
Abstract
In patients with idiopathic pulmonary fibrosis (IPF), the effects of antifibrotic agents on the prognosis remain unclear. This study aimed to investigate the impact of antifibrotic treatment on the risks of mortality, hospitalisation, and acute exacerbation in real-world patients with IPF. A total of 1213 IPF patients (biopsy-proven cases: 405) were included in this retrospective study. Propensity score matching was used to adjust for differences in baseline characteristics between patients who received antifibrotic treatment and who did not. A Cox proportional hazard model was used to compare the risks of all-cause mortality, hospitalisation, acute exacerbation, and mortality following acute exacerbation between the two groups. From the 1213 patients, 474 matched pairs were generated. The mean age of the patients in the matched cohort was 65.8 years and 82.8% were men. The median follow-up duration was 27 months. Antifibrotic treatment significantly reduced the risks of mortality [hazard ratio (HR), 0.59; 95% confidence interval (CI), 0.48–0.72; p < 0.001], all-cause hospitalisation (HR 0.71), respiratory-related hospitalisation (HR 0.67), acute exacerbation (HR 0.69), and mortality after acute exacerbation (HR 0.60). Our results suggest that antifibrotic treatment may reduce the risks of all-cause mortality, hospitalisation, acute exacerbation, and mortality after acute exacerbation in patients with IPF.
Similar content being viewed by others

Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrosing interstitial lung disease with a poor prognosis1. Most patients with IPF experience frequent hospitalisations for respiratory and/or non-respiratory causes during the course of the disease2,3,4. As a result, this condition is associated with a high economic burden3 and short-term mortality5. Notably, acute exacerbation (AE) results in in-hospital deaths in approximately 50% of the cases, and seriously impacts the prognosis of IPF patients6. Therefore, assessing how a drug affects the risks of hospitalisation, and AE, in addition to mortality is important when evaluating the efficacy of treatments in patients with IPF.
Both pirfenidone and nintedanib were shown to significantly reduce the rate of decline in forced vital capacity (FVC) in IPF patients in previous clinical trials7,8,9. Since this discovery, several pooled analyses have been conducted to evaluate the effect of antifibrotic agents on clinical outcomes aside from FVC. Pirfenidone was associated with a significant reduction in the risks of mortality10, respiratory-related hospitalisation, and death after hospitalisation11, whereas nintedanib was associated with lower risks of on-treatment mortality and AE rate12. However, these results bear a fundamental limitation in that they were based on clinical trial data; the patients evaluated were a pharmaceutical cohort selected according to strict inclusion criteria and may not represent real-world patients with various comorbidities and different levels of disease severity.
So far, observational studies have evaluated the effect of antifibrotic treatment in a real-world setting13,14,15,16,17,18,19. Correlating with the findings of the previously mentioned clinical trials, these studies showed that antifibrotic treatment reduced FVC decline rate13,14,15,16 and mortality17,18,19, but most of the studies included a small number of patients and were underpowered. Recently, Dempsey et al. reported a mortality benefit of antifibrotic treatment in a large cohort of IPF patients based on an insurance database. However, the study included mostly white patients and Asian population comprised only 3.0% of the total subject. More importantly, evidence demonstrating that antifibrotic treatment reduces the risk of AE was not present. In this study, we aimed to analyse the impact of antifibrotic agents on the risks of all-cause mortality, hospitalisation, and AE in IPF patients in clinical practice.
Methods
Study subjects
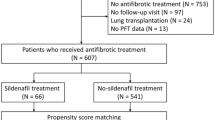
Between January 2004 and December 2017, a total of 1494 patients with IPF were diagnosed at Asan Medical Center, Seoul, Republic of Korea. Patients were excluded if they (1) received concomitant sildenafil, (2) did not attend a follow-up visit after diagnosis, (3) underwent lung transplantation, or (4) did not have baseline pulmonary function test results. Finally, 1213 IPF patients (biopsy performed in 405 patients) were included in this study (Fig. 1). Some of these patients had been included in previous studies6,20,21,22. All patients fulfilled the IPF diagnostic criteria of the American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society, and Latin American Thoracic Association1. The diagnosis of each patient was made through a multidisciplinary discussion.

Flowchart of patient selection. IPF idiopathic pulmonary fibrosis, PFT pulmonary function test.
To assess the effect of antifibrotic agents, patients were classified into two groups: an antifibrotic group and a no-antifibrotic group. The antifibrotic group comprised patients who received either pirfenidone or nintedanib for the treatment of IPF at least once and the no-antifibrotic group comprised patients who did not receive antifibrotic agents during the study period. The index date was set as the date of the first prescription of an antifibrotic agent in the antifibrotic group and the date of IPF diagnosis in the no-antifibrotic group. Patients were followed from the index date until the occurrence of the study outcome or June 30, 2019. This study was approved by the Institutional Review Board of Asan Medical Center (No.: 2019-0735), and the requirement for informed consent was waived due to the retrospective nature of the study. All methods were performed in accordance with the relevant guidelines and regulations of the journal.
Study data and outcomes
Clinical and survival data for all patients were retrospectively obtained from medical records, telephone interviews, and/or the records of the National Health Insurance of Korea. Spirometric parameters23, diffusing capacity of the lung for carbon monoxide (DLCO)24,25, and total lung capacity26 were measured according to the ERS/ATS recommendations, and the results were presented as percentages of the normal predicted values.
The study outcomes included the risks of all-cause mortality, all-cause hospitalisation, respiratory- and non-respiratory-related hospitalisation, AE, and mortality after AE. Respiratory-related hospitalisation was defined as an unexpected admission due to acute respiratory worsening such as pneumonia, pneumothorax, pulmonary embolism, and AE. Non-respiratory-related hospitalisation was defined as an unexpected admission because of a non-respiratory problem such as acute coronary syndrome. AE was defined according to the criteria suggested by Collard et al.27 Follow-up visit usually every 3–6 months and hospitalisation records were reviewed to identify the development of the study outcomes.
Statistical analysis
All values are expressed as mean ± standard deviation for continuous variables or as percentages for categorical variables. The student’s t-test was used for continuous data, and Pearson’s chi-squared test or Fisher’s exact test was used for categorical data.
We performed propensity score matching to adjust for differences in baseline characteristics between the antifibrotic and no-antifibrotic groups. The matched variables were age, sex, body mass index (BMI), FVC, DLCO, corticosteroid use in the 6 months prior to the index date. Survival was evaluated by Kaplan–Meier survival analysis and the log rank test. The relative risks of mortality, hospitalisation, and AE were analysed using a Cox proportional hazards model.
We also performed additional analyses in study subjects that did not exclude those treated with sildenafil. Sildenafil treatment and Charlson Comorbidity Index were matched in addition to the aforementioned variables. Study outcomes were investigated in these patients to see whether the inclusion of sildenafil users alter the study results. All p-values were two-tailed, and statistical significance was set at p < 0.05. All statistical analyses were performed using SPSS software (version 22.0; IBM Corporation, Somers, NY, USA) or R version 3.3.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Baseline characteristics
The median follow-up duration of all 1213 patients was 27 months (interquartile range: 17–44 months). The mean age of the patients was 66.4 years and 82.0% were men. Antifibrotic treatment was administered to 541 patients (44.6%) (Fig. 1). In the unmatched cohort, patients without antifibrotic treatment were older and had a lower BMI, FVC, and DLCO than those who received antifibrotic treatment (Table 1). Propensity score matching was performed to adjust for these differences, and 474 matched pairs were created. The baseline characteristics of the patients included in the matched cohort and those excluded are shown in Supplementary Table S1. The patients excluded from the matched cohort were older and showed greater lung function than those included.
The mean age of the total patients in the matched cohort was 65.8 years and 82.8% were men (Supplementary Table S1). The median follow-up duration was 27 months (antifibrotic: 24 months vs. no-antifibrotic: 33 months, p < 0.001) and the median time from diagnosis to start of antifibrotic treatment was 11 months (interquartile range, 1–43 months). The median duration of antifibrotic treatment was 16 months. In the antifibrotic group, pirfenidone was the most commonly used medication and was prescribed to 429 patients (90.5%). Nintedanib was administered to 85 patients (17.9%). Forty patients (8.4%) were administered pirfenidone and nintedanib in sequence; treatment was switched from pirfenidone to nintedanib (6.8%) or vice versa (1.7%) due to intolerance. High-dose N-acetylcysteine (NAC) was prescribed to 290 patients (61.2%) in the no-antifibrotic group, whereas 1 patient (0.2%) in the antifibrotic group received high-dose NAC alongside pirfenidone.
Clinical course and survival
Table 2 shows the number of patients in the matched cohort who experienced at least one study outcome. A total of 522 patients (55.1%) died during the study period, and the median survival duration was 42 months (95% confidence interval [CI], 39.0–45.0). The antifibrotic group exhibited a significantly longer survival duration (median: 52 vs. 36 months; p < 0.001) than the no-antifibrotic group (Fig. 2). The annual mortality rates were also significantly lower in the antifibrotic group (1-year: 10.7% vs. 19.4%; 3-year: 34.2% vs. 50.3%; 5-year: 50.3% vs. 70.9%, all p < 0.001).

Comparison of survival curves between the antifibrotic and no-antifibrotic groups of patients with idiopathic pulmonary fibrosis. Kaplan–Meier survival curves are shown for the antifibrotic (solid line) and no-antifibrotic (dotted line) groups.
A total of 362 patients (38.2%) experienced unexpected hospitalisation at least once during follow-up. Overall, 243 patients (25.6%) experienced respiratory-related hospitalisations, 82 (8.6%) experienced non-respiratory-related hospitalisations, and 37 (3.9%) experienced both. Furthermore, 195 patients (20.6%) experienced AE, and 149 of them (90.3%) died after the development of AE during follow-up. As shown in Table 2, significantly fewer patients experienced hospitalisation (respiratory- and non-respiratory- related) and AE in the antifibrotic group. In addition, the number of deaths after the development of AE was significantly lower in the antifibrotic group. The median survival duration after AE was 4 months in the antifibrotic group and 1 month in the no-antifibrotic group (p < 0.001, Fig. 3).

Comparison of survival after the development of acute exacerbation between the antifibrotic and no-antifibrotic groups. Kaplan–Meier survival curves are shown for the antifibrotic (solid line) and no-antifibrotic (dotted line) groups.
The causes of the first respiratory-related hospitalisation of the patients (n = 280) in the matched cohort are shown in Supplementary Table S2. In both the antifibrotic and no-antifibrotic groups, the most common cause was AE (44.0% and 43.8%) followed by focal pneumonia (32.0% and 37.1%). There was no statistically significant difference in the cause of respiratory-related hospitalisation between the antifibrotic and no-antifibrotic groups (p = 0.381).
Impact of antifibrotic treatment on prognosis
In the univariate Cox analysis, antifibrotic treatment was associated with a significantly reduced risks of mortality [hazard ratio (HR), 0.59; 95% CI, 0.48–0.72; p < 0.001], all-cause hospitalisation (HR 0.71; 95% CI 0.57–0.88; p = 0.002), respiratory-related hospitalisation (HR 0.67; 95% CI 0.52–0.86; p = 0.002), AE (HR 0.69; 95% CI 0.50–0.96; p = 0.026), and mortality following AE (HR 0.60; 95% CI 0.42–0.85; p = 0.004) in IPF patients (Fig. 4). The risk of non-respiratory-related hospitalisation was lower in the antifibrotic group, although this difference was not statistically significant (HR 0.68; 95% CI 0.46–1.01; p = 0.055).

Forest plot demonstrating the risk of clinical outcomes in the antifibrotic and no-antifibrotic groups. Hazard ratios were calculated from univariable Cox proportional hazard analyses. HR hazard ratio.
Impact of antifibrotic treatment in the study subjects that include sildenafil users
Additional analyses were performed in study subjects including those treated with sildenafil. Propensity score matching yielded 530 matched pairs and their baseline characteristics are described in Supplementary Table 3. Patients who received sildenafil comprised 11.1% and 11.5% of the antifibrotic and no-antifibrotic group, respectively. As compared with the main analysis, inclusion of patients treated with sildenafil did not change the trend of beneficial effects of antifibrotic treatment. All-cause mortality (HR 0.63; 95% CI 0.52–0.75; p < 0.001), all-cause hospitalisation (HR 0.78; 95% CI 0.64–0.95; p = 0.013), respiratory-related hospitalisation (HR 0.76; 95% CI 0.61–0.95; p = 0.015), and mortality after acute exacerbation (HR 0.67; 95% CI 0.49–0.91; p = 0.011) were significantly lower with antifibrotic treatment as shown in Supplementary Figure S1. The risk of acute exacerbation tended to be lower in patients with antifibrotic treatment than in those treated without antifibrotic agents although it was not statistically significant (HR 0.77; 95% CI 0.58–1.04; p = 0.084).
Discussion
Our study results suggest that antifibrotic treatment reduces the risks of all-cause mortality, hospitalisation (all-cause and respiratory-related), AE, and mortality after AE in the real-world cohort with IPF. The risk of all-cause mortality was approximately 40% lower in patients treated with antifibrotic agents than in those not administered antifibrotic agents.
The mortality benefit of antifibrotic agents is thought to persist during follow-up according to the results of our study and previous studies28,29. In open-label extension studies in which patients who finished the phase 3 trials were included to examine the long-term effect and safety profiles of antifibrotic medications, the effect of both pirfenidone and nintedanib on slowing the annual rate of FVC decline persisted beyond the clinical trial periods28,29. Therefore, it seems acceptable to assume that antifibrotic treatment also improves long-term clinical outcomes such as the risks of mortality and hospitalisation, as shown in our study. Interestingly, a recent study which analysed the clinical effectiveness of antifibrotic agents using an insurance database in the US found that the mortality benefit of antifibrotic treatment was observed only during the first 2 years of treatment30. The authors suggested some plausible reasons for this finding, one of which was that lung fibrosis continues despite antifibrotic treatment and vascular remodelling diminishes drug deposition in the lung parenchyma. It is unclear why a difference exists between the US data and our study, but the persistent mortality benefit shown in our study seems reasonable given the results of previous clinical trials and pooled analyses10, 12.
Patients with IPF experience frequent unexpected respiratory-related hospitalisations31, and AE is the most common cause of acute deterioration requiring hospitalisation6. In our study, the proportion of patients who experienced AE was significantly lower in the antifibrotic group. Previous studies have shown conflicting results with regards to AE. In the two replicate randomised phase 3 trials evaluating the efficacy of nintedanib, one showed that nintedanib significantly increased the time to the first AE, whereas the other found that the drug did not significantly affect this outcome8. However, in the pooled analysis, the number of patients who developed AE was lower in the nintedanib group than in the placebo group, and the time to the first AE also significantly decreased with nintedanib treatment (HR 0.53; 95% CI 0.34–0.83; p = 0.0047)12. In a recent Japanese study, perioperative administration of prophylactic pirfenidone was shown to be effective in preventing postoperative AE in lung cancer patients32,33. These data suggest that antifibrotic treatment may prevent the development of AE and support the findings of our study.
Aside from the incidence of AE, we found that the incidence of mortality following AE was also significantly lower in the antifibrotic group. To date, a few studies have suggested that antifibrotic treatment may reduce the risk of mortality after AE6,11,34,35. In a previous pooled analysis that showed that pirfenidone reduced the risk of unexpected hospitalisation, pirfenidone treatment was also found to significantly reduce the risk of mortality after hospitalisation (HR 0.56; 95% CI 0.32–0.99; p = 0.047)11. Given that AE is the most common cause of hospitalisation in IPF patients6, this finding may suggest that pirfenidone reduces the risk of mortality after AE, although this was not specifically investigated in that study. In a small study involving 20 IPF patients admitted to an intensive care unit (ICU) due to severe AE, pirfenidone was associated with a survival benefit34; patients who were taking pirfenidone at the time of admission (n = 11) had a significantly longer median survival after AE (137 days vs. 16 days, p = 0.009) than the control group (n = 9) and tended to have a lower ICU mortality rate (27.3 vs. 77.8%, p = 0.070). With regards to nintedanib, in the post-hoc analysis of the INPULSIS trials (n = 69), the 30-, 90-, and 180-day mortality rates after AE were numerically lower in patients treated with nintedanib than in those who received placebo (30-day: 20.6% vs. 40.0%; 90-day: 29.4% vs. 42.9%; 180-day: 35.3% vs. 57.1%)35. Along with the results of our study, these findings suggest that antifibrotic treatment reduces the risk of not only AE but also mortality after AE. Further studies are required to confirm this promising effect of antifibrotic treatment.
It should be noted that this study has some limitations. First, this was a retrospective observational study performed at a single centre, and this may limit the generalisability of our results. However, the baseline characteristics of our patients were similar to those of the patients included in previous reports28,36. In addition, single centre data could be advantageous in that patient management is less variable than in multicentre cohorts. Second, the follow-up duration was shorter in the antifibrotic group. The index date was the date of first prescription of an antifibrotic agent in the antifibrotic group, whereas it was the date of diagnosis in the no-antifibrotic group. Therefore, the shorter follow-up duration in the antifibrotic group was inevitable. Nonetheless, the favourable effects of antifibrotic agents remained the same even after adjustment for the follow-up period using a Cox proportional hazard model. Third, patients who were treated with sildenafil were not included in this study. In a previous study in which combination treatment with nintedanib and sildenafil was compared with nintedanib alone, combination treatment tended to improve the patients’ health-related quality of life and slow the rate of lung function decline37. Although these benefits were not statistically significant, sildenafil may provide an additional effect to antifibrotic agents, especially in patients with right heart dysfunction38. To eliminate any possible influence of sildenafil, we therefore excluded patients who received sildenafil. Instead, we performed additional analyses including patients who received sildenafil and the results were in favour of antifibrotic treatment. All-cause mortality, all-cause hospitalisation, respiratory-related hospitalisation, and mortality after exacerbation were significantly lower in the antifibrotic group than in the no-antifibrotic group. Acute exacerbation also tended to be lower with antifibrotic treatment. Lastly, more patients treated with pirfenidone than nintedanib were included in this study due to the limited accessibility to nintedanib. In South Korea, unlike pirfenidone, nintedanib is not yet covered by National Health Insurance. Although no clinical trial has directly compared the effectiveness of pirfenidone and nintedanib, it is assumed that pirfenidone and nintedanib exert similar beneficial effects given that both agents were demonstrated to reduce the decline rate in FVC by approximately 50%7,8,9.
In conclusion, our results suggest that in addition to reducing the rate of lung function decline, antifibrotic treatment may also reduce the risks of all-cause mortality, hospitalisation, AE, and mortality after AE in the real-world cohort with IPF.
References
Raghu, G. et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 (2011).
Collard, H.R., Chen, S.Y., Yeh, W.S., Li, Q., Lee, Y.C., Wang, A., Raghu, G. Health care utilization and costs of idiopathic pulmonary fibrosis in U.S. Medicare beneficiaries aged 65 years and older. Ann. Am. Thorac. Soc. 12, 981–987 (2015).
Raimundo, K. et al. Clinical and economic burden of idiopathic pulmonary fibrosis: A retrospective cohort study. BMC Pulm. Med. 16, 2 (2016).
Nishiyama, O. et al. Characteristics and association with survival of respiratory-related hospitalization in Japanese idiopathic pulmonary fibrosis patients. Respir. Investig. 57, 415–421 (2019).
Brown, A. W. et al. Outcomes after hospitalization in idiopathic pulmonary fibrosis: A cohort study. Chest 147, 173–179 (2015).
Song, J. W., Hong, S. B., Lim, C. M., Koh, Y. & Kim, D. S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 37, 356–363 (2011).
King, T. E. Jr. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 (2014).
Richeldi, L. et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2071–2082 (2014).
Noble, P. W. et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet (London, England). 377, 1760–1769 (2011).
Nathan, S. D. et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 5, 33–41 (2017).
Ley, B. et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 196, 756–761 (2017).
Richeldi, L. et al. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir. Med. 113, 74–79 (2016).
Oltmanns, U. et al. Pirfenidone in idiopathic pulmonary fibrosis: real-life experience from a German tertiary referral center for interstitial lung diseases. Respiration. 88, 199–207 (2014).
Salih, G. N., Shaker, S. B., Madsen, H. D. & Bendstrup, E. Pirfenidone treatment in idiopathic pulmonary fibrosis: Nationwide Danish results. Eur. Clin. Respir. J. 3, 32608 (2016).
Skold, C. M. et al. A retrospective chart review of pirfenidone-treated patients in Sweden: The REPRIS study. Eur. Clin. Respir. J. 3, 32035 (2016).
Harari, S. et al. Efficacy of pirfenidone for idiopathic pulmonary fibrosis: An Italian real life study. Respir. Med. 109, 904–913 (2015).
Jouneau, S. et al. A 2-year observational study in patients suffering from idiopathic pulmonary fibrosis and treated with pirfenidone: A French ancillary study of PASSPORT. Respiration. 98, 19–28 (2019).
Zurkova, M. et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir. Res. 20, 16 (2019).
Margaritopoulos, G. A. et al. Pirfenidone improves survival in IPF: Results from a real-life study. BMC Pulm. Med. 18, 177 (2018).
Yoon, H. Y., Park, S., Kim, D. S. & Song, J. W. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir. Res. 19, 203 (2018).
Yoon, H. Y., Kim, D. S. & Song, J. W. Efficacy and safety of pirfenidone in advanced idiopathic pulmonary fibrosis. Respiration. 97, 242–251 (2019).
Song, J. W. et al. Blood biomarkers MMP-7 and SP-A: Predictors of outcome in idiopathic pulmonary fibrosis. Chest 143, 1422–1429 (2013).
Miller, M. R. et al. Standardisation of spirometry. Eur. Respir. J. 26, 319–338 (2005).
Macintyre, N. et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur. Respir. J. 26, 720–735 (2005).
Graham, B. L. et al. ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur. Respir. J. 2017, 49 (2017).
Wanger, J. et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 26, 511–522 (2005).
Collard, H.R., Ryerson, C.J., Corte, T.J., Jenkins, G., Kondoh, Y., Lederer, D.J., Lee, J.S., Maher, T.M., Wells, A.U., Antoniou, K.M. et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 194, 265–75 (2016).
Costabel, U. et al. An open-label study of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis (RECAP). Respir. Int. Rev. Thorac. Dis. 94, 408–415 (2017).
Crestani, B. et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 7, 60–68 (2019).
Dempsey, T. M. et al. Clinical effectiveness of antifibrotic medications for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 200, 168–174 (2019).
Ley, B., Collard, H. R. & King, T. E. Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 183, 431–440 (2011).
Iwata, T. et al. A phase II trial evaluating the efficacy and safety of perioperative pirfenidone for prevention of acute exacerbation of idiopathic pulmonary fibrosis in lung cancer patients undergoing pulmonary resection: West Japan Oncology Group 6711 L (PEOPLE Study). Respir. Res. 17, 90 (2016).
Iwata, T. et al. Effect of perioperative pirfenidone treatment in lung cancer patients with idiopathic pulmonary fibrosis. Ann. Thorac. Surg. 102, 1905–1910 (2016).
Vianello, A. et al. Pirfenidone improves the survival of patients with idiopathic pulmonary fibrosis hospitalized for acute exacerbation. Curr. Med. Res. Opin. 35, 1187–1190 (2019).
Collard, H.R., Richeldi, L., Kim, D.S., Taniguchi, H., Tschoepe, I., Luisetti, M., Roman, J., Tino, G., Schlenker-Herceg, R., Hallmann, C. et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 49 (2017).
Taniguchi, H. et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 35, 821–829 (2010).
Kolb, M. et al. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 379, 1722–1731 (2018).
Behr, J., Kolb, M., Song, J.W., Luppi, F., Schinzel, B., Stowasser, S., Quaresma, M., Martinez, F.J. Nintedanib and sildenafil in patients with idiopathic pulmonary fibrosis and right heart dysfunction. A prespecified subgroup analysis of a double-blind randomized clinical trial (INSTAGE). Am. J. Respir. Crit. Care Med. 200, 1505–1512 (2019).
Funding
This study was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science and Technology (NRF-2019R1A2C2008541).
Author information
Authors and Affiliations
Contributions
Conceptualization: J.W.S. Data acquisition: J.K., J.W.S. Investigation: J.K. Formal analysis: J.K., M.H. Writing—original draft: J.K. Writing—review & editing: J.K., J.W.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, J., Han, M. & Song, J.W. Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: a propensity score matching analysis. Sci Rep 10, 15620 (2020). https://doi.org/10.1038/s41598-020-72607-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72607-1
This article is cited by
-
The impact of antifibrotic use on long-term clinical outcomes in the pulmonary fibrosis foundation registry
Respiratory Research (2024)
-
Antifibrotics and mortality in idiopathic pulmonary fibrosis: external validity and avoidance of immortal time bias
Respiratory Research (2024)
-
Utility of the 52-Gene Risk Score to Identify Patients with Idiopathic Pulmonary Fibrosis at Greater Risk of Mortality in the Era of Antifibrotic Therapy
Lung (2024)
-
Flt1 produced by lung endothelial cells impairs ATII cell transdifferentiation and repair in pulmonary fibrosis
Cell Death & Disease (2023)
-
Interstitielle Lungenerkrankungen und andere progressive pulmonale Fibrosen
Zeitschrift für Pneumologie (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
