
Abstract
The gram-negative, aerobic, rod-shaped bacterium Aeromonas hydrophila, the causative agent of motile aeromonad septicaemia, has attracted increasing attention due to its high pathogenicity. Here, we constructed the complete genome sequence of a virulent strain, A. hydrophila HX-3 isolated from Pseudosciaena crocea and performed comparative genomics to investigate its virulence factors and quorum sensing features in comparison with those of other Aeromonas isolates. HX-3 has a circular chromosome of 4,941,513 bp with a 61.0% G + C content encoding 4483 genes, including 4318 protein-coding genes, and 31 rRNA, 127 tRNA and 7 ncRNA operons. Seventy interspersed repeat and 153 tandem repeat sequences, 7 transposons, 8 clustered regularly interspaced short palindromic repeats, and 39 genomic islands were predicted in the A. hydrophila HX-3 genome. Phylogeny and pan-genome were also analyzed herein to confirm the evolutionary relationships on the basis of comparisons with other fully sequenced Aeromonas genomes. In addition, the assembled HX-3 genome was successfully annotated against the Cluster of Orthologous Groups of proteins database (76.03%), Gene Ontology database (18.13%), and Kyoto Encyclopedia of Genes and Genome pathway database (59.68%). Two-component regulatory systems in the HX-3 genome and virulence factors profiles through comparative analysis were predicted, providing insights into pathogenicity. A large number of genes related to the AHL-type 1 (ahyI, ahyR), LuxS-type 2 (luxS, pfs, metEHK, litR, luxOQU) and QseBC-type 3 (qseB, qseC) autoinducer systems were also identified. As a result of the expression of the ahyI gene in Escherichia coli BL21 (DE3), combined UPLC-MS/MS profiling led to the identification of several new N-acyl-homoserine lactone compounds synthesized by AhyI. This genomic analysis determined the comprehensive QS systems of A. hydrophila, which might provide novel information regarding the mechanisms of virulence signatures correlated with QS.
Similar content being viewed by others


Introduction
The motile gram-negative bacterium Aeromonas hydrophila, recognized as an opportunistic pathogen, has been widely found in a variety of aquatic environments and can infect fish, shrimps, crabs, and other aquatic animals, even possibly causing human diseases resulting from infections by consuming A. hydrophila-contaminated animals1,2,3,4,5. Motile aeromonad septicaemia (MAS) in different fish species is often involved in cases for which A. hydrophila is believed to be the causative agent6, and appears to be distributed globally. MAS disease outbreaks in the fish industry caused a yield reduction of approximately 30–40 thousand tons of fish per year between 1989 and 1993 in southeast China7,8, and led to an estimated economic loss of more than $3 million pounds in 2009 in the southeastern United States9,10,11. Hence, A. hydrophila linked to a variety of fish diseases has generated wide concern in the aquaculture industry.
The pathogenicity of A. hydrophila is mediated by multiple virulence factors, including haemolysin, cytotoxic enterotoxin, aerolysin, adhesins, the S-layer, exoprotease, lipase, elastase, the flagellum and lipopolysaccharides12,13,14,15. In fact, quorum sensing (QS), which regulates gene expression and diverse physiological changes, is usually considered to be the other player in the bacterial infection process16,17. Three typic autoinducer systems, the N-acyl-homoserine lactone (AHL)-based AI-1 system18, the S-ribosylhomocysteinase (LuxS)-based AI-2 system19, and the QseB/QseC-dependent autoinducer AI-3 system20 have been demonstrated to exist in A. hydrophila and to have positive or negative effects on biofilm formation, motility and virulence.
The AI-1 QS system in A. hydrophila based on the AHL is composed of two core proteins, an AHL synthase (AhyI) and a transcriptional regulator (AhyR) encoded by the genes ahyI and ahyR, respectively18,21. A schematic diagram of the AI-1 quorum sensing system in A. hydrophila is presented in Fig. 1a. First, AhyI protein synthetizes AHL molecules, which freely diffuse across cellular membranes and accumulate in both the cell and surrounding environment22. When AHL concentrations exceed a threshold, they are recognized by the AhyR protein to form an AHL-AhyR complex that binds to the lux box region in the intergenic region 10 bp upstream of the ahyI promoter and induces the transcription of the downstream operon23. In a previous study, AhyI was recognized as an acyl-ACP-dependent AHL synthase that utilizes S-adenosyl-L-methionine (SAM) as an amino donor and acyl-acyl carrier protein (acyl-ACP) as an acyl donor to synthetize AHL and release methylthioadenosine (MTA) via acylation and lactonization reactions (Fig. 1b), while acyl-ACP is derived from fatty acid biosynthesis24.

Models of the autoinducer AI-1, AI-2 and AI-3 quorum sensing system of A. hydrophila. (a) Schematic representation of QS regulation by AhyI/AhyR in A. hydrophila. AhyR* represents the active AhyR. (b) Synthesis of AHL catalysed by AhyI protein using acyl-ACP and SAM. Fas II represents type II fatty acid synthesis reactions. (c) A putative regulatory mechanism of the LuxS-type 2 QS system in A. hydrophila. (d) AI-2 synthetic routes based on LuxS protein. DPD undergoes further reactions to form distinct biologically active signal molecules (AI-2), including (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuryl borate (S-THMF-borate) and (2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran (R-THMF). (e) A putative regulatory mechanism of the QseBC-based AI-3 system in A. hydrophila. The “P” surrounded by a circle represents phosphorylation.
To date, AI-2 synthetic pathways in A. hydrophila have been proposed (Fig. 1d)25. First, SAM is catalysed by methyltransferase to produce S-adenosylhomocysteine (SAH), and the degradation occurs by S-adenosylhomocysteine nucleosidase (Pfs) to form S-ribosylhomocysteine (SRH). Then, LuxS catalyses the cleavage of SRH into 4,5-dihydroxy-2,3-pentanedione (DPD), which spontaneously cyclizes to form a furanone and finally forms AI-2 molecules by possibly reacting with borate (-B). AI-2 molecules can freely diffuse across cell membranes and bind to a periplasmic receptor for indirect transcriptional regulation when the concentration reaches a threshold26,27. However, the AI-2 internalization step of A. hydrophila is not yet known (discontinuous traits). In the current study, a putative AI-2 quorum sensing system in A. hydrophila is presented in Fig. 1c. Transduction of the AI-2 signal and gene regulation in A. hydrophila may be involved in the phosphorylation cascade (-P) via LuxQ, LuxU, and ultimately LuxO, which subsequently activates the transcriptional regulator LitR, thereby repressing lux operon-related gene transcription16.
The AI-3 QS system involved in the regulation of flagella and motility was initially described in enterohaemorrhagic E. coli28, and contains two core components, the response regulator QseB and the sensor kinase QseC. AI-3 molecules are presumed to be one group of eukaryotic hormone-like signals (e.g. epinephrine, Epi, or norepinephrine, NE) associated with interkingdom cross-signalling, and are usually produced by other bacterial cells or other bacterial species28,29. Recently, this system was also found in A. hydrophila20,30, but the synthesis of AI-3 molecules is not yet known. A putative regulatory mechanism of the AI-3 QS system based on the E. coli autoinducer-3 model is presented in Fig. 1e25. First, QseC is activated by AI-3 signals and undergoes autophosphorylation (-P) and then transmits the signal by phosphorylation (-P) to the QseB. Finally, phosphorylated QseB activates the transcription of virulence-related genes and autoregulates the operon of qseBC genes.
Herein, a new member of A. hydrophila HX-3 isolated from Pseudosciaena crocea was identified with QS systems. Its complete genome was compared to those of seven closely related fish-epidemic A. hydrophila strains31,32,33,34,35,36, or other fully sequenced Aeromonas spp. More importantly, this study revealed a complete list of potential virulence-related genes and QS-related genes. Novel insights into the virulence factors/mechanisms of QS regulation contributing to the pathogenicity of A. hydrophila were investigated by functional genomic analysis.
Materials and methods
Bacterial strains and DNA extraction
A. hydrophila HX-3 was previously isolated from spoiled Pseudosciaena crocea24. Bacterial cultures of A. hydrophila HX-3 were grown in Luria–Bertani (LB) broth at 28 °C overnight, and then the genomic DNA of bacteria in the exponential growth phase was extracted using HiPure Bacterial DNA Kits (Magen, Guangzhou, China). The DNA concentration was detected using Qubit (Thermo Fisher Scientific, Waltham, MA, USA) and NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA) instruments, and the integrity of the genomic DNA was analysed by 1% agarose gel electrophoresis.
Whole-genome sequencing
The whole genome was sequenced by Illumina Hiseq combined with third-generation sequencing technology (Pacific Biosciences, Menlo Park, CA, USA). The high-quality genomic DNA of A. hydrophila HX-3 was fragmented with G-tubes and end-repaired to prepare SMRTbell DNA template libraries. Continuous long reads attained from single-molecule real-time sequencing runs (PacBio RS II) were used for de novo genome assembly in Falcon version 0.3.037. A total of 133,453 raw reads with an average length of 8592.1 bp and total size of 1146.64 Mbp were obtained. Furthermore, qualified genomic DNA was converted to 300–400 bp insert-size Illumina libraries using the NEBNext Ultra DNA Library Prep Kit (NEB, USA). The libraries were sequenced on an Illumina HiSeq 4000 sequencer using paired-end technology (PE 150). Raw data generated from the HiSeq Illumina platform were filtered using a quality control analysis by FASTP version 0.20.038. After filtering, clean reads were obtained by (1) removing reads with ≥ 10% unidentified nucleotides; (2) removing reads with ≥ 50% bases having phred quality scores ≤ 20; and (3) removing reads aligned to the barcode adapter. Then, the resulting clean reads were mapped to the A. hydrophila HX-3 genome sequences from the PacBio platform, and the genome assembly above was corrected using Pilon version 1.2339.
Phylogenetic and comparative genomic analysis
Genome-wide comparisons of all 26 Aeromonas organisms were performed by OrthoMCL tools40 to identify orthologous genes, with a similarity cutoff of 30% and an E-value of 1e−5 using DIAMOND BLASTP alignments. The phylogenetic tree based on conserved core single-copy orthologous genes was constructed by Neighbor-joining method with 500 bootstrap replicates statistical support, using MEGA version X. The ANI values between two genome sequences were calculated by the Python module pyani (https://widdowquinn.github.io/pyani/)41. A heatmap based on ANI values was generated by using the package heatmap in statistical software R (version 3.6.3$2020). Pan-genome analysis was carried out using Panseq (https://github.com/chadlaing/Panseq)42. A Venn diagram of the unique/core gene content was generated with a custom pl script using the package VennDiagram.
Functional annotations
Open reading frames (ORFs) were predicted using NCBI prokaryotic genome annotation pipeline43. The rRNAs, tRNAs and ncRNAs were predicted by using rRNAmmer (version 1.2)44, tRNAscan (version 1.3.1)45 and cmscan (version 1.1.2)46, respectively. Repeat elements such as interspersed repeat elements were identified by RepeatMasker (version 4.0.5)47, and tandem repeat elements were identified by TRF (version 4.09)48. Transposon prediction was carried out using TransposonPSI (version 20,100,822)49. Clustered regularly interspaced short palindromic repeats (CRISPR) elements were identified by CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr)50. Genomic islands (Gls) were determined with the web tool IslandViewer (version 4) using four independent methods IslandPick, SIGI-HMM, IslandPath-DIMOB and Islander51. Prophages were identified using the PHASTER web server (https://phaster.ca/)52. The genome sequence was annotated by BLAST against the NCBI non-redundant protein (Nr), Cluster of Orthologous Groups of proteins (COG), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), UniProt/Swiss-Prot, protein families (Pfam), Carbohydrate-Active enZymes (CAZy), Pathogen-Host Interactions (PHI), Virulence Factors of Pathogenic Bacteria (VFDB) and Comprehensive Antibiotic Resistance Database (CARD) databases. Two-component systems (TCSs) were predicted based on their structural characteristics53. Details of the Nr, COG, GO, KEGG and Swiss-Prot annotations can be found in Supplementary Information C. The web bTSSfinder server (https://www.cbrc.kaust.edu.sa/btssfinder) was used to predict promoter regions and Shine-Dalgarno (SD) sequences54. The complete genome sequence has been deposited in GenBank under accession number CP046954.
Cloning and expression of the ahyI gene
The ahyI gene was amplified from genomic DNA of strain HX-3 using the following primer set: 5ʹ-CCGAATTCATGTTGGTTTTCAAAGGAAAATTAAAAGAAC-3ʹ (forward) and 5ʹ-TGCTCGAGTTATTCTGTGACCAGTTCGC-3ʹ (reverse). The PCR products with EcoRI and XhoI restriction sites (underlined) were cloned into pET-30a (+) to generate the recombinant plasmid pET30a-ahyI, and then transformed into E. coli BL21 (DE3). Kanamycin (20 μg/mL) was added to select the transformants. The AHL products of the screened transformants were detected using the Chromobacterium violaceum CV026 biosensor in response to short-chain AHLs with four to eight carbons and further analysed by UPLC-MS/MS.
AHL extraction and detection
E. coli BL21 (DE3) with the recombinant plasmid pET30a-ahyI was cultured in 20 mL of LB broth supplemented with kanamycin (20 μg/mL) at 28 °C, induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at an OD600 of 0.6, and incubated for an additional 12 h at 28 °C. The AHL extraction method was performed as described previously55. UPLC-MS/MS analysis was carried out according to the methods described by Jin et al.24, with slight modification. A detailed description of the UPLC-MS/MS methods is presented in Supplementary Information A “Supplementary Methods”.
Results and discussion
Genomic features of A. hydrophila HX-3
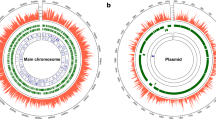
The complete genome of A. hydrophila HX-3 comprises one circular chromosome of 4,941,513 bp with a G + C content of 61.0% (Fig. 2). The genome contains 4483 predicted genes, of which 4318 are coding DNA sequences (CDSs). No plasmids were found during the HX-3 genome analysis. Previous studies have shown that the antimicrobial resistance and virulence in pathogenic A. hydrophila may be plasmid-mediated. For instance, a 21-kb plasmid plays a pivotal role in the specific virulence and pathogenicity of A. hydrophila VB21 when injected in Clarias batrachus56. A 165,906-bp circular plasmid, pR148, confers on A. hydrophila isolates the antimicrobial resistance against streptomycin, chloramphenicol, mpicillin, tetracycline, and sulfamonomethoxine57. However, plasmids as the mobile elements were absent in the HX-3 genome sequence, which indicated that they could be dispensable elements to impact the overall environmental fitness and virulence in a plasmid-negative A. hydrophila infection.

Circular map for the whole genome of A. hydrophila HX-3. From the outside to the centre: genome sequence coordinates, genes encoded on forward and reverse chains (different colours based on COG categories), ncRNA (red: rRNA, black: tRNA), GC content, and GC skew (G − C/G + C).
Comparative analysis of HX-3 genome and 25 fully sequenced Aeromonas species genomes was performed to confirm the evolutionary relationships. A phylogenetic tree constructed based on conserved core single-copy orthologous gene alignment showed the clear lineage divergence, and Aeromonas species all formed monophyletic branches (Fig. 3a). Four terminal branches were clearly separated for eight virulent A. hyrophila strains. Further, five A. hyrophila members (NJ-35, ML09-119, AL09-71, JBN2301 and D4) clustered together with a high bootstrap value support (100%), which suggested that they had a close evolutionary relationship. A. hyrophila HX-3 formed an independent branch and was distinct from other seven A. hyrophila isolates tested. In order to further confirm evolutionary relationship, ANI phylogenetic analysis was performed to estimate genomic differences and relatedness between two genomes. As a result (Fig. 3b and Supplementary Information B “ANI values”), the genomes of eight A. hydrophila strains shared ANI values ranging from 96.64 to 99.99%, which are values above the threshold of 94–96% identity usually used to serve as a speciation boundary58. On the other hand, the A. hydrophila HX-3 genome was found to share identities with the other seven A. hyrophila strains of the ANI group of less than 97%, which indicated that HX-3 appeared to be a distinct strain from those currently classified within the A. hydrophila group.

Genomic comparison of the A. hydrophila HX-3 with other Aeromonas species. GenBank assembly accession for 26 strains showed in the Supplementary Information B “ANI values”. (a) Phylogenetic tree constructed based on conserved core single-copy orthologous genes of 26 Aeromonas strains complete genomes. Eight A. hydrophila strains are colored in blue, red and yellow, respectively. The same color indicates A. hyrophila members clustered on a branch. (b) Heatmap of 26 strains based on the average nucleotide identity (ANI) values between two Aeromonas species. (c) Venn diagram of core and unique genes shared by comparison of eight closely related A. hydrophila strains.
As shown in the Venn diagram constructed for 8 closely related A. hyrophila genomes (Fig. 3c), the pan-genome consisted of 5778 genes, including 3674 core genes (63.59%), 1226 accessory genes (21.22%), and 878 unique genes (15.19%). Variability in the number of unique genes was observed from 3 to 360 genes, and the least and most number of unique genes were identified in A. hyrophila AL09-71 and AL06-06, respectively. Notably, the strain HX-3 contains 229 unique genes and shares higher number of core genes with strains D4 (3918), ML09-119 (3902) and JBN2301 (3889). These results confirmed the high conservation and diversity of genome structure of the eight virulent A. hyrophila strains. Additionally, RNAmmer predicted HX-3 contains ten rRNA operons (encoding 11, 10, and 10 copies of 5S rRNA, 16S rRNA, and 23S rRNA genes, respectively) consistent with those in the genomes of seven other A. hydrophila strains (Table 1). A total of 127 tRNA genes were predicted by tRNAscan-SE, equal to the numbe in the genome of A. hydrophila ATCC 7966T. Intriguingly, the numbers of tRNAs in strain HX-3, ATCC 7966T and JBN2301 were clearly higher than those in the other five strains. There are at least two copies of tRNA genes for each specific amino acid, and the tRNA genes with the highest copy number of thirteen for methionine are encoded in the genome of A. hydrophila HX-3. Based on comparisons with the Rfam database, a total of 7 non-coding RNAs (ncRNAs), otherwise known as regulatory RNAs, were predicted by the software cmscan, consistent with the results for the genomes of A. hydrophila D4 and ML09-119, except that more were obtained than in the other four strains.
Genetic repeat elements (interspersed or tandem repeats) are components of gene regulatory networks and have diverse functional roles in evolution, heredity and variation, such as mismatch repair and damage repair of nucleic acid bases59. DNA replication origins for many bacteria and viruses often contain direct repeat, palindrome and simple repeat sequences60. For the repeat element analysis, the results (Fig S2) showed that the repeat elements identified in A. hydrophila HX-3 constituted 1.18% of the whole genome, including 0.20% as interspersed repeat sequences and 0.98% as tandem repeats. Among the interspersed repeats, 47 SINEs (short interspersed nuclear elements), 17 LINEs (long interspersed nuclear elements) and 6 DNA elements were identified, while no LTR (long terminal repeat) or unclassified elements were detected. Among the tandem repeats, a total of 153 repeats were identified, including 146 minisatellite DNA repeats and 7 microsatellite DNA repeats, but no satellite DNA was found.
A total of 7 types of transposons were identified in the A. hydrophila HX-3 genome, including 4 full-length Ty1/copia retrotransposons, 2 Ty3/gypsy LTR-retrotransposons and 1 LINE retrotransposon (see Supplementary Information B “Transposon”). Moreover, the strain HX-3 genome contains 8 CRISPR loci with a total of 15 spacers and only one CRISPR-associated (cas) gene (cas5/casD, GQR50_07335), which could confer resistance against the intrusion of mobile elements such as phages and plasmids61 (Supplementary Information B “CRISPR”). A total of 39 GIs with 385 genes were detected in the genome (Supplementary Information B “Gls”). In the GIs, 20 encoded transposases belonged to the IS5, IS630, IS66, IS3, IS5/IS1182, and IS1595 families, and four belonging to GL_017. A total of 116 and 18 genes were identified that encoded hypothetical proteins and tyrosine-type recombinase/integrase, respectively. Two genes in GL_024 and GL_032 were predicted to encode the T6SS proteins VgrG and one in GL_009 was predicted to encode another T6SS effector, Hcp1. Another two genes in GL_006 and GL_037 were predicted to encode T2SS-secreted toxins. Clearly, no prophage sequences were identified.
Gene functional analysis
The COG categories were generated by comparing predicted and known proteins in all completely sequenced genomes of Aeromonas and other bacteria to infer sets of orthologues. The results revealed that a total of 3283 genes were annotated in the COG database and distributed in 22 different functional categories (from A to Z, Fig. 4a). In the COG classification, the five most abundant annotated functions were as follows: (1) general function, encoded by 490 genes, accounting for 12.8% of the total functional annotations; (2) amino acid transport and metabolism function, with 390 classified genes, accounting for 9.61%; (3) transcription function, with 305 classified genes, accounting for 7.52%; (4) signal transduction mechanism function, with 295 classified genes, accounting for 7.27%; (5) energy production and conversion function, with 273 classified genes, accounting for 6.73%. Furthermore, 299 unique genes accounting for 7.37% of the total annotations were classified as having unknown functions, which may be revealed by further functional studies. In addition, only one gene and two genes were involved in RNA processing and modification, and chromatin structure and dynamics, respectively. Genes related to extracellular structure, nuclear structure, and the cytoskeleton were not detected in the genome of HX-3.

Gene functional annotation of the A. hydrophila HX-3 genome in the COG, GO and KEGG databases. (a) COG annotation classifications. The COG functional annotations were divided into 25 categories. The COG categories are shown on the X-axis as alphabets, with category names on the right. (b) GO annotation distribution (level 2). The GO assignments were divided into three categories (level 1) namely, biological process (red), cellular process (green), and molecular function (blue). (c) KEGG annotation distribution. The KEGG orthologies were categorized into five major categories: metabolism (green), genetic information processing (purple), environmental information processing (blue), cellular processes (pink), and organismal systems (orange). (d) Genes encoding two-component systems in A. hydrophila HX-3. TCSs could be categorized into four groups: HK/RR protein pairs (27 gene pairs), HK/RR fusion (17 genes), HK orphans (7 genes) and RR orphans (29 genes).
Gene Ontology (GO) is a functional classification system regarding the functions of genes and gene products. In this study, 783 protein-coding genes were categorized by GO analysis (Fig. 4b) (for details, see Supplementary Information B “GO”). The differentially expressed genes were mainly found in 32 subfunctional items of three major categories: biological process (15 subfunctions), cellular composition (8 subfunctions) and molecular function (9 subfunctions). A total of 1876 differentially expressed genes were annotated as belonging to the biological process category, with most involved in metabolic process (GO:0008152), cellular process (GO:0009987), and single-organism process (GO:0044699). A total of 888 genes were annotated as belonging to the cell component category, with most involved in the subfunctions cell (GO:0005623) and cell part (GO:0044464). A total of 915 genes were annotated as belonging to the molecular function category, of which the differentially expressed genes with the subfunctions catalytic activity (GO:0003824) and binding (GO:0005488) were the most numerous. Altogether, genes annotated to the subfunctions metabolic process, cellular process and catalytic activity were the most numerous.
The KEGG database is an integrated database resource for the functions of gene products and cell metabolic pathways. A total of 2577 genes were annotated in the KEGG orthology (KO) database and divided into five categories: metabolism (1864 genes), genetic information processing (193 genes), environmental information processing (387 genes), cellular processes (245 genes), and organismal systems (12 genes) (Fig. 4c). The KO distribution results showed that the most abundant orthology was global and overview maps from the “Metabolism” category, with 711 genes. The second most abundant was carbohydrate metabolism from the “Metabolism” category (233 genes), followed by membrane transport (213 genes) and amino acid metabolism (207 genes) from the “Environmental information processing” and “Metabolism” categories, respectively. In epidemic A. hydrophila ST25162, three carbohydrate metabolic pathways utilizing myo-inositol, sialic acid and L-fucose are predicted to be the key factors causing the strain to be epidemic by helping it overcome nutritional limitations in vivo and thereby increasing its fitness during infection. However, only two specific metabolic pathways utilizing myo-inositol (GQR50_13900) and sialic acid (GQR50_20745) were identified in our study, which indicated that A. hydrophila HX-3 exhibits less virulence than the clinical isolates.
The analysis of the genome suggests that A. hydrophila HX-3 responds efficiently to environmental changes due to the encoded systems for gene expression regulation, such as bacterial two-component systems (TCSs), composed of a sensor histidine kinase (HK) and a response regulator (RR)63,64,65,66. Upon signal perception, the HK first autophosphorylates on a histidine residue and then transfers the phosphoryl group to the cognate RR, which binds target promoters and thereby regulates gene expression67,68. In the genome of A. hydrophila HX-3, a total of 54 genes encode twenty-seven HK/RR protein pairs and 17 genes encode the independent HK/RR fusion proteins (Fig. 4d). In addition, single histidine kinase and response regulator proteins are encoded by 7 and 29 genes, respectively. TCSs are often the critical regulators of pathogenicity and virulence in A. hydrophila. For instance, one TCS (QseB/QseC) can modulate the in vitro and in vivo virulence of A. hydrophila by affecting motility, protease production, exotoxin secretion and biofilm formation20. The PhoP/PhoQ two-component system that mediates adaptation to Mg2+-limiting environments has a negative regulatory effect on the expression of T3SS and virulence factors69.
Virulence factor analysis
The type II secretion system (T2SS) is a major virulence factor, responsible for the extracellular secretion of protein toxins and degradative enzymes that mediate pathogenic effects70,71. Several T2SS component genes encoding ExeAB (GQR50_02545-02550), ExeN-C (GQR50_19875-19935) and TapD (GQR50_02110) were identified in the genome of A. hydrophila, and these genes were also present in seven other A. hydrophila strains (Table 2). The type III secretion system (T3SS) utilized by some gram-negative bacteria to inject effector proteins into the cytosol of host cells72 was present only in strain AL06-06, which indicates that the T3SS makes a small contribution to the virulence of A. hydrophila HX-3 and other strains. Conversely, a functional type VI secretion system (T6SS) with several T6SS effectors73, including three haemolysin-coregulated proteins (Hcp, ID: GQR50_04035, GQR50_13180, GQR50_17010), three valine-glycine repeats G (VgrG, ID: GQR50_13070, GQR50_13175, GQR50_17005) and one proline-alanine-alanine-arginine repeat (PAAR, ID: GQR50_13075), was identified in the A. hydrophila HX-3 genome (Table 2). In contrast to the T3SS, a gene cluster encoding a T6SS that was present in 7 of the 8 A. hydrophila genomes (except for strain AL09-17) was more widely distributed, which suggested that T6SS can play a significant role in the pathogenicity of these strains.
The role of motility and adhesion in A. hydrophila, enabled by flagella or pili, may facilitate the invasion of fish cell lines74,75. Genes for the polar flagella were found in four gene clusters dispersed throughout the genome of HX-3 (GQR50_07735-07810, 07840–07850, 15535–15655, and 15575–15655), which had the same distribution pattern in the other seven strains, whereas lateral flagella were absent in all strains. Both TEM and SEM analysis (Fig. 5) confirmed that A. hydrophila HX-3 possessed a single polar flagellum and lacked lateral flagella. The genes for three different pili, including the Msh type IV pilus (GQR50_20800-20880), Tap type IV pilus (GQR50_02125-02110, 03235-03240, 05870-05890, 08610, 13680, and 03160) and Type I pilus (GQR50_20150-20175), were found in the A. hydrophila HX-3 genome, and was present in all eight genomes. Interestingly, the Flp type IV pilus was absent in A. hydrophila HX-3 and AL06-06. In contrast to the Tap pilus, the Flp type IV pilus was confirmed to make little or no contribution to the virulence of Aeromonas species against Atlantic salmon, while the Flp pilus is an important factor for adherence to host surfaces76.

Electron micrograph of A. hydrophila HX-3: SEM (a, b) and TEM (c, d).
Other classes of virulence factors are toxins and secreted enzymes, which are also associated with the pathogenic potential of A. hydrophila62,75,77,78. Several typical toxin-encoding genes were identified in the A. hydrophila HX-3 genome, including the cytotoxic enterotoxin Act (GQR50_20590), the two cytotonic enterotoxins Ast (GQR50_18705) and Alt (GQR50_22295), haemolysin AHH1 (GQR50_14970), haemolysin III (GQR50_04325), thermostable haemolysin (GQR50_05755) and RtxA (GQR50_15680). The act and rtxA genes were absent in the AL06-06 genome. In addition, a number of genes encoding secreted enzymes were also detected in the A. hydrophila HX-3 genome and apparently conserved in all of the genomes of A. hydrophila; however, the RTX toxin activator gene rtxC found in the A. hydrophila HX-3 genome (GQR50_15685) was absent in the genomes of A. hydrophila D4, AL09-71, AL06-06, JBN2301 and NJ-35.
Antibiotic and multidrug resistance, two additional classes of virulence factors75,77,79, were well represented in the A. hydrophila HX-3 genome and distributed similarly among all of the A. hydrophila strains. Three β-lactamase genes, ampC (GQR50_06175), ampS (GQR50_22500) and cphA (GQR50_19040), are located on the HX-3 chromosome that also carries genes encoding DsbA (GQR50_21040), polymyxin B ArnA (GQR50_17695), organic hydroperoxide (GQR50_03510-03520) and fosmidomycin (GQR50_00245) resistance proteins. Genes for several multidrug efflux systems were also identified, including members of the RND (GQR50_00105, 07065), DMT (GQR50_10960), SMR (GQR50_03945), MFS (GQR50_09275, 11,130, 13,885, 18,395), and ABC (GQR50_22590) superfamilies.
Iron acquisition is typically recognized as an essential factor for bacterial pathogen survival in the host, and it significantly contributes to A. hydrophila virulence and is a key factor for infectious fish disease development80. In the genome of A. hydrophila HX-3, the genes involved in the synthesis and uptake of amonabactin (well known as a phenolate siderophore) were located within a cluster (GQR50_09810-09840), which confers to the HX-3 isolate the capacity to obtain iron for growth during a systemic A. hydrophila infection81. The amonabactin synthesis gene cluster is widespread in other Aeromonas species, such as the fish pathogen A. salmonicida82, and importantly, the latest study by Balado et al.83 revealed the biosynthetic pathway for amonabactin in A. salmonicida subsp salmonicida. However, genes encoding the synthesis and uptake of anguibactin were not found in any of the A. hydrophila strains. An additional gene cluster for siderophore synthesis (GQR50_05430-05435) was characterized in the HX-3 genome and a gene cluster (GQR50_22585-22605) encoding a hydroxamate siderophore receptor and an ABC transporter system, indicated that HX-3 may use a hydroxamate-type ferric siderophore for iron acquisition. Genes for ferrichrome iron uptake (GQR50_12415-12430), haeme uptake (GQR50_17805-17825), haeme receptor (GQR50_17785), and a ferric uptake regulator (GQR50_14875) were identified in the genomes of HX-3 and other strains.
Quorum sensing system in A. hydrophila
QS is a bacterial communication system involving the secretion and detection of specific signal molecules to regulate gene expression and diverse physiological changes84,85. Recent studies have focused on the relationship between pathogenicity and QS in A. hydrophila. Three different QS systems have been described in A. hydrophila, including the AHL-type 1, LuxS-type 2 and QseBC-type 3 autoinducer systems18,19,20.
Type 1 autoinducer (AI-1) system
Based on the genome analysis, the core genes of the type I QS system encoding AhyR protein (GQR50_19990) and AhyI protein (GQR50_19995) were identified in the HX-3 genome, and the ahyI gene (red arrow) was located downstream of the ahyR gene (blue arrow) in reversed orientation, with an intergenic region of 62 bp (Fig S3b). The ahyI/R and adjacent genes were analysed in comparison with those of the other members of A. hydrophila. The analysis showed that these genes were distributed similarly among all of the A. hydrophila strains and highly homologous to each gene of strain HX-3 (Fig S3a). Based on nucleotide sequence alignment, ahyI and ahyR of strain HX-3 shared 99.04% and 99.36% identity with the homologous genes of A. hydrophila ATCC 7966T, respectively. In addition, a promoter region (highlighted in red) located at the − 29 bp central position upstream of ahyI was predicted, and a lux box sequence (highlighted in green) was identified at position − 52 to − 41 bp position based on the conserved sequences upstream of the promoter region (Fig S3b)23. An SD sequence (highlighted in the underlined bases) was also predicted to be responsible for transcription of ahyI.
To assess the dominant AHL products of the enzyme AhyI, E. coli BL21 (DE3) harbouring pET30a-ahyI was grown in LB medium (kanamycin, 20 μg/mL), and the supernatant extracts were analysed by UPLC-MS/MS. Previous studies confirmed that an increase in retention can be observed as the acyl-chain length increases86,87, which was used to predict the unknown compounds in combination with MS data. Thus, a total of 10 types of AHL compounds were identified based on the retention time and precursor ion m/z 102, and Fig. 6 shows predominantly the following ions: m/z 172 for C4-HSL, m/z 200 for C6-HSL, m/z 228 for C8-HSL, m/z 256 for C10-HSL, m/z 270 for C11-HSL, m/z 284 for C12-HSL, m/z 298 for C13-HSL, m/z 312 for C14-HSL, m/z 326 for C15-HSL and m/z 340 for C16-HSL (mass data shown in Supplementary information A “Fig. S1”). In terms of AHL profiles, diverse acyl-chain lengths of AHLs (C4 ~ C14-HSLs) and the level of saturation and side-chain modification by 3-oxo substituents have been observed in Aeromonas species in culture supernatants24,25. However, most A. hydrophila were observed to produce C4-HSL, C6-HSL or both25, and other members isolated from milk produce three types of HSL, C8-HSL, C12-HSL and C14-HSL88. Moreover, AhyI proteins from A. hydrophila have been previously verified to produce only two types of short-chain AHLs, i.e., C4-HSL and C6-HSL21. In this study, a transformant harbouring pET30a-ahyI was shown to produce 10 types of short- and long-chain AHLs, and the results may indicate the high flexibility of the choice of acyl-ACP utilized by AhyI, consistent with the other AHL synthases RhlI89, BmaI190 and BjaI91 using E. coli ACP as acyl substrates.

Total ion current (TIC) UPLC-MS/MS chromatograms of extracts from recombinant E. coli BL21 (DE3) harbouring pET30a-ahyI. The identified compounds with retention times and principal product ions are listed in the table.
For many years, the functions of AI-1 in A. hydrophila have been studied by constructing ΔahyI and ΔahyR mutants. As shown in Table 3, mutagenesis studies demonstrated that the AI-1 QS system influences the expression of numerous virulence factors in A. hydrophila, including the production of exoprotease, haemolysin, amylase, DNase, the S-layer, and T6SS-associated effectors (Hcp, VgrG) and biofilm formation. Interestingly, many studies reported that haemolysin is upregulated by the AI-1 QS system93,96, but other studies observed downregulation of the production of haemolysin18,95. Overall, AI-1 system-associated genes and AHL compounds produced by AhyI were identified in the present study, suggesting that the AhyRI solo system in A. hydrophila HX-3 plays an important role in pathogenic potential. However, the evidence for the underlying mechanism between the AI-1 QS system and virulence factors in A. hydrophila needs to be reinforced because of conflicting data, i.e., haemolysin production.
Type 2 autoinducer (AI-2) system
The genomic analysis of A. hydrophila HX-3 revealed core genes predicted to be involved in the AI-2 synthesis of the strain. These genes encoded MtnN (homologue of Pfs, GQR50_13920), LuxS (GQR50_19260), MetH (GQR50_20625), MetE (GQR50_12405), MetK (GQR50_06220) and SAM methyltransferase (GQR50_12270), which suggested that A. hydrophila HX-3 could produce AI-2 through the LuxS solo system. In addition, genes for AI-2-related QS regulation were present in the genome of A. hydrophila HX-3. GQR50_03840 encodes a transcriptional regulator of the LitR family, probably controlling the expression of LitR-regulated genes, and GQR50_14630, GQR50_14645 and GQR50_17625 encode three putative signal transduction proteins (AI-2 sensor kinase/phosphatase LuxQ, phosphorelay protein LuxU, regulatory protein LuxO) probably involved in the export of AI-2 molecules. These genes are known to be probably its closing into AI-2 quorum sensing regulation16, while the regulatory mechanism remains unclear, and the level of proof is so far only genetic. Kozlova et al.19 demonstrated that the luxS isogenic mutant of A. hydrophila SSU enhanced biofilm formation and bacterial virulence in a septicemic mouse model and showed a decrease in swimming motility. In addition, the work of Cui et al.97, by constructing a ΔluxS mutant of A. hydrophila ATCC 7966, indicated that the AI-2 QS system upregulates biofilm formation and extracellular protease production. However, the regulatory mechanism of the AI-2 QS system in A. hydrophila remains unclear, and future studies are needed to investigate the detailed relationships between AI-2 system-associated genes and virulence factors.
Type 3 autoinducer (AI-3) system
Based on the genomic analysis, open reading frames for the qseB (GQR50_05730) and qseC (GQR50_05725) genes overlapping by 4 bp at the ATGA motif were found in the A. hydrophila HX-3 genome, which was consistent with the finding of Khajanchi et al.20. A promoter region with an SD sequence was also identified to be responsible for transcription of qseBC (Fig S3c). Compared with the ΔqseB mutant, in A. hydrophila SSU20, the AI-3 QS system enhances virulence, swarming and swimming motility and inhibits biofilm maturation. The study by Meng et al.98 suggested that biofilm formation and haemolytic activity were remarkably decreased in a △qseC mutant of Aeromonas hydrophila NJ-35 and showed no effect on motility, lipase activity or protease activity. In addition, interplay between AI-1 and AI-3 QS systems in A. hydrophila has already been demonstrated, and the transcription of the qseBC locus is negatively regulated by the AI-1 QS system30. While information on AI-3 metabolism in A. hydrophila is limited, future studies will address different functions of the AI-3 QS system in more detail.
Conclusion
In summary, the complete genome of A. hydrophila HX-3 was sequenced and a total of 4318 CDSs were annotated. Comparative genomic analysis of HX-3 genome and 7 fully sequenced A. hydrophila strains revealed the core and pan genomes consisting of 3674 and 5778 genes, respectively. Analysis of putative virulence factors in comparison with the genomes of eight closely related A. hydrophila strains revealed conserved and unique virulence genes of this pathogenic species, including genes related to secretion systems, motility and adhesion, toxins, antibiotic and multidrug resistance, and iron acquisition, among others. In contrast, genes encoding lateral flagella and the synthesis and uptake of anguibactin were absent in all of the A. hydrophila strains. T3SS was not found in seven strains (with A. hydrophila AL06-06 being an exception). However, the Flp type IV pilus gene was missing only from the A. hydrophila HX-3 and AL06-06 genomes. In particular, the RTX toxin activator gene rtxC was absent in the five A. hydrophila genomes (D4, AL09-71, AL06-06, JBN2301, and NJ-35). In addition, genes related to the AI-1(ahyI, ahyR), AI-2 (luxS, pfs, metEHK, litR, luxOQU) and AI-3 (qseB, qseC) regulatory systems were identified to illustrate the comprehensive QS systems of A. hydrophila at the genetic level. Moreover, several new AHL compounds were detected by recombinant AhyI protein using UPLC-MS/MS analysis. Overall, the genomic information of A. hydrophila HX-3 provided with valuable data suggesting a relationship between pathogenicity and QS systems; and a framework for infection and the prevention of quorum sensing.
References
Awan, F. et al. The fight for invincibility: environmental stress response mechanisms and Aeromonas hydrophila. Microb. Pathog. 116, 135–145 (2018).
Janda, J. M. & Abbott, S. L. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23(1), 35–73 (2010).
Parker, J. L. & Shaw, J. G. Aeromonas spp. clinical microbiology and disease. J. Infect. 62(2), 109–118 (2011).
Daskalov, H. The importance of Aeromonas hydrophila in food safety. Food Control 17(6), 474–483 (2006).
Li, J., Ni, X. D., Liu, Y. J. & Lu, C. P. Detection of three virulence genes alt, ahp and aerA in Aeromonas hydrophila and their relationship with actual virulence to zebrafish. J. Appl. Microbiol. 110(3), 823–830 (2011).
Hossain, M. J. et al. Implication of lateral genetic transfer in the emergence of Aeromonas hydrophila isolates of epidemic outbreaks in channel catfish. PLoS ONE 8(11), 1–26 (2013).
Zhang, X., Yang, W., Wu, H., Gong, X. & Li, A. Multilocus sequence typing revealed a clonal lineage of Aeromonas hydrophila caused motile Aeromonas septicemia outbreaks in pond-cultured cyprinid fish in an epidemic area in central China. Aquaculture 432, 1–6 (2014).
Nielsen, M. E. et al. Is Aeromonas hydrophila the dominant motile aeromonas species that causes disease outbreaks in aquaculture production in the Zhejiang province of China?. Dis. Aquat. Org. 46(1), 23–29 (2001).
Griffin, M. J. et al. Rapid quantitative detection of Aeromonas hydrophila strains associated with disease outbreaks in catfish aquaculture. J. Vet. Diagn. Investig. 25(4), 473–481 (2013).
Pridgeon, J. W. & Klesius, P. H. Molecular identification and virulence of three Aeromonas hydrophila isolates cultured from infected channel catfish during a disease outbreak in west Alabama (USA) in 2009. Dis. Aquat. Org. 94(3), 249–253 (2011).
Hossain, M. J. et al. An Asian origin of virulent Aeromonas hydrophila responsible for disease epidemics in United States-farmed catfish. MBio 5(3), e00848-e914 (2014).
Samayanpaulraj, V., Velu, V. & Uthandakalaipandiyan, R. Determination of lethal dose of Aeromonas hydrophila Ah17 strain in snake head fish Channa striata. Microb. Pathog. 127, 7–11 (2019).
Sen, K. & Rodgers, M. Distribution of six virulence factors in Aeromonas species isolated from US drinking water utilities: a PCR identification. J. Appl. Microbiol. 97(5), 1077–1086 (2004).
Sha, J., Kozlova, E. V. & Chopra, A. K. Role of various enterotoxins in Aeromonas hydrophila-induced gastroenteritis: generation of enterotoxin gene-deficient mutants and evaluation of their enterotoxic activity. Infect. Immun. 70(4), 1924–1935 (2002).
Sen, K. & Lye, D. Importance of flagella and enterotoxins for Aeromonas virulence in a mouse model. Can. J. Microbiol. 53(2), 261–269 (2007).
Kozlova, E. V., Khajanchi, B. K., Sha, J. & Chopra, A. K. Quorum sensing and c-di-GMP-dependent alterations in gene transcripts and virulence-associated phenotypes in a clinical isolate of Aeromonas hydrophila. Microb. Pathog. 50(5), 213–223 (2011).
Khajanchi, B. K. et al. N-acylhomoserine lactones involved in quorum sensing control the type VI secretion system, biofilm formation, protease production, and in vivo virulence in a clinical isolate of Aeromonas hydrophila. Microbiol. Read. Engl. 155, 3518–3531 (2009).
Swift, S. et al. Quorum sensing-dependent regulation and blockade of exoprotease production in Aeromonas hydrophila. Infect. Immun. 67(10), 5192–5199 (1999).
Kozlova, E. V. et al. Mutation in the S-ribosylhomocysteinase (luxS) gene involved in quorum sensing affects biofilm formation and virulence in a clinical isolate of Aeromonas hydrophila. Microb. Pathog. 45, 343–354 (2008).
Khajanchi, B. K., Kozlova, E. V., Sha, J., Popov, V. L. & Chopra, A. K. The two-component QseBC signalling system regulates in vitro and in vivo virulence of Aeromonas hydrophila. Microbiol. Read. Engl. 158, 259–271 (2012).
Swift, S. et al. Quorum sensing in Aeromonas hydrophila and Aeromonas salmonicida: identification of the LuxRI homologs AhyRI and AsaRI and their cognate N-acylhomoserine lactone signal molecules. J. Bacteriol. 179(17), 5271–5281 (1997).
Garde, C. et al. Quorum sensing regulation in Aeromonas hydrophila. J. Mol. Biol. 396(4), 849–857 (2010).
Kirke, D. F., Swift, S., Lynch, M. J. & Williams, P. The Aeromonas hydrophila LuxR homologue AhyR regulates the N-acyl homoserine lactone synthase, AhyI positively and negatively in a growth phase-dependent manner. FEMS Microbiol. Lett. 241(1), 109–117 (2004).
Jin, L. et al. Identification of a novel N-acyl-homoserine lactone synthase (AhyI) in Aeromonas hydrophila and structural basis for its substrate specificity. J. Agric. Food Chem. 68(8), 2516–2527 (2020).
Talagrand-Reboul, E., Jumas-Bilak, E. & Lamy, B. The social life of Aeromonas through biofilm and quorum sensing systems. Front. Microbiol. 8, 37 (2017).
Henke, J. M. & Bassler, B. L. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J. Bacteriol. 186(20), 6902–6914 (2004).
Xavier, K. B. & Bassler, B. L. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 187(1), 238–248 (2005).
Sperandio, V., Torres, A. G. & Kaper, J. B. Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol. Microbiol. 43(3), 809–821 (2002).
Clarke, M. B., Hughes, D. T., Zhu, C., Boedeker, E. C. & Sperandio, V. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. USA 103(27), 10420–10425 (2006).
Kozlova, E. V., Khajanchi, B. K., Popov, V. L., Wen, J. & Chopra, A. K. Impact of QseBC system in c-di-GMP-dependent quorum sensing regulatory network in a clinical isolate SSU of Aeromonas hydrophila. Microb. Pathog. 53(3–4), 115–124 (2012).
Zhu, L., Zheng, J. S., Wang, W. M. & Luo, Y. Complete genome sequence of highly virulent Aeromonas hydrophila strain D4, isolated from a diseased blunt-snout bream in China. Microbiol. Resour. Announc. 8(4), e01035-e1118 (2019).
Pridgeon, J. W., Zhang, D. & Zhang, L. Complete genome sequence of the highly virulent Aeromonas hydrophila AL09-71 isolated from diseased channel catfish in West Alabama. Genome Announc. 2(3), e00450-e514 (2014).
Tekedar, H. C. et al. Complete genome sequence of fish pathogen Aeromonas hydrophila AL06-06. Genome Announc. 3(2), e00368-e415 (2015).
Tekedar, H. C. et al. Complete genome sequence of a channel catfish epidemic isolate, Aeromonas hydrophila strain ML09-119. Genome Announc. 1(5), e00755-e813 (2013).
Yang, W., Li, N., Li, M., Zhang, D. & An, G. Complete genome sequence of fish pathogen Aeromonas hydrophila JBN2301. Genome Announc. 4(1), e01615-e1715 (2016).
Seshadri, R. et al. Genome sequence of Aeromonas hydrophila ATCC 7966T: jack of all trades. J. Bacteriol. 188(23), 8272–8282 (2006).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13(12), 1050 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884–i890 (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9(11), 1–14 (2014).
Chen, F., Mackey, A. J., Stoeckert, C. J. & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34(1), D363–D368 (2006).
Pritchard, L. pyani: python module for average nucleotide identity analyses. https://widdowquinn.github.io/pyani/ (2017).
Laing, C. et al. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 11(1), 461 (2010).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44(14), 6614–6624 (2016).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35(9), 3100–3108 (2007).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1), W54–W57 (2016).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22), 2933–2935 (2013).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25(1), 4–10 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2), 573–580 (1999).
Haas, B. TransposonPSI: An Application of PSI-Blast to Mine (retro-) Transposon ORF Homologies (Broad Institute, Cambridge, 2007).
Couvin, D. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46(W1), W246–W251 (2018).
Bertelli, C. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45(W1), W30–W35 (2017).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44(W1), W16–W21 (2016).
Cheung, J. & Hendrickson, W. A. Sensor domains of two-component regulatory systems. Curr. Opin. Microbiol. 13(2), 116–123 (2010).
Shahmuradov, I. A., Mohamad Razali, R., Bougouffa, S., Radovanovic, A. & Bajic, V. B. bTSSfinder: a novel tool for the prediction of promoters in cyanobacteria and Escherichia coli. Bioinformatics 33(3), 334–340 (2017).
Tang, X., Liu, S., Zhang, Z. & Zhuang, G. Identification of the release and effects of AHLs in anammox culture for bacteria communication. Chem. Eng. J. 273, 184–191 (2015).
Majumdar, T. et al. Role of virulence plasmid of Aeromonas hydrophila in the pathogenesis of ulcerative disease syndrome in Clarias batrachus. Indian. J. Biochem. Biophys. 44, 401–406 (2007).
del Castillo, C. S. et al. Comparative sequence analysis of a multidrug-resistant plasmid from Aeromonas hydrophila. Antimicrob. Agents Chemother. 57(1), 120–129 (2013).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106(45), 19126–19131 (2009).
Zhou, K., Aertsen, A. & Michiels, C. W. The role of variable DNA tandem repeats in bacterial adaptation. FEMS Microbiol. Rev. 38(1), 119–141 (2014).
Weber, P., Rausch, C., Scholl, A. & Cardoso, M. C. Repli-FISH (Fluorescence in Situ Hybridization): application of 3D-(Immuno)-FISH for the study of DNA replication timing of genetic repeat elements. OBM Genet. 3(1), 1–31 (2019).
Koonin, E. V. & Makarova, K. S. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol. Rep. 1, 95 (2009).
Pang, M. et al. Novel insights into the pathogenicity of epidemic Aeromonas hydrophila ST251 clones from comparative genomics. Sci. Rep. 5, 9833 (2015).
Galperin, M. Y. A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 5(1), 35 (2005).
Zschiedrich, C. P., Keidel, V. & Szurmant, H. Molecular mechanisms of two-component signal transduction. J. Mol. Biol. 428(19), 3752–3775 (2016).
Wuichet, K., Cantwell, B. J. & Zhulin, I. B. Evolution and phyletic distribution of two-component signal transduction systems. Curr. Opin. Microbiol. 13(2), 219–225 (2010).
Li, J. et al. Complete genome sequence provides insights into the quorum sensing-related spoilage potential of Shewanella baltica 128 isolated from spoiled shrimp. Genomics 112(1), 736–748 (2020).
Mira-Rodado, V. New insights into multistep-phosphorelay (MSP)/Two-component system (TCS) regulation: are plants and bacteria that different?. Plants 8(12), 590 (2019).
Branscum, K. M., Menon, S. K., Foster, C. A. & West, A. H. Insights revealed by the co-crystal structure of the Saccharomyces cerevisiae histidine phosphotransfer protein Ypd1 and the receiver domain of its downstream response regulator Ssk1. Protein Sci. 28(12), 2099–2111 (2019).
Vilches, S., Jimenez, N., Tomás, J. M. & Merino, S. Aeromonas hydrophila AH-3 type III secretion system expression and regulatory network. Appl. Environ. Microbiol. 75(19), 6382–6392 (2009).
Tseng, T. T., Tyler, B. M. & Setubal, J. C. Protein secretion systems in bacterial-host associations, and their description in the gene ontology. BMC Microbiol. 9(S1), S2 (2009).
Rasmussen-Ivey, C. R., Figueras, M. J., McGarey, D. & Liles, M. R. Virulence factors of Aeromonas hydrophila: in the wake of reclassification. Front. Microbiol. 7, 1337 (2016).
Vilches, S. et al. Complete type III secretion system of a mesophilic Aeromonas hydrophila strain. Appl. Environ. Microbiol. 70(11), 6914–6919 (2004).
Pissaridou, P. et al. The Pseudomonas aeruginosa T6SS-VgrG1b spike is topped by a PAAR protein eliciting DNA damage to bacterial competitors. Proc. Natl. Acad. Sci. USA 115(49), 12519–12524 (2018).
Merino, S., Rubires, X., Aguilar, A. & Tomás, J. M. The role of flagella and motility in the adherence and invasion to fish cell lines by Aeromonas hydrophila serogroup O: 34 strains. FEMS Microbiol. Lett. 151(2), 213–217 (1997).
Grim, C. J. et al. Characterization of Aeromonas hydrophila wound pathotypes by comparative genomic and functional analyses of virulence genes. MBio 4(2), e00064-e113 (2013).
Boyd, J. M. et al. Contribution of type IV pili to the virulence of Aeromonas salmonicida subsp. salmonicida in Atlantic salmon (Salmo salar L.). Infect. Immun. 76(4), 1445–1455 (2008).
Grim, C. J. et al. Functional genomic characterization of virulence factors from necrotizing fasciitis-causing strains of Aeromonas hydrophila. Appl. Environ. Microbiol. 80(14), 4162–4183 (2014).
Gao, X. et al. Genomic study of polyhydroxyalkanoates producing Aeromonas hydrophila 4AK4. Appl. Microbiol. Biotechnol. 97(20), 9099–9109 (2013).
Palu, A. P. et al. Antimicrobial resistance in food and clinical Aeromonas isolates. Food Microbiol. 23(5), 504–509 (2006).
Massad, G., Arceneaux, J. E. & Byers, B. R. Acquisition of iron from host sources by mesophilic Aeromonas species. Microbiology 137(2), 237–241 (1991).
Barghouthi, S., Payne, S. M., Arceneaux, J. E. & Byers, B. R. Cloning, mutagenesis, and nucleotide sequence of a siderophore biosynthetic gene (amoA) from Aeromonas hydrophila. J. Bacteriol. 173(16), 5121–5128 (1991).
Reith, M. E. et al. The genome of Aeromonas salmonicida subsp. salmonicida A449: insights into the evolution of a fish pathogen. BMC Genom. 9(1), 427 (2008).
Balado, M. et al. Two catechol siderophores, acinetobactin and amonabactin, are simultaneously produced by Aeromonas salmonicida subsp. salmonicida sharing part of the biosynthetic pathway. ACS Chem. Boil. 10(12), 2850–2860 (2015).
Schuster, M., Joseph Sexton, D., Diggle, S. P. & Peter Greenberg, E. Acyl-homoserine lactone quorum sensing: from evolution to application. Annu. Rev. Microbiol. 67, 43–63 (2013).
Waters, C. M. & Bassler, B. L. Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21, 319–346 (2005).
Gould, T. A., Herman, J., Krank, J., Murphy, R. C. & Churchill, M. E. Specificity of acyl-homoserine lactone synthases examined by mass spectrometry. J. Bacteriol. 188(2), 773–783 (2006).
Ortori, C. A. et al. Comprehensive profiling of N-acylhomoserine lactones produced by Yersinia pseudotuberculosis using liquid chromatography coupled to hybrid quadrupole-linear ion trap mass spectrometry. Anal. Bioanal. Chem. 387(2), 497–511 (2007).
Cataldi, T. R., Bianco, G., Palazzo, L. & Quaranta, V. Occurrence of N-acyl-L-homoserine lactones in extracts of some Gram-negative bacteria evaluated by gas chromatography–mass spectrometry. Anal. Biochem. 361(2), 226–235 (2007).
Raychaudhuri, A., Jerga, A. & Tipton, P. A. Chemical mechanism and substrate specificity of RhlI, an acylhomoserine lactone synthase from Pseudomonas aeruginosa. Biochemistry 44(8), 2974–2981 (2005).
Montebello, A. N. et al. Acyl-ACP substrate recognition in Burkholderia mallei BmaI1 acyl-homoserine lactone synthase. Biochemistry 53(39), 6231–6242 (2014).
Dong, S. H. et al. Molecular basis for the substrate specificity of quorum signal synthases. Proc. Natl. Acad. Sci. USA 114(34), 9092–9097 (2017).
Lynch, M. J. et al. The regulation of biofilm development by quorum sensing in Aeromonas hydrophila. Environ. Microbiol. 4(1), 18–28 (2002).
Bi, Z. X., Liu, Y. J. & Lu, C. P. Contribution of AhyR to virulence of Aeromonas hydrophila J-1. Res. Vet. Sci. 83(2), 150–156 (2007).
Khajanchi, B. Evidence of Water to Human Transmission of Aeromonas Hydrophila: Critical Role of Quorum Sensing in Bacterial Virulence and Host Response. Doctoral dissertation (2016).
Li, D. et al. Acetylation of lysine 7 of AhyI affects the biological function in Aeromonas hydrophila. Microb. Pathog. 140, 103952 (2020).
Chu, W., Jiang, Y., Yongwang, L. & Zhu, W. Role of the quorum-sensing system in biofilm formation and virulence of Aeromonas hydrophila. Afr. J. Microbiol. Res. 5(32), 5819–5825 (2011).
Cui, Y. et al. Construction of LuxS gene deletion mutant strain in Aeromonas hydrophila ATCC7966. Chin. J. Zoo. 32, 462–468 (2016).
Meng, Y. X. et al. Role of sensor histidine kinase QseC of Aeromonas hydrophila in response to norepinephrine. J. Fish Sci. China 80, 1–11 (2020).
Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFD0901105), and the Public Interest Science and Technology Plan of Ningbo (2019C10007).
Author information
Authors and Affiliations
Contributions
The authors' contribution to this manuscript is as follows: J.L. is the frst author on the manuscript; he completed all the experimental work and drafed the manuscript. C.Y. is the second author on the manuscript; she assisted on the AHL products analysis and prepared figures S3. Y.W.G. is the third author on the manuscript; she is the genome project principal investigator, performed the experimental design and assisted on writing of the manuscript. Q.Z.H. is the fourth author on the manuscript; she provided minor comments and prepared figures 1. Z.X.J. is the corresponding author on the manuscript; he provided significant input on the data interpretation, comparative genome analysis and major comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, L., Chen, Y., Yang, W. et al. Complete genome sequence of fish-pathogenic Aeromonas hydrophila HX-3 and a comparative analysis: insights into virulence factors and quorum sensing. Sci Rep 10, 15479 (2020). https://doi.org/10.1038/s41598-020-72484-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72484-8
This article is cited by
-
Heterologous expression of quorum sensing transcriptional regulator LitR and its function in virulence-related gene regulation in foodborne pathogen Aeromonas hydrophila
Molecular Biology Reports (2023)
-
Black pepper and tarragon essential oils suppress the lipolytic potential and the type II secretion system of P. psychrophila KM02
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
