percentage of one specific ARG in each RTE food is equal to its corresponding abundance divided by the sum of its abundance in the three types of RTE food. The results showed that the abundance of genes related to tetracycline (average = 1.8E−1), sulphonamides (average = 3.7E−2), beta-lactams (average = 5.6E−2), aminoglycosides (average = 1.6E−1) and quinolone (average = 0.18) was much higher in RTE meat than in RTE vegetables or fruit. However, the abundance of multidrug resistance genes (average = 5.7E−1) was much higher in RTE vegetables and fruit than in RTE meat.

The composition and distribution of shared antibiotic resistance gene (ARG) in ready-to-eat (RTE) meat, vegetable and fruit samples. (a) Venn diagram showing the number of shared ARG subtypes among RTE meat, RTE vegetable and RTE fruit samples. (b) Composition of shared ARGs in RTE meat, RTE vegetable and RTE fruit. (c) Ternary plot comparing the abundance of the 82 shared ARG subtypes in RTE meat, RTE vegetable and RTE fruit. The sum of the abundance of one specific ARG in the three types of RTE foods was set as 100%. In the ternary plot, the percentage (%) of one specific ARG in each type of food was equal to its corresponding abundance divided by the sum of its abundance in the three types of RTE food. The symbol size indicates the abundance of ARGs (copies of ARG/copies of 16S rRNA gene).
Co-occurrence between ARG subtypes and microbial taxa in the RTE foods
The diversity of microbial community and ARGs in representative RTE food were displayed in Supplementary Table S9. There was a significant Spearman’s rank correlation (Spearman’s ρ = 0.83 ~ 0.86, P value < 1.0E−5) between the microbial diversity and the ARGs diversity (Fig. 5a,b). The network analysis approach was applied to explore the co-occurrence patterns between ARG subtypes and microbial taxa (Fig. 5c). According to a previous study23,24, it was hypothesized that the non-random co-occurrence patterns between ARGs and microbial taxa could indicate possible host information for AGRs if the ARGs and co-existing microbial taxa possessed significantly similar abundance trends among the different environments (Spearman’s ρ > 0.8, P value < 0.01). Forsberg verified that some specific microbial taxa carrying some specific ARGs led to the corresponding similar abundance trends25. The modularity index of 0.359 indicates that the co-occurrence pattern network has a modular structure26,27. The detailed co-occurrences between ARG subtypes and microbial taxa are summarized in Supplementary Table S10. A total of 8 genera were identified as possible host ARGs based on the co-occurrence results, and Vibrio, Enhydrobacter, Acinetobacter, Klebsiella, Pseudomonas and Enterobacter were present in all samples. Klebsiella was the host of multidrug resistance genes (Acr, macAB, emrD, mdtG, mdtH, mdtL, mdtK), the fosmidomycin resistant gene rosab, the aminoglycoside resistance gene ksgA and other resistance gene bcr_mfs. Enterobacter appeared to possess diverse resistance genes, including multidrug resistance genes (mdtK, emrD, mdtG, mdtL, mdtH, macAB, mdfA and Acr), the fosmidomycin resistance gene rosab, the aminoglycoside resistance gene ksgA and other resistance genes (bcr_mfs). By contrast, Pseudomonas mainly possessed multidrug resistance genes (mexEF, mexVW and mexAB), whereas the multidrug resistance gene (norM) was only associated with Vibrio. Acinetobacter was found to be the host of multidrug resistance gene ADeABC. Eubacterium rectal group appeared to carry the MLS resistance gene erm while Enhydrobacter was found to be the host of the fosmidomycin resistance gene rosab. Empedobacter harboured the multidrug resistance genes (Acr, mdtK and emrD) and other resistant gene bcr_mfs.

The correlation analysis and network analysis between antibiotic resistance genes (ARGs) and microbial taxa in ready-to-eat (RTE) foods. (a) Correlation analysis of the ARGs diversity with microbial diversity at subtype level. (b) Correlation analysis of the ARGs diversity with microbial diversity at reference sequence level. (c) Network analysis showing the co-occurrence patterns between antibiotic resistance gene subtypes and microbial taxa in RTE foods. The nodes were colored according to antibiotic resistance gene types and genus. A connection represents a strong (Spearman’s correlation coefficient ρ > 0.8) and significant (P value < 0.01) correlation. The size of each node is proportional to the number of connections.
Correlations among bacterial community, MGEs and ARGs in the RTE foods
Canonical correspondence analysis (CCA) and variation partitioning analysis (VPA) were performed to further analyze the potential links among bacterial community structure, MGEs and ARGs distribution. CCA showed that Methylotenera, Trichococcus, Prevotella, Osillibacter, Integrons, Comamonas, Bacteroides, transposons (ISs), plasmid, Acholeplasmia, Pseudomonas and Treponema played important roles in shaping the ARG profiles in the RTE food samples, which contributed 98.82% of ARG shifts (Fig. 6a,b). Methylotenera, Trichococcus, Prevotella and Osillibacter positively correlated with RTE vegetables.1 and RTE meat.3, which indicated that their importance in determining ARG compositions in these two samples. Integrons, Comamonas, Bacteroides, transposons and plasmid positively correlated with RTE meat.1. Acholeplasma, Pseudomonas and Treponema were positively correlated with the remaining 15 samples. VPA further revealed that the joint effects of bacterial community shift and MGEs change contributed more (79.37%) to the resistome alteration than bacterial community shift (18.2%) and MGEs change (1.25%; Fig. 6b).

The correlation analysis among bacterial community, mobile genetic elements (MGEs) and antibiotic resistance genes in the ready-to-eat (RTE) foods. (a) Canonical correspondence analysis (CCA) illustrating relationships between the genera and MGEs in RTE foods. (b) Variation partitioning analysis differentiating effects of bacterial community and MGEs on the resistome alteration.
Discussion
The food chain is considered to be an important contributor to the development and dissemination of antibiotic resistant microbes28,29. There have been many reports on microbial community in raw and RTE foods, but most were based on traditional culture or PCR methods20,30,31. In recent years, although metagenomic sequencing technology has been applied to investigate microbial community in food, most studies have focused on tracking changes in complex microbial community in fermented foods32,33. Moreover, RTE foods have not yet been thoroughly analysed, which are directly eaten without further treatment. The present study showed that the most abundant phyla composition in RTE foods were similar, which were Proteobacteria, Firmicutes, Cyanobacteria, Bacteroidetes and Actinobacteria. By comparison, many previous studies have reported the dominant phyla was Proteobacteria in commercial salad leaf vegetables34, RTE salad35, fresh spinach stored at low temperature35, and RTE lettuce36. However, the bacterial community at the family and genus levels varied differently among the RTE food samples. This indicated that the lower the classification level of microbial community structure in RTE food, the higher the richness of microbial groups was observed. The differences might be due to differences in the food types, growing conditions of food, food processing, or preservation conditions37. In contrast, the microbial community in RTE food at family and genus level was previously less studied. HTS-based metagenomic approach can comprehensively reflect microbial community structure. Additionally, although the 18 samples in this study were collected from six different supermarkets, most of the samples (17/18) were well clustered according to the sample type. The reason why RTE vegetables.1 was clustered with RTE. meat samples may be due to the deviation caused by the food processing. For example, the staffs in the supermarket that have treated the meat have also processed RTE vegetables.1.
To our knowledge, this is also the first comprehensive analysis of the bacterial composition in the RTE foods by the metagenomic approach. 16S rDNA sequencing is another widely used method for identifying food microbial communities. The cost is relatively lower and bioinformatic tools designed for sequencing data analysis are free and easy to operate. Nevertheless, the selection of the hypervariable region for 16S rDNA has been more dependent on published or in-house designed protocols38.By contrast; the metagenomic approach can provide the comprehensive information on genes, structure and organization of the genomes, microbial community structure and evolutionary relationships present in the sample17.
In this study, the top 10 most abundant ARG subtypes in each sample type were considered to be representative ARGs (Table 1). The relative abundances of the 10 ARG subtypes ranged from 7.55 × 10–5 to 3.42 × 10–3 copy of ARG/ copy of 16S rRNA gene. Typically, the relative abundance of ARGs in unpolluted environments ranges from 10–8 to 10–6 copies/16S rRNA, whereas concentrations in highly contaminated sites are often several orders of magnitude higher (e.g., 10–4)39. Previous study revealed that the relative abundance of ARGs (sulI, sulII, blaTEM-1,tetA, tetO, tetQ, and tetW) ranged from 10–6 to 10–1 gene copies per 16S rRNA in the four major Chinese carp, while the relative abundance of ARGs (qac, sul, erm and tetM) ranged from 1.46 × 10–7 to 4.6 × 101 copies/ 16S rRNA in raw and fresh foods16,40. Yao et al. also reported that the relative abundances of qnrD, qnrS, mexF, ermA, ermB, mefA, sul1, and sul2 in most landfills were > 10–4 copies per 16S rRNA, suggesting the presence of highly contaminated ARGs39. Therefore, we should pay more attention on the ARGs with relative abundance > 10–4 copies per 16S rRNA. However, according to previous reports based on PCR methods, well-studied ARG types mainly include resistance genes related to tetracycline, sulphonamides, β-lactams, macrolides, methicillin and aminoglycosides (Supplementary Table S11). Tetracycline and macrolide resistance genes account for nearly 80% of previous studies (Supplementary Table S11). Meanwhile, no more than 50 ARG subtypes are listed in previous studies (Supplementary Table S11). Due to the limited availability of primers used in traditional PCR-based methods, a mere snapshot of possible ARG profiles has been determined for RTE foods, such as tetM, ermB and aac(6′)Ie‐aph(2″) in RTE dishes41. However, HTS-based metagenomic analysis could capture a more comprehensive picture of the correlations among ARG profiles without PCR bias, and can simultaneously analyse 18 ARG types, consisting of 204 ARG subtypes. Furthermore, the results of metagenome would provide the new research perspective in the future. For example, our study revealed that the multidrug resistance genes acrB (relative abundance = 1.95 × 10–4, 4.72 × 10–4, respectively) and ksgA (relative abundance = 1 × 10–4, 3.48 × 10–4, respectively) were both among the top 10 most abundant gene types in RTE vegetables and fruit, which has been barely reported based on PCR-based approaches and could be researched deeply in future.
Multidrug-ARGs were found to be the most dominant in RTE foods with an abundance of 3.7 × 10−2–5.8 × 10−1 ratios. By contrast, environmental data showed that the dominant ARG types in drinking water, animal faeces and activated sludge were multidrug-ARGs, tetracycline-ARGs and aminoglycoside-ARGs, respectively23,24. Many previous studies have reported the prevalence of multidrug-ARGs in RTE foods442,43,44,45,46. Multidrug resistance has been detected in RTE turkey meat products, and multidrug resistance was higher than in raw meat44. Diverse multidrug resistant strains were isolated from RTE Paan, including Salmonella Teko and Salmonella Virchow38. Moreover, fishery products such as RTE raw fish and shrimp were once considered to be a vehicle for transmission of multidrug resistant pathogens42,43. Carvalheira et al. reported that lettuce and fruits were the source of multidrug resistant Acinetobacter spp. and 29.8% of the strains were classified as multidrug-resistant47. Due to the long-term use of antibiotics in human and veterinary medicine, the problem of microbial resistance has become increasingly significant. Many studies have demonstrated that antibiotic resistance genes could spread by horizontal gene transfer between different bacterial communities, leading to the widespread prevalence of drug resistance genes and the emergence of multidrug resistance48,49. The multidrug-resistant strains in the foods may be a threat to public health, which may transmit these pathogens to human beings and to the environments that surround them44,45. The multidrug resistance gens identified in the present research may be intrinsic to bacteria associated with the food itself or result from environmental contamination or transmission by food handlers. However, further systematic studies are needed to track contamination pathways of AR bacteria and ARGs in food production and to determine whether these resistance genes can cause potential harm to humans.
By studying the components in the shared ARGs, the results showed that resistance genes related to multidrug resistance, bacitracin, aminoglycosides and tetracycline were most abundant in RTE foods. The shared ARGs might be caused by bacterial community shift or MGEs alteration among food, the environment, and human manipulation50. Tetracycline, sulphonamides, beta-lactams, aminoglycosides and quinolone were found to be much higher in RTE meat than in RTE vegetables or fruit, which may be caused by the overuse and misuse of antibiotics in the livestock breeding51.
We utilized a correlation-based network approach to explore the ARG-species co-occurrence patterns in RTE foods, which helped to propose a useful reference for future studies on risk management of ARGs in RTE foods. Based on the network analysis data obtained in this study, some of the identified ARG hosts have been reported in previous studies. For instance, the resistance gene MexVW is commonly carried by Pseudomonas52, and the resistance gene emrD has been reported in Enterobacter53. Some hosts have been rarely reported previously and should receive more attention in further studies, such as Empedobacter. The dominant genera, Acinetobacter (average = 9.0%), Pseudomonas (average = 3.6%) and Vibrio (average = 2.9%) were found to carry multidrug resistance genes (MexVW, MexEF, AdeABC and so on), which have been always investigated in the RTE foods54,55,56 (Supplementary Tables S2 and S10). Some Acinetobacter spp. (e.g., A. baumannii), Pseudomonas spp. (e.g., P. aeruginosa) and Vibrio spp. (e.g., V. parahaemolyticus) have been considered as opportunistic pathogens, and their capacity of thriving in RTE foods could increase the risks of the exposure and spread of ARGs between the RTE foods and human beings. Thus, we should focus on theses ARG-carrying dominant bacteria observed in RTE foods and how to develop the effective strategies to control AR.
The results from the VPA analysis showed that the joint effects of bacterial community shift and MGEs mainly explain the resistome alteration in the RTE food samples. It is known that MGEs alteration were found to affect the ARG profiles directly in commensal and/or potential pathogens found in the food, such as chicken and vegetable57,58. However, the role of the bacterial community shift for ARGs change in the food was few explored. By contrast, many previous study revealed that the bacterial community played an important role in the shaping the profiles of ARGs among different microorganisms in the environments. Han et al. reported that community composition was the main factor driving changes in ARG abundance in a mariculture sediment59. Liao et al. found that shifts in bacterial community composition were associated with the maintenance of ARGs in food waste composting60. Thus, further work is needed to better understand how bacterial community shift drive ARG alteration in the food samples. Additionally, the CCA analysis explained that Acholeplasma, Pseudomonas and Treponema were positively correlated with the most of the studied RTE food samples. Pseudomonas was the dominant genus in RTE food samples, harboring the multidrug resistant genes, which was presented in all RTE samples (Supplementary Table S10). This may illustrate the phenomena that multidrug-ARGs were the most abundant in RTE foods. Similarity, it was also reported that Pseudomonas carried the multidrug resistant genes52,61.
This is the first study of the distribution of the microbial community and ARGs between RTE foods using the metagenomic approach. These findings could provide a broader perspective on the ARGs and AR bacteria in RTE foods for the government regulation and risk assessment. Future study with larger sample size would help to validate the findings of the current study and reveal more information and biological significance.
Methods
Sampling information and DNA extraction
Basic information on the 18 RTE food samples included in this study is summarized in Supplementary Table S1, including 8 RTE meat (roast pork, roast duck, roast chicken and salmon sushi), 7 RTE vegetables (cucumber, kelp, lotus root and so on) and 3 RTE fruit (fresh-cut hami melon). These samples were randomly purchased from 6 local supermarkets in southern China. The samples were placed in separate sterile plastic bags to prevent cross-contamination and immediately transported to the laboratory in a cooler with ice packs and processed within two hours. Each sample unit was at least 500 g. Each sample was placed in a sterile culture dish and then wiped off the food surface with a disposable sterile specimen collection swab (Copan, Italy) dipped in saline. The wiped sterile specimen collection swabs were placed in sterile 50 ml centrifuge tubes. All these samples were stored at -80 °C, and then transported to Novogene (Beijing, China) with dry ice.
Genomic DNA was extracted using the cetyltriethylammnonium bromide standardization operation protocol (Supplementary Text S1) at Novogene (Beijing, China). DNA degradation degree and potential contamination was monitored on 1% agarose gels. Meanwhile, DNA concentration was measured using Qubit dsDNA Assay Kit in Qubit 2.0 Flurometer (Life Technologies, CA, USA). OD value is between 1.8 and 2.0, DNA contents above 1 μg are used to construct library.
Metagenomic sequencing and bioinformatics analysis
Metagenomic sequencing was performed on an Illumina Novaseq platform (paired-end 150 bp reads) by Novogene (Beijing, China). The generated raw sequences containing 10 or more unknown nucleotides (`N`) or low-quality bases (default quality threshold value ≤ 38) or contaminated by adapters (15-bp overlap) were removed for each sample (FASTQ format) using the quality control pipeline recommended by the sequencing institution. Considering the possibility of host pollution in samples, clean data needed to be blasted against the host database with default parameters using Bowtie 2.2.4 software to filter the reads that were of host origin. The parameters were as follows: –end-to-end, –sensitive, -I 200, -X 40062,63. In addition, trimmed metagenome reads were assembled into scaffolds using SOAPdenovo software (B2.04)64, and the assembled scaffolds from N connections were then interrupted, leaving scaftigs without N65,66,67. All scaftigs were subjected to prediction of open reading frames (ORFs) using MetaGeneMark (V2.10) software65,66,68,69. The complete data totalled 270 Gb, which is the largest sequence dataset reported to date for a study on ARGs in RTE food samples.
The SILVA_128_SSURef_Nr99 database was used to characterize the structure of the bacterial community, where the reads was blasted using the BLAST software (V2.2.25) and the parameters were as follows: -p blastn -m 8 -a 4 -e 1e-20 -b 50, identity ≥ 90, alignment ≥ 7570,71. Briefly, 16S rRNA gene-like sequences from the BLAST results were assigned to NCBI taxonomies, as implemented in the R “Vegan” package, using the unweighted Pair-group Method with Arithmetic Means (UPGMA) algorithm72. All metagenomic sequencing data were searched for ARGs against the Comprehensive Antibiotic Resistance Database (CARD) using the RGI software with an E-value ≤ 1e−3071,73. A package of customized scripts was developed for automatic classification of identified ARG-like sequences into 18 “ARG types” (e.g., tetracycline resistance gene) and 204 “ARG subtypes” (e.g., tetA, tetB, etc.)23,24. The sequence identity was ≥ 90%, and the alignment length was ≥ 25 amino acids24. To assess the ARG distribution in all samples, the abundance of ARGs was expressed as “copy of ARG/copy of 16S rRNA gene” and calculated using the following equation:
where NARG-like sequence is the number of the ARG-like sequence annotated as one specific ARG reference sequence; LARG reference sequence is the sequence length of the corresponding specific ARG reference sequence; N16S sequence is the number of the 16S sequence identified from the metagenomic data; L16S sequence is the average length of 16S rRNA genes (1432 bp) in Greengenes database; n is the number of the mapped ARG reference sequence belonging to the ARG type or subtype and Lreads is the sequence length of the Illumina reads used in the present study24.
To determine the diversity and abundance of MGEs, BLAST tool was applied to annotate sequencing reads against offline databases of MGEs (including integrons, plasmids and transposons), and the parameters were as follows: –identity 90 –alignlen 7544,24,71. The referenced MGEs databases were NCBI NR (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr.gz), NCBI plasmids (ftp://ftp.ncbi.nih.gov/genomes/Plasmids/), and Isfinder (https://www-is.biotoul.fr/), respectively.
Statistical and network analyses
In order to calculate the abundance of genes, the ORF prediction results assembled by each sample were de-redundant using the CD-HIT software to obtain the non-redundant initial gene catalogue. By default, clustering was performed with indentify 95% and coverage 90%, and the longest sequence was selected as the representative sequence. The Bowtie2 was used to compare the clean data of each sample to the initial gene catalogue, and calculate the number of reads for the gene comparison in each sample. Filter out the genes that support the number of reads ≤ 2 in each sample to obtain the gene catalogue (unigenes) for subsequent analysis. The abundance of each gene in each sample was calculated based on the number of reads and the length of genes on the alignment. The calculation equation was shown as follows:
where r is the number of reads annotated to genes, L is the length of genes.
Then, the DIAMOND software was used to compare the unigenes with the MicroNR database to obtain the species annotation. The parameters were as follows: blastp, evalue ≤ 1e−5. For the alignment results of each unigenes, the alignment results of evalue ≤ min evalue × 10 were selected for the further analysis. After filtering, as each unigenes may have many comparison results and obtained different information of species classification. In order to ensure its biological significance, LCA algorithm was applied to take the first branch of classification level as the species annotation information of the unigenes. The abundance of the species was obtained by combing the abundance of the unigenes. In order to analysis the taxonomic affiliation of ARGs, the unigenes was firstly blasted against the CARD using the RGI software to search for ARGs, and the parameters were as follows: evalue ≤ 1e−3074. According to the comparison result of RGI, combined with the abundance of unigenes that annotated to antibiotic resistance ontology (ARO), the relative abundance of each ARO was calculated to obtain the abundance of ARGs. The species annotation information of ARO was obtained from the unigenes annotated ARO. Finally, we used the script software to draw the picture of svg at phylum level.
Ternary plots were implemented in the R “ggtern” package based on the abundance matrix of shared ARG subtypes in all RTE food samples24.
CCA was performed to determine the potential links among bacterial community structure, MGEs and ARGs distribution. VPA was further conducted to determine relative contribution of bacterial community shift and MGEs changes to the resistome alteration. CCA and VPA were conducted using the R “vegan” package75,76.
A co-occurrence network was employed to visualize the correlation between ARG subtypes and microbial taxa. A connection indicated a strong (ρ > 0.8) and significant (P value < 0.01) Spearman’s correlation. The size of each node was proportional to the number of connections, and the thickness of each connection (i.e., edge) between two nodes was proportional to the absolute value of the Spearman’s correlation coefficient. Co-occurrence networks were visualized using the Gephi interactive platform77,78. Various parameters (e.g., number of nodes and edges, average path length, clustering and modularity coefficient) were calculated using the R “igraph” package79.
Data availability
All sequencing data has been deposited in the European Nucleotide Archive (ENA) under accession code PRJEB33440.
References
Allen, H. K. et al. Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259 (2010).
He, L. Y. et al. Discharge of swine wastes risks water quality and food safety: antibiotics and antibiotic resistance genes from swine sources to the receiving environments. Environ. Int. 92–93, 210–219 (2016).
Chen, Q. et al. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ. Int. 92–93, 1–10 (2016).
Ju, F. et al. Antibiotic resistance genes and human bacterial pathogens: co-occurrence, removal, and enrichment in municipal sewage sludge digesters. Water Res. 91, 1–10 (2016).
Founou, L. L., Founou, R. C. & Essack, S. Y. Antibiotic resistance in the food chain: a developing country-perspective. Front. Microbiol. 7, 1881 (2016).
Rolain, J. Food and human gut as reservoirs of transferable antibiotic resistance encoding genes. Front. Microbiol. 4, 173 (2013).
Igbinosa, I. H. Prevalence and detection of antibiotic-resistant determinant in salmonella isolated from food-producing animals. Trop. Anim. Health Prod. 47, 37–43 (2015).
Kirbis, A. & Krizman, M. Spread of antibiotic resistant bacteria from food of animal origin to humans and vice versa. Proc. Food Sci. 5, 148–151 (2015).
Verraes, C. et al. Antimicrobial resistance in the food chain: a review. Int. J. Environ. Res. Public Health 10, 2643–2669 (2013).
Zhang, S., Wu, Q., Zhang, J., Lai, Z. & Zhu, X. Prevalence, genetic diversity, and antibiotic resistance of enterotoxigenic Escherichia coli in retail ready-to-eat foods in China. Food Control 68, 236–243 (2016).
Zhang, T., Zhang, X. & Ye, L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS ONE 6, e26041 (2011).
Guo, S. et al. Phenotypic and genotypic characterization of antimicrobial resistant Escherichia coli isolated from ready-to-eat food in Singapore using disk diffusion, broth microdilution and whole genome sequencing methods. Food Control 99, 89–97 (2019).
Somda, N. S. et al. Safety of ready-to-eat chicken in Burkina Faso: microbiological quality, antibiotic resistance, and virulence genes in Escherichia coli isolated from chicken samples of ouagadougou. Food Sci. Nutr. 6, 1077–1084 (2018).
Yang, J. et al. Prevalence and characterization of antimicrobial resistance in Salmonella enterica isolates from retail foods in Shanghai, China. Foodborne Pathog. Dis. 17, 35–43 (2019).
Yang, X. et al. Prevalence of staphylococcus aureus and methicillin-resistant Staphylococcus aureus in retail ready-to-eat foods in China. Front. Microbiol. 7, 816 (2016).
Xiong, L., Sun, Y., Shi, L. & Yan, H. Characterization of antimicrobial resistance genes and class 1 integrase gene in raw meat and aquatic product, fresh vegetable and fruit, and swine manure in Southern China. Food Control 104, 240–246 (2019).
Cao, Y., Fanning, S., Proos, S., Jordan, K. & Srikumar, S. A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Front. Microbiol. 8, 1829 (2017).
Stetca, G., Mocuta, N., Chintoanu, M. & Miclean, M. Prevalence of Listeria monocytogenes in food, detected by PCR. Curr. Opin. Biotech. 24, S89–S90 (2013).
Wang, H. H. et al. Food commensal microbes as a potentially important avenue in transmitting antibiotic resistance genes. FEMS Microbiol. Lett. 254, 226–231 (2006).
Losio, M. N. et al. Microbiological survey of raw and ready-to-eat leafy green vegetables marketed in Italy. Int. J. Food Microbiol. 210, 88–91 (2015).
Wang, M. et al. Comparison between digital PCR and real-time PCR in detection of Salmonella typhimurium in milk. Int. J. Food Microbiol. 266, 251–256 (2017).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 9, 811–814 (2012).
Ma, L. et al. Catalogue of antibiotic resistome and host-tracking in drinking water deciphered by a large scale survey. Microbiome 5, 154 (2017).
Li, B. et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. Isme J. 9, 2490–2502 (2015).
Forsberg, K. J. et al. Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–616 (2014).
Newman, M. E. & Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E. Stat. Nonlinear Soft Matter Phys. 69, 26113 (2004).
Blondel, V. D., Guillaume, J. L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, 155–168 (2008).
Wang, H., Mcentire, J. C., Zhang, L., Li, X. & Doyle, M. The transfer of antibiotic resistance from food to humans: facts, implications and future directions. Rev. Sci. Tech. 31, 249 (2012).
Gelbíčová, T. et al. Dissemination and comparison of genetic determinants of MCR-mediated colistin resistance in enterobacteriaceae via retailed raw meat products. Front. Microbiol. 10, 2824 (2019).
Tanaka, Y., Watanabe, J. & Mogi, Y. Monitoring of the microbial communities involved in the soy sauce manufacturing process by PCR-denaturing gradient gel electrophoresis. Food Microbiol. 31, 100–106 (2012).
Baert, L., Uyttendaele, M. & Debevere, J. Evaluation of viral extraction methods on a broad range of ready-to-eat foods with conventional and real-time RT-PCR for norovirus GII detection. Int. J. Food Microbiol. 123, 101–108 (2008).
Ji, Y. J. et al. Metagenomic analysis of kimchi, a traditional korean fermented food. Appl. Environ. Microbiol. 77, 2264–2274 (2011).
Wolfe, B. E., Button, J. E., Santarelli, M. & Dutton, R. J. Cheese rind communities provide tractable systems for in situ and in-vitro studies of microbial diversity. Cell 158, 422–433 (2014).
Jackson, C. R., Randolph, K. C., Osborn, S. L. & Tyler, H. L. Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol. 13, 1–12 (2013).
Söderqvist, K. et al. Emerging microbiota during cold storage and temperature abuse of ready-to-eat salad. Infect. Ecol. Epidemiol. 7, 1328963 (2017).
Weiss, A. et al. Analysis of the bacterial epiphytic microbiota of oak leaf lettuce with 16s ribosomal RNA gene analysis. J. Microbiol. Biotechnol. Food Sci. 05, 271–276 (2016).
Bengtsson-Palme, J. Antibiotic resistance in the food supply chain: where can sequencing and metagenomics aid risk assessment?. Curr. Opin. Food Sci. 14, 66–71 (2017).
Claesson, M. J. et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucl. Acids Res. 38, e200 (2010).
Yao, L. et al. Prevalence of fluoroquinolone, macrolide and sulfonamide-related resistance genes in landfills from East China, mainly driven by MGEs. Ecotox. Environ. Safe. 190, 110131 (2020).
Yuan, L. et al. Antibiotic resistance and microbiota in the gut of Chinese four major freshwater carp from retail markets. Environ. Pollut. 255, 113327 (2019).
Chajęcka-Wierzchowska, W., Zadernowska, A., Zarzecka, U., Zakrzewski, A. & Gajewska, J. Enterococci from ready-to-eat food–horizontal gene transfer of antibiotic resistance genes and genotypic characterization by PCR melting profile. J. Sci. Food Agric. 99(3), 1172–1179 (2019).
Duran, G. M. & Marshall, D. L. Ready-to-eat shrimp as an international vehicle of antibiotic-resistant bacteria. J. Food Protect. 68, 2395–2401 (2005).
Hammad, A. M., Watanabe, W., Fujii, T. & Shimamoto, T. Occurrence and characteristics of methicillin-resistant and -susceptible Staphylococcus aureus and methicillin-resistant coagulase-negative staphylococci from Japanese retail ready-to-eat raw fish. Int. J. Food Microbiol. 156, 286–289 (2012).
Khaitsa, M. L., Kegode, R. B. & Doetkott, D. K. Occurrence of antimicrobial-resistant salmonella species in raw and ready to eat turkey meat products from retail outlets in the Midwestern United States. Foodborne Pathog. Dis. 4, 517 (2007).
Singh, B. R. et al. Prevalence of multidrug-resistant salmonella on ready-to-eat betel leaves (Paan) and in water used for soaking betel leaves in North Indian Cities. J. Food Prot. 69, 288–292 (2006).
Campos, J. et al. Microbiological quality of ready-to-eat salads: an underestimated vehicle of bacteria and clinically relevant antibiotic resistance genes. Int. J. Food Microbiol. 166, 464–470 (2013).
Carvalheira, A., Silva, J. & Teixeira, P. Lettuce and fruits as a source of multidrug resistant Acinetobacter Spp.. Food Microbiol. 64, 119–125 (2017).
Levinreisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Martínez, J. L. Antibiotics and antibiotic resistance genes in natural environments. Science 321, 365–367 (2008).
Hudson, J. A. et al. The agri-food chain and antimicrobial resistance: a review. Trends Food Sci. Tech. 69, 131–147 (2017).
Nhung, N. T., Cuong, N. V., Thwaites, G. & Carrique-Mas, J. Antimicrobial usage and antimicrobial resistance in animal production in Southeast Asia: a review. Antibiotics 5(4), 37 (2016).
Li, Y. et al. A new member of the tripartite multidrug efflux pumps, MexVW-OprM, in Pseudomonas aeruginosa. J. Antimicrob. Chemoth. 52, 572–575 (2003).
Alfaresi, M. Whole genome sequencing of Klebsiella pneumoniaestrain unravels a new model for the development of extensive drug resistance in enterobacteriaceae. Open Microbiol. J. 12, 195–199 (2018).
Nyenje, M. E., Odjadjare, C. E., Tanih, N. F., Green, E. & Ndip, R. N. Foodborne pathogens recovered from ready-to-eat foods from roadside cafeterias and retail outlets in Alice, Eastern Cape Province, South Africa: public health implications. Int. J. Environ. Res. Pub. Health. 9, 2608–2619 (2012).
Oranusi, S. U. & Braide, W. A study of microbial safety of ready-to-eat foods vended on highways: Onitsha-Owerri, South East Nigeria. Int. Res. J. Microbiol. (IRJM) 3(2), 066–071 (2012).
Xie, T., Xu, X., Wu, Q., Zhang, J. & Cheng, J. Prevalence, molecular characterization, and antibiotic susceptibility of vibrio parahaemolyticus from ready-to-eat foods in China. Front. Microbiol. 7, 549 (2016).
Manson, A. L. et al. Chicken meat-associated enterococci: influence of agricultural antibiotic use and connection to the clinic. Appl. Environ. Microb. 85, e1519–e1559 (2019).
Caniça, M., Manageiro, V., Abriouel, H., Moran-Gilad, J. & Franz, C. M. A. P. Antibiotic resistance in foodborne bacteria. Trends Food Sci. Tech. 84, 41–44 (2019).
Han, Y. et al. Fishmeal application induces antibiotic resistance gene propagation in mariculture sediment. Environ. Sci. Technol. 51, 10850–10860 (2017).
Liao, H. et al. Horizontal gene transfer and shifts in linked bacterial community composition are associated with maintenance of antibiotic resistance genes during food waste composting. Sci. Total Environ. 660, 841–850 (2019).
Sekiya, H. et al. Functional cloning and characterization of a multidrug efflux pump, MexHI-OpmD, from a pseudomonas aeruginosa mutant. Antimicrob. Agents Ch. 47, 2990 (2003).
Karlsson, F. H. et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245 (2012).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Mende, D. R. et al. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE 7, e31386 (2012).
Nielsen, H. B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Qin, N. et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 (2014).
Oh, J. et al. Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64 (2014).
Zhu, W., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucl. Acids Res. 38, e132 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261 (2007).
Hua, F., Wang, H., Lin, C. & Yu, Y. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ. Sci. Technol. 49, 1095–1104 (2015).
Chen, B. et al. Metagenomic analysis revealing antibiotic resistance genes (ARGs) and their genetic compartments in the tibetan environment. Environ. Sci. Technol. 50, 6670–6679 (2016).
Yang, Y., Li, B., Ju, F. & Zhang, T. Exploring variation of antibiotic resistance genes in activated sludge over a four-year period through a metagenomic approach. Environ. Sci. Technol. 47, 10197–10205 (2013).
McArthur, A. G. et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Ch. 57, 3348 (2013).
Yang, S. L., Zhang, J. & Xu, X. J. Influence of the three gorges dam on downstream delivery of sediment and its environmental implications, Yangtze River. Geophys. Res. Lett. 34 (2007).
Sheik, C. S. et al. Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS ONE 7, e40059 (2012).
Newman, M. Modularity and community structure in networks. APS March Meet. 2006, 8577–8582 (2006).
Newman, M. E. J. The structure and function of complex networks. SIAM Rev. 45, 167–256 (2003).
Csardi, G. The igraph software package for complex network research. Interjournal Complex Syst. 1695 (2006).
Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province of China (Grant No. 2017A030310175); the National Key Research and Development Program of China (Grant No. 2016YFD0500600) and the Guangdong provincial science and technology plan project (Grant No. 2017B020207004).
Author information
Authors and Affiliations
Contributions
L.Y. and L.S. conceived, organized, and supervised the project. Y.M.L, W.W.C and S.L.L interpreted the results and prepared the manuscript. Y.M.L, W.W.C, S.Y and X.C, performed experiments and analyzed the data. All of the authors read and approved the final version of the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Cao, W., Liang, S. et al. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci Rep 10, 15175 (2020). https://doi.org/10.1038/s41598-020-72620-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72620-4
This article is cited by
-
Antimicrobial susceptibility and genotypic characterization of Escherichia coli isolated from foods controlled by the National Food Safety Agency in Burkina Faso
Journal of Consumer Protection and Food Safety (2024)
-
Microbial hitchhikers harbouring antimicrobial-resistance genes in the riverine plastisphere
Microbiome (2023)
-
Natural selenium stress influences the changes of antibiotic resistome in seleniferous forest soils
Environmental Microbiome (2022)
-
Characteristics of antibiotic resistance gene distribution in rainfall runoff and combined sewer overflow
Environmental Science and Pollution Research (2022)
-
Characteristics of fungal communities and the sources of mold contamination in mildewed tobacco leaves stored under different climatic conditions
Applied Microbiology and Biotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
