
Abstract
Extrinsic black dental staining is an external dental discoloration of bacterial origin, considered a special form of dental plaque. Currently, there is no definitive therapeutic option for eliminating black stain. This study employed 16S rRNA metagenomics to analyze black stain and white-plaque samples from 27 adult volunteers. Study objectives were to: describe the microbial diversity of adult black stain samples; characterize their taxonomic profile; compare the microbiomes of black stain versus white-plaque from adult volunteers and propose a functional map of the black stain microbiome using PICRUSt2. The black stain microbiome was poorer in species diversity as compared to white-plaque. The five most abundant genera in black stain were Capnocytophaga, Leptotrichia, Fusobacterium, Corynebacterium and Streptococcus. Functional analysis of microbial species revealed conserved and consistent clustering of functional pathways within and between black stain and white-plaque microbiomes. We describe enrichment of heme biosynthetic pathways in black stain. Our results suggest that the dysbiosis in black stain resembles “orally healthy” communities. The increased abundance of heme biosynthetic pathways suggests that heme-dependent iron sequestration and subsequent metabolism are key for black stain formation. Further research should decipher the regulation of heme biosynthetic genes and characterize the temporal sequence leading to colonization and dysbiosis.
Similar content being viewed by others
Introduction
The oral microbiome is the second most complex after the gut microbiome, with an estimated 687 predominant bacterial species1,2. Specific microbial communities can be found for each oral cavity site: tongue, buccal mucosa, tooth surfaces, soft and hard palate, tonsils and lip vestibule. In addition, the salivary microbiome is considered as comprising of a mixture of all those sites’ microbiomes3. Healthy dental plaque on tooth surfaces and gingival margin is considered as being amongst the most stable and diverse microbial communities in the mouth4.
Dental plaque has been clinically defined as “the microbial community that develops as a structurally- and functionally-organized biofilm on the tooth surface, embedded in a matrix of polymers of bacterial and host salivary origin”5. Recent advances in high-throughput sequencing and metagenomics have permitted a deeper understanding of the microbial the composition of dental plaque in healthy individuals. Ten leading (abundance > 1%) genera have been shown to be commonly found in supragingival dental plaque: Streptococcus, Veillonella, Granulicatella, Fusobacterium, Neisseria, Campylobacter, Gemella, Abiotrophia, Selenomonas and Capnocytophaga6. When sub- and supra-gingival plaque microbiomes are analyzed jointly, eleven genera with a relative abundance over 2% have been described: Streptococcus, Veillonella, Prevotella, Neisseria, Fusobacterium, Actinomyces, Leptotrichia, Corynebacterium, Capnocytophaga, Rothia and Porphyromonas7.
It is well established that changes in the composition and structure of the oral microbiome are related to diseases, such as caries, periodontal disease or odontogenic infections3,8. According to the “keystone pathogen” hypothesis by Hajishengallis, oral diseases arise as a consequence of disruptions in the oral microbiome, initiated by low-abundant pathogens, which finally drive an ecological imbalance or dysbiosis9. It was seen than when these ecological shifts occur in dental plaque they lead to caries10. In this context, several recent studies have focused on the study of microbiome changes in dental plaque in individuals with caries6,11,12.
We hypothesize that similar changes may promote the formation of extrinsic black dental staining (BS). The exact origin and nature of these microbial changes currently remain elusive13. BS is considered a special type of dental plaque, formed by microorganisms within an organic matrix14. BS is characterized by the formation of dots or black lines distributed in parallel to the gingival margin of teeth and firmly attached to the enamel15. Children are mainly affected, but it can also be observed in adults, with a prevalence ranging from 1 to 20%16. Currently, there is no long-term therapeutic solution for patients with BS, who commonly undergo repeated rounds of dental prophylactic cleaning in order to maintain a stain-free oral cavity. The observed dark pigment in BS has been described by some authors, as a black insoluble ferric compound formed by bacteria in the dental plaque17. This has been recently confirmed by Zhang et al. who observed higher iron levels in BS patients than in white plaque patients using ICP-MS analysis18. A microbiological analysis of BS samples from children’s teeth highlighted the presence and importance of chromogenic bacteria such as Prevotella melaninogenica, Actinomyces israelii and Actinomyces naeslundii19. Application of PCR techniques have contributed to broaden the knowledge regarding the bacterial composition of BS, nevertheless results remain controversial. Saba et al. found Porphyromonas gingivalis, P. melaninogenica, Actinomyces spp, and Aggregatibacter actinomycemcomitans to be prevalent in BS, while Li et al. revealed a different core community composed of A. actinomycemcomitans, Prevotella intermedia, Cardiobacterium spp, Haemophilus spp, Corynebacterium spp, Tannerella spp and Treponema spp as the main species20,21.
To our knowledge, currently there are only two publications presenting BS microbiome analysis based on metagenomics approaches. Significantly, both studies used dental plaque samples from pediatric patients and reported few or no differences in community diversity between BS and non-BS samples. Differences in taxa relative abundances were reported, with Actinomyces naeslundii found to be more abundant in BS samples and Candidate_division_TM7 more abundant in non-BS samples22. Moreover, Leptotrichia and Fusobacterium were suggested to play an important role in the formation of pigment in primary dentition23.
The main objectives of this study are to: describe the microbial diversity of adult BS; characterize the microbial community found in adult BS; compare the taxonomic profiles of adult BS and white-plaque; and develop a functional map of the adult BS microbiome with a view to advancing the knowledge related to microbial dysbiosis and BS in the oral cavity. To our knowledge, this is the first study to investigate the dysbiosis hypothesis in adult patients with BS using a metagenomics approaches.
Results
Demographic characteristics of the study volunteers
A total of 27 adult volunteers participated in the study. All participants were free of caries, gingivitis or periodontitis, with a DMFT index of < 4.424. Nine were orally healthy individuals whilst 18 had BS in two or more teeth (Fig. 1). The mean age in the control group was (42.9 ± 10.1) whereas the mean age of the patients with BS was (43.8 ± 15.8) (Table 1).

Dental black stain in an adult patient. BS appears as dots or black lines distributed in parallel to the gingival margin of teeth in the cervical third.
Diversity of the BS microbiome
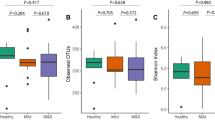
Amplicon sequencing results revealed higher microbial richness in white-plaque biofilm samples as compared to BS (p = 0.01 Fig. 2A). However, we observed no significant difference in Pielou’s evenness index between BS and white-plaque samples (Fig. 2B).

Alpha diversity analysis, comparison between BS and white-plaque samples. (A) Diversity richness expressed as the number of observed OTUs (i.e. phylotypes) and (B) Pielou’s evenness index.
Beta diversity analyses based on Unifrac phylogenetic-based distances showed differences in the structure of microbial communities between BS and white-plaque biofilms (Fig. 3). Differences were observed for both unweighted (Fig. 3A) and weighted Unifrac (Fig. 3B) distances (Permanova p = 0.001 for both distances) revealing that black and white biofilms differ in quality (i.e. presence/absence) and in abundance of phyloypes. Jaccard qualitative distance and Bray Curtis quantitative distance were also used, results also showed differences between BS and white-plaque samples (Supplementary Fig. 1).

Beta diversity analysis. Principal coordinate analysis based on unweighted Unifrac (A) and weighted Unifrac (B) distance matrices. The differences in community structure between BS and white-plaque samples were confirmed by Permanova test (P = 0.001).
Taxonomic composition of the BS microbiome
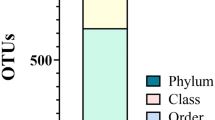
Eleven bacterial phyla were detected in BS samples. The top five most abundant phyla were found to be Bacteroidetes 26.5% [95% CI (24.9–28.1)], Actinobacteria 17.4% [95%CI (14.2–20.3)], Proteobacteria 16.9% [95%CI (14.0–19.8)], Fusobacteria 15.6% [95%CI (13.1–18.0)] and Firmicutes 15.0% 95%CI (13.2–16.8)] (Fig. 4A). These 11 phyla constituted 91.4% of the average relative abundance of the reads (Fig. 4A). A total of 109 distinct genera were observed in BS samples. The five most abundant genera, constituting 38.34% average relative abundance were found to be Capnocytophaga 11.7% [95%CI (10.0–13.4)], Leptotrichia 8.5% [95%CI (6.8–10.3)], Fusobacterium 7.1% [95%CI (6.2–7.9)], Corynebacterium 5.7% [95%CI (4.7–6.8)], and Streptococcus 5.3% [95%CI (4.0–6.7)] (Fig. 4B).

Top five most abundant phyla (A) and genera (B) in the BS biofilm. Average relative abundances are shown.
Comparison of the taxonomic profiles of BS and white-plaque
A comparative analysis between BS and white-plaque samples was carried out. Differences in relative abundances of phyla between BS and white plaque samples were characterized using a Kruskal Wallis non-parametric test. Of the top 5 phyla in terms of abundance, we found Actinobacteria, and Proteobacteria to be more abundant in BS samples than in white plaque samples (Fig. 5a,b), whilst Bacteriodetes and Firmicutes were more abundant in white plaque samples as compared to BS samples (Fig. 5d,e). Amongst the bacteria of lesser abundance, we found that the relative abundances of Patescibacteria, Epsilonbaceraeota, Spirochaetes, Synergistetes and Tenericutes were found to be higher in white plaque samples as compared to BS samples (Fig. 5c,f–i), Table 2).

Differences in abundance of key phyla between BS (black) and white-plaque (white) samples.
Overall, at the genus level, we observed 42 microbial genera with differential abundance between BS and white-plaque samples (Supplementary Table 1). Of the top 5 most abundant genera identified in BS, comparative analysis with white-plaque samples showed that Capnocytophaga and Corynebacterium were found in higher relative abundance in BS samples (Fig. 6a,b) while Fusobacterium was present in higher abundance in white-plaque samples (Fig. 6c).

Differences in abundance of key genera between BS (black) and white-plaque (white) samples.
Functional analysis of the BS microbiome
The metagenomic analysis of the BS and white-plaque samples allowed a deeper analysis of the potential functional composition of the microbiome using PICRUSt2 (Fig. 7). The functional analysis was based on two annotations: the Clusters of Orthologous Groups of proteins (COG) and the Enzyme Commission numbers (EC) to further predict Metacyc reactions and infer pathway abundances.

Average relative abundances (%) of COG functional categories in the BS microbiome.
A comparison of the predicted metagenomes of BS and white-plaque samples revealed the differential abundance of 2.222 COGs (Supplementary Table 2) and 183 Metacyc pathways (Supplementary Table 3). Hierarchical clustering of samples according to the abundance of COG and Metacyc pathways were observed (Supplementary Figs. 2 and 3). These results demonstrate conserved and consistent clustering of functional pathways within and between BS and white-plaque microbiomes.
The COG functional groups detected in BS, in rank order were “metabolism”, “information storage and processing”, followed by “cellular processes and signaling” (Fig. 7). Specifically, the following top 5 known pathways were found in greater relative abundance in BS; “translation, ribosomal structure and biogenesis”, “amino acid transport and metabolism”, “Cell wall/membrane/envelope biogenesis”, “carbohydrate transport and metabolism” and finally “Replication, recombination and repair”.
Amongst the 183 Metacyc pathways identified when comparing BS with white-plaque samples, we observe a statistically significant enrichment in the abundance of heme biosynthetic pathways in BS samples (Fig. 8). The relative abundances of the superpathway of heme biosynthesis from uroporphyrinogen-III (PWY0-1415; Fig. 8A), tetrapyrrole biosynthesis-I from glutamate (PWY-5188; Fig. 8B), anaerobic heme biosynthesis (HEMESYN2-PWY; Fig. 8C), and aerobic heme biosynthesis I (HEME-BIOSYNTHESIS-II; Fig. 8D) pathways were all found to be higher in BS as compared with white-plaque samples. The mycolyl-arabinogalactan-peptidoglycan complex biosynthesis pathway was also found in greater abundance in BS (data not shown). This is a pathway involved in the synthesis of cell wall components in Corynebacterium, supporting the increased abundance of the genera detected in BS samples during the taxonomic characterization analysis. These data demonstrate the pathways differentially abundant in BS as compared to white-plaque, which suggest a significant change in the overall microbial community functions between the two sample types.

Box-and-whisker plots showing the relative abundances of four pathways statistically significantly enriched in BS samples; (A) the superpathway of heme biosynthesis from uroporphyrinogen-III (PWY0-1415), (B) tetrapyrrole biosynthesis- I from glutamate (PWY-5188), (C) anaerobic heme biosynthesis (HEMESYN2-PWY), and (D) aerobic heme biosynthesis I (HEME-BIOSYNTHESIS-II).
Discussion
This study aimed to employ metagenomics to characterize the microbiome associated with supragingival dental plaque in adult patients with BS, as compared with white-plaque. We also attempted to develop a functional map of the bulk microbial metabolism through PICRUSt2 analysis. Thus far, existing investigations have centered on BS samples taken from pediatric patients. This is the first study characterizing the microbial community in BS and comparing it to white-plaque in adult volunteers.
With regard to the alpha diversity, we found that BS had lower species richness and evenness as compared to white-plaque. The differences were statistically significant (Fig. 2). Li et al., using salivary samples from pediatric volunteers, described results similar to those in the current study (lower diversity in BS versus control), however there were no statistically significant differences between samples of BS and white-plaque23. Chen et al. found no differences in alpha diversity between BS and white-plaque samples from pediatric patients22. The difference in results between studies using samples from pediatric volunteers and our study using adult volunteers demonstrates a clear distinction in the composition and diversity of the oral microbiome. Whilst pediatric samples demonstrate little or no difference in species diversity in plaque samples and richness, our results from adult patients suggest that a strong dysbiosis may develop over time in these patients.
We showed that, in comparison with white-plaque, patients with BS harbor a statistically significantly different microbial community in terms of presence of individual species and their relative abundances (Fig. 3). The difference between BS and white-plaque demonstrates conclusively the presence of a dysbiosis. There is currently no data available for comparison regarding the beta diversity of BS compared to white-plaque in either adult or pediatric samples.
We were able to detect a total of 11 distinct bacterial phyla (Fig. 4A) and 109 different genera (Fig. 4B) within BS samples. Previous studies highlighted the presence of Prevotella and Actinomyces species, which were not amongst the top 5 genera identified in our study. It should be noted that this research was carried out using culture-based techniques, which have since been superceded by modern molecular methodology19. More recently Saba et al. and Li et al. used PCR and next-generation sequencing-based detection to identify the composition of BS in pediatric samples. Only one genus was found in common with the current study (Corynebacterium), suggesting that the composition of pediatric and adult BS is significantly different20,21. It is notable that there was only one genus (Aggregatibacter) in common between the Saba and Li studies.
Interestingly, genera such as Capnocytophaga and Corynebacterium are commonly found in greater abundance in individuals with healthy oral cavities (lower caries experience). Overall, all members are commonly found in the oral cavity, however, are not amongst the most common. There are significant differences amongst the top 5 bacterial phyla when comparing BS with white-plaque samples (Fig. 5). Actinobacteria and Proteobacteria are enriched in BS as compared to white-plaque, whilst Bacteroides and Firmicutes are found in greater abundance in white-plaque as compared to BS in adult volunteers. In pediatric samples, Chen et al. obtained similar results to our study, although the differences were not statistically significant22. Likewise, Li et al. showed an increase in abundance of Actinobacteria in plaque samples from children with black stain, however did not describe significant differences in Proteobacteria, Bacteriodes or Firmicutes23. This could be due to differences between the study populations, since the participants in the Chen and Li studies were pediatric, whilst our research included adult participants with a mean age of 43.8 years.
At the genus level (Fig. 6), data published by Chen et al. contradicts our results, as they show a relatively higher abundance (although not statistically significant) of Capnocytophaga and Corynebacterium in white-plaque as compared to BS samples. They coincide with our results in that they observed an enrichment of Fusobacterium in white-plaque samples22. Li et al. on the other hand, coincide partly with our data, showing an increased abundance of Capnocytophaga and Corynebacterium in saliva samples of children with black stain. They did not describe statistically significant differences in the relative abundance of Fusobacterium23. It is important to bear in mind that the existing studies have been carried out on pediatric samples and therefore any variations must be evaluated in the context of the age difference.
Earlier research has linked the increased presence of Corynebacterium, Capnocytophaga, Fusobacterium and Leptotrichia amongst others, with improved oral health and lower caries experience25. The top 5 phyla identified as part of the BS microbiome in this study coincide completely with the healthy “core microbiome” described by Zaura et al., although the rank order of relative abundances vary between the two studies. This suggests that the dysbiosis represented by BS may play a protective role against caries26. Similar results were published by Belstrøm et al. who compared saliva from healthy patients with samples active caries and periodontitis. Bacteriodes, Actinobacteria, and Fusobacteria were all found to be more abundant in saliva from healthy patients as compared to those with oral disease27. When comparing the results of our study with supragingival plaque sample analysis of patients with periodontitis, we observe that Bacteroides and Fusobacteria are enriched in both BS and periodontitis patients, although we concurrently observe an increased abundance of Firmicutes and Actinobacteria in BS, both of which are associated with healthy oral microbiomes28.
The progression of the oral dysbiosis leading to BS is as yet unclear, however we propose a mechanism based on the ecological plaque hypothesis in which local oral environmental conditions promote the growth and enrichment of key species leading whilst inhibiting the accumulation of others29. Our study shows that BS has a composition resembling mature dental plaque biofilm, with several key differences; the reduced abundances of Porphyromonas and Prevotella30. This could explain the lack of gingival inflammation and the generally better oral health observed in patients with BS as compared to white-plaque. We find a relative decrease in abundance of “yellow complex” and “red complex” bacterial species, both of which are associated with oral disease states, with a concurrent increase in species associated with oral health31. Our findings suggest that the microbial community found in BS acts as a driver for a supragingival biofilm characterized by low oral inflammation and thus promoting lower periodontal disease. Previous research from our group has shown that individuals with BS have higher salivary pH, thus confirming the promotion of an anti-caries environment in the oral cavity16.
Our PICRUSt2 analysis reveals significant alterations in the abundances of functional pathways between BS and white-plaque samples (Figs. 7 and 8). We observed an enrichment of heme-biosynthesis pathways, supporting the theory that the black stain may be the black insoluble ferric deposits described by Reid et al.17. High iron levels were detected in black stain by ICP-MS analysis18. Preliminary ICP-MS data from our group suggest that BS is also associated with high iron levels in supragingival BS (data not shown). It is not possible to determine the temporal sequence of events resulting in the formation of BS from the current study. The heme and iron compound biosynthesis may precede the formation of BS or constitute an integral part of the development process as a result of the actions of the predominant genera present.
Microbiome analysis of BS from adult volunteers reveals a profound dysbiosis when compared with white-plaque samples. This dysbiosis is not observed in pediatric patients and there is a lack of comparative studies using adult volunteers. The microbiome composition in BS aligns with a “orally healthy” community as identified in previous studies. The temporal sequence resulting in dysbiosis (colonizers and enrichment) and the pathway employed for the generation and deposition of black pigment remains elusive. PICRUSt2 analysis confirms an increased relative abundance of heme biosynthetic pathways in BS, which, when combined with evidence demonstrating increased iron abundance in BS, suggest that heme-dependent iron sequestration and subsequent metabolism may play a role in BS generation. Further research could be targeted at confirming the upregulation of heme biosynthetic genes in BS, identifying the microbial species responsible for BS and the temporal sequence leading to colonization and dysbiosis. It remains to be determined whether the heme biosynthesis pathway would constitute a valid therapeutic target for the treatment of BS.
Materials and methods
Dental plaque sample collection
Fresh supragingival dental plaque samples (n = 27 [18 black plaque samples and 9 white plaque samples]) were obtained from adult patients attending the university dental clinic at CEU Cardenal Herrera University. The power of a one-sided test, under the assumptions that the responses are distributed according to a Laplace parent distribution, with 18 participants in one group and 9 in the other, α = 0.05 and an effect size, d = 1, was calculated, and found to be 90.3%, indicating that the number of volunteers participating in the study was statistically significant. Volunteers were selected on the basis of the inclusion and exclusion criteria shown in Table 3. Plaque samples were collected by MDMJ, RGM and ICB, using plastic dental curettes and following standard plaque sample removal procedures. All samples were obtained after informed consent, and this research was reviewed and approved by the University Ethics Committee. Samples were stored at 80 °C until further processing.
DNA extraction and PCR amplification
DNA Extraction and Library preparation were performed by Microomics Systems S.L. DNA was extracted from plaque samples using the DNeasy PowerLyzer PowerSoil Kit (Qiagen) following manufacturer’s instructions. The extraction tubes were agitated twice using Tissue lyser II (Qiagen) at 30 Hz/s for 5 min. A negative control containing no template DNA was included from purification steps. As a positive control for downstream procedures, ZymoBIOMICS Microbial Community DNA Standard (ZymoResearch) was used. This “mock community” comprises a proportioned mixture of bacterial and fungi DNA from selected species, designed to deliver reproducible and predictable results upon amplification. Samples were amplified using specific primers for the V3-V4 regions of the 16S rRNA gene . (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and V3-V4-Reverse (5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′). The PCR was performed in 10-μL final volume with 0.2-μM primer concentration. The PCR included: 3 min at 95 °C (initial denaturation) followed by 25 cycles: 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C, and a final elongation step of 5 min at 72 °C. PCR products were purified using AMPure XP beads (Beckman Coulter, Nyon, Switzerland) with a 0.9 × ratio according to the manufacturer’s instructions. PCR products were eluted from the magnetic beads with 30 μL of Milli-Q water. The above-described primers contain overhangs allowing the addition of full-length Nextera barcoded adapters for multiplex sequencing in a second PCR step, resulting in sequencing ready libraries with approximately 450 bp insert sizes. In brief, 5 μL of the first PCR purified product were used as the template for a second PCR with Nextera XT v2 adaptor primers in a final volume of 30 μL using the same PCR mix and thermal profile as for the first PCR but with only 8 cycles. 25 μL of the second PCR product were purified with SequalPrep normalization kit (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. Libraries were eluted in 20 μL final volume and pooled for sequencing.
Sequencing was performed in an Illumina MiSeq with 2 × 300 bp reads using v3 chemistry with a loading concentration of 10 pM. In all cases, 15% of PhIX control.
libraries was spiked in to increase the diversity of the sequenced sample. Negative controls included sample collection buffer, DNA extraction, and PCR amplification steps, PRC products after both PCR steps were visualized using an electrophoresis gel (1.5% agarose) with SYBR Safe (Applied Biosystems, ThermoFisher Scientific, Waltham, MA, USA). No visible bands were observed.
Sequence and analysis
Raw demultiplexed forward and reverse reads were processed using the following methods and pipelines as implemented in QIIME2 version 2019.4 with default parameters unless stated33. DADA2 was used for quality filtering, denoising, pair- end merging and amplicon sequence variant calling (ASV, i.e. phylotypes) using qiime dada2 denoise-paired method34. Q20 was used as quality threshold to define read sizes for trimming before merging (parameters: -p-trunc-len-f and -p-trunc-len-r). Reads were truncated at the position when the 75th percentile Phred score felt below Q20: 273 nt for Forward and 225 nt for Reverse reads. After quality filtering steps, average sample size was 28,611 reads (min: 18,887 reads, max: 47,572 reads) and 2,653 phylotypes were detected. ASVs were aligned using the qiime alignment mafft method35. The alignment was used to create a tree and to calculate phylogenetic relations between ASVs using qiime phylogeny fasttree method36. ASV tables were subsampled without replacement in order to even sample sizes for diversity analysis using qiime diversity core-metrics-phylogenetic pipeline. The smallest sample size was chosen for subsampling . Jaccrad, Bary Curtis and unweighted and weighted Unifrac distances were calculated to compare community structure37. The following alpha diversity metrics were calculated: observed OTU number (i.e. richness) and Pielou’s evenness index. Taxonomic assignment of ASVs was performed using a Bayesian Classifier trained with Silva database (i.e. 99% OTUs database) using the qiime feature-classifier classify-sklearn method38. Phylotypes were filtered to discard contaminant Eukariota DNA-derived amplicons using Blast against the mentioned database with a 90% identity cutoff.
Differential abundance of taxa was tested using Kruskal Wallis non-parametric test. FDR Benjamini–Hochberg correction was used to correct for multiple comparisons. Alpha diversity comparisons were performed using Kruskal–Wallis non-parametric test. Unifrac, Jaccard and Bray Curtis distance matrices and ASV count tables were used to calculate principal coordinates and construct ordination plots. The significance of groups (i.e. black vs. White plaque samples) in community structure was tested using Permanova. Permdisp test was used to identify location vs. dispersion effects. Significant threshold was set at p ≤ 0.05. ‘BiodiversityR’ version 2.11-1, ‘PMCMR’ version 4.3 and ‘vegan’ version 2.5-5 packages of the R software package version 36.6.0 (www.R-project.org) were used.
Potential functional profiles for sequenced samples were predicted using PICRUSt239. In summary, phylotypes were placed into a reference tree containing 20,000 full 16S rRNA genes from prokaryotic genomes in the Integrated Microbial Genomes (IMG) database.
Functional annotation of these genomes was based on Clusters of Orthologous Groups of proteins (COG) and the Enzyme Commission numbers (EC) databases. MetaCyc ontology was used for inference of pathway abundances using MinPath40. In order to infer Metacyc pathways, EC numbers were first regrouped to MetaCyc reactions. Pathway abundances were calculated as the harmonic mean of the key reaction abundances in each sample. To infer the abudance of each gene family per sample: the abundances of phylotypes were corrected by their 16S rRNA gene copy number and then multiplied by their functional predictions. The analysis of differential relative abundances of Metacyc Pathways and COGs was perfomed with Kruskal Wallis test. FDR Benjamini–Hochberg correction was used to correct for multiple comparisons. Package ‘pheatmap’ version 1.0.12 of the R software package version 36.6.0 was used for hierarchical clustering and heatmap ploting.
The metagenome data have been deposited in the SRA (sequence read archive) database, belonging to NCBI (National Centre for Biotechnology Information), with accession number: PRJNA644495.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Ethics Committee of CEU Cardenal Herrera University (authorization number CEI16 / 019).
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
Huttenhower, C. et al. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Dewhirts, F. E. et al. The human oral microbiome. J. Bacteriol. 192, 5002–5017 (2012).
Krishnan, K., Chen, T. & Paster, B. J. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 23, 276–286 (2017).
Edlund, A. et al. Uncovering complex microbiome activities via transcriptomics during 24 hours of biofilm assembly and maturation. Microbiome 6, 217 (2018).
Marsh, P.D. et al. in Marsh and Martin´s Oral Microbiology (ed Marsh P.D. & Martin M.) 81-109-000 (Elsevier, 2016).
Peterson, S. N. et al. The dental plaque microbiome in health and disease. PLoS ONE 8(3), e58487. https://doi.org/10.1371/journal.pone.0058487 (2013).
Segata, N. et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13, R42 (2012).
Solbiati, J. & Frias-Lopez, J. Metatranscriptome of the oral microbiome in health and disease. J. Dent. Res. 97(5), 492–500 (2018).
Hajishengallis, G., Darveau, R. P. & Curtis, M. A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10(10), 717–725 (2012).
Marsh, P. D. In Sickness and in health—What does the oral microbiome mean to us? an ecological perspective. Adv. Dent. Res. 29(1), 60–65 (2018).
Struzycka, I. The oral microbiome in dental caries. Pol. J. Microbiol. 63(2), 127–135 (2014).
Fakhruddin, K. S., Ngo, H. C. & Samaranayake, L. P. Cariogenic microbiome and microbiota of the early primary dentition: A contemporary overview. Oral Dis. 25, 982–995 (2019).
Theilade, D. E., Slots, J. & Fejerskov, O. The ultrastructure of black stain on human primary teeth. Scan. J. Dent. Res. 81, 528–352 (1973).
Żyła, T., Kawala, B., Antoszewska-Smith, J. & Kawala, M. Black stain and dental caries: a review of the literature. Biomed. Res. Int. 2015, 469392 (2015).
Albelda-Bernardo, M. A., Jovani-Sancho, M. D. M., Veses, V. & Sheth, C. C. Remediation of adult black dental stains by phototherapy. BDJ Open 4, 17035 (2018).
Ortiz-López, C. S., Veses, V., Garcia-Bautista, J. A. & Jovani-Sancho, M. D. M. Risk factors for the presence of dental black plaque. Sci. Rep. 8(1), 16752 (2018).
Reid, J. S., Beeley, J. A. & MacDonald, D. G. Investigations into black extrinsic tooth stain. J. Dent. Res. 56, 895–899 (1977).
Zhang, F. et al. A preliminary study on the relationship between iron and black extrinsic tooth stain in children. Lett. Appl. Microbiol. 64(6), 424–429 (2017).
Slots, J. The microflora of black stain of human primary teeth. Scan. J. Dent. Res. 82, 484–490 (1974).
Saba, C. et al. Black stains in the mixed dentition: a PCR microbiological study of the etiopathogenic bacteria. J. Clin. Pediatr. Dent. 30, 219–224 (2006).
Li, Y. et al. Analysis of the microbiota of black stain in the primary dentition. PLoS ONE 10(9), e0137030 (2015).
Chen, L., Zhang, Q., Wang, Y., Zhang, K. & Zou, J. Comparing dental plaque microbiome diversity of extrinsic black stain in the primary dentition using Illumina MiSeq sequencing technique. BMC Oral Health 19(1), 269 (2019).
Li, Y. et al. Oral Microbial community typing of caries and pigment in primary dentition. BMC Genomics 17, 558 (2016).
Petersen, P. E. Global policy for improvement of oral health in the 21st century–implications to oral health research of World Health Assembly 2007, World Health Organization. Comm. Dent. Oral Epidemiol. 37(1), 1–8 (2009).
Schoiler, K. et al. Bacterial biofilm composition in healthy subjects with and without caries experience. J. Oral Microbiol. 11(1), 1633194 (2019).
Zaura, E., Keijser, B. J. F., Huse, S. M. & Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9, 259 (2009).
Belstrøm, D. et al. Metagenomic and metatranscriptomic analysis of saliva reveals disease-associated microbiota in patients with periodontitis and dental caries. NPJ Biofilms Microbiomes 3, 23 (2017).
Pozhitkov, A. E. et al. Towards microbiome transplant as a therapy for periodontitis: an exploratory study of periodontitis microbial signature contrasted by oral health, caries and edentulism. BMC Oral Health 15, 125 (2014).
Jakubovics, N. S. Intermicrobial interactions as a driver for community composition and stratification of oral biofilms. J. Mol. Biol. 427(23), 3662–3675 (2015).
Takeshita, T. et al. Dental plaque development on a hydroxyapatite disk in young adults observed by using a barcoded pyrosequencing approach. Sci. Rep. 5, 8136 (2015).
Aruni, W. A., Dou, Y., Mishra, A. & Fletcher, H. M. The biofilm community: rebels with a cause. Curr. Oral Health Rep. 2, 48–56 (2015).
Gasparetto, A. et al. Prevalence of black tooth stains and dental caries in Brazilian schoolchildren. Braz. Dent. J. 14, 157–161 (2003).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 1091. https://doi.org/10.1038/s41587-019-0252-6 (2019).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. https://doi.org/10.1093/molbev/msp077 (2009).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73(16), 5261–5267 (2007).
Douglas, G.M. et al. PICRUSt2: An improved and customizable approach for metagenome inference. bioRxiv 672295; 10.1101/672295 (2020)
Ye, Y. & Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 5(8), e1000465 (2009).
Funding
This study was funded by Universidad CEU Cardenal Herrera: Banco Santander 2017-2019 to VV, Banco Santander 2019-2020 to CCS and INDI19/50.
Author information
Authors and Affiliations
Contributions
M.M.J.-S., R.M.-G. and I.C.-B. performed the clinical examinations, data and sample collection. V.V. and C.C.S. participated in the experimental design, design and analyzed the metagenomics experiments and wrote the manuscript. P.G.-T. and B.C. performed and analyzed the metagenomics experiments, performed the PICRUSt2 analysis, and contributed to manuscript preparation. All authors reviewed the data and the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Veses, V., González-Torres, P., Carbonetto, B. et al. Dental black plaque: metagenomic characterization and comparative analysis with white-plaque. Sci Rep 10, 15962 (2020). https://doi.org/10.1038/s41598-020-72460-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72460-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
