
Abstract
Ribosome inactivating proteins (RIPs) are RNA N-glycosidases that depurinate a specific adenine residue in the conserved sarcin/ricin loop of 28S rRNA. These enzymes are widely distributed among plants and bacteria. Previously, we have described for the first time RIP genes in mosquitoes belonging to the Culicidae family. We showed that these genes are derived from a single event of horizontal gene transfer (HGT) from a prokaryotic donor. Mosquito RIP genes are evolving under purifying selection, strongly suggesting that these toxins have acquired a functional role. In this work, we show the existence of two RIP encoding genes in the genome of the whitefly Bemisia tabaci, a hemiptera species belonging to the Aleyrodidae family distantly related to mosquitoes. Contamination artifacts were ruled out analyzing three independent B. tabaci genome databases. In contrast to mosquito RIPs, whitefly genes harbor introns and according to transcriptomic evidence are transcribed and spliced. Phylogeny and the taxonomic distribution strongly support that whitefly RIP genes are derived from an independent HGT event from a plant source. These results, along with our previous description of RIPs in Diptera, suggest that the acquired genes are functional in these insects and confer some fitness advantage.
Similar content being viewed by others


Introduction
Horizontal gene transfer (HGT) is the reproduction-independent transmission of genetic material between organisms of different species. HGT has been reported to have occurred in all the three domains of life and is accepted as an important evolutionary force in prokaryotes1,2,3. On the contrary, its impact on multicellular eukaryotes (e.g. metazoans) is largely controversial4. In arthropods, many well-supported HGT events from bacterial or fungal sources have been described5,6. Moreover, it has been suggested that HGT played a role in the herbivory of several arthropods and nematodes (see7,8 for a review).
Ribosome inactivating proteins (RIPs, EC 3.2.2.22) are RNA N-glycosidases depurinating ribosomes in the conserved alpha-sarcin/ricin loop of 28S rRNA, leading to irreversible arresting of protein synthesis9,10,11. RIP encoding genes are widely distributed in plants but scarce within bacterial and fungal lineages12. Previously, we demonstrated the presence of RIP genes in genomes of mosquitoes belonging to the subfamily Culicinae13. This was the first description of the existence of RIPs in metazoans. Our research also indicated that these genes are derived from a single HGT event from a bacterial donor species14. Moreover, we have shown that, in Aedes aegypti, these RIP genes are transcribed and their expression levels are modulated across the developmental stages15. Recently, evidence has been accumulated on the role of Spiroplasma spp RIPs in the protective mechanisms generated by these endosymbiotic species against arthropod infection by natural enemies (see16 for a review).
Here we demonstrate that RIP genes are also present in genomes of a second lineage of insects: the hemiptera whiteflies (Aleyrodidae family). Notably, these genes are not closely related to mosquito homologues, instead they form a clade along with plant-derived RIPs. Altogether our results are consistent with two independent HGT events of RIP encoding genes to different insect lineages. Our results suggest that RIP genes may fulfill an important functional niche in insects leading to the recurrent selection either from horizontally transferred genetic material or by a symbiotic interaction.
Results and discussion
Bemisia tabaci genome harbors two RIP encoding genes
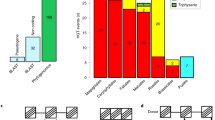
In the course of routine database searches, we found two RIP encoding sequences in the hemiptera B. tabaci MEAM 1 (henceforth named BtRIP1 and BtRIP2). Figure 1A shows a schematic representation of the genomic scaffold (NW_017547285) harboring these genes. In contrast to RIP genes from mosquitoes, BtRIP1 and BtRIP2 contain one and two introns, respectively. This excludes the chance of bacterial contamination, a possible artifact of genome sequencing projects17,18. BLAST analyses of all encoding protein sequences surrounding the RIP genes yielded maximum scores with arthropod annotated proteins (Supplementary Table S1). In contrast, BtRIPs showed maximum sequence identity to plant RIPs, and marginal identity to their mosquito homologues (around 24%).

(A) Scheme depicting the B. tabaci MEAM1 genomic region harboring BtRIP1 (XM_019046661) and BtRIP2 (XM_019046743) genes. Arrows depict the genes in the genomic scaffold. The empty arrows show genes surrounding the BtRIPs that encode proteins with highest BLAST score to arthropod sequences (see arrow code in Supplementary Table S1). The untranslated (UTRs) and coding regions of mRNA in BtRIP1 and BtRIP2 genes are represented with light blue and blue colors, respectively. (B) Unrooted phylogeny of RIP genes. Branches are colored according to taxonomy: bacteria (light blue), plants (green), fungi (blue), metazoan (red). TBE support values of relevant divergences are shown at nodes. Mosquito and whitefly clades are marked with silhouettes. Fully annotated phylogeny is available as Supplementary Figure S4. (C) Phylogeny of selected species from Neoptera orders. The tree including species from Diptera (17), Hemiptera (12) and Psocodea (1) orders with fully sequenced genomes was constructed with the TimeTree knowledge-base33. Insects harboring RIP genes are shown in red branches. The two independent HGT events are graphically represented at the estimated time windows, with the hypothetical donors shown as silhouettes. Time in million years ago (MYA) is indicated at the bottom.
We further confirmed the existence of these Bemisia newly discovered genes by analyzing two additional, independent genome contigs belonging to the B. tabaci SSA-ECA19 (PGTP01000858) and B. tabaci MED/Q20 (ML134445) assemblies. Supplementary Figure S1 shows synteny analysis of the genomic regions including RIP genes in these three subspecies of B. tabaci (with the exception of BtRIP1 in the Bemisia tabaci SSA1 due to a 3 kb gap in the corresponding scaffold), ruling out possible artifacts arising from sequence assembly. Supplementary Table S1 summarizes this information.
BtRIP1 gene is 3,466 bp and includes two exons of 273 bp and 517 bp and a single intron of 2,730 bp. The mRNA (790 bp) shows 5′ and 3′UTRs of 92 bp and 86 bp, respectively. The mature mRNA encodes a protein of 185 amino acids (Fig. 1A). The BtRIP2 gene extends over 5,974 bp: three exons (156 bp, 354 bp and 563 bp) and two introns (3,156 bp and 1,745 bp). The 5´ UTR (186 bp) is formed by the whole first exon and 30 bp of the second exon (Fig. 1A). The 3´UTR has 65 bp of the last exon. The mature mRNA encodes a protein of 273 amino acids. Sequence alignment and structural modeling of B. tabaci predicted proteins revealed that all the characterized residues forming the active site are conserved. Interestingly, BtRIP1 is remarkably smaller than average RIPs. This is caused by a N-terminal shortening. The functional relevance of this shortening is not clear from the structural model since although a big open cavity is generated, the active site architecture is conserved. Other shorter N-terminal deletions are observed in functional RIPs as lychinin (Protein Data Bank; PDB 2G5X). BtRIP2 is predicted to conserve the architecture and all secondary structure elements canonical of functional RIPs such as momordin (PDB 5CF9) or trichosantin (PDB 1QD2) (Supplementary Figure S2).
By using transcriptomic data (kindly provided by Dr. Zhangjun Fei21), we analyzed the expression level of BtRIPs in comparison with the full set of genes. Transcripts for both genes were found. In particular, BtRIP2 expression level is in the top quartile of B. tabaci genes, suggesting a relevant functional role for this protein (Supplementary Figure S3).
Trialeurodes vaporariorum draft genome harbors three non-annotated RIP encoding sequences
In December 2019, the draft genome sequence of T. vaporariorum, a whitefly closely related to B. tabaci was released. We downloaded the genome data and performed tBLASTn searches using B. tabaci RIPs as queries. We found three contiguous RIP encoding sequences in the same scaffold (VJOP01000134); namely TvRIP1 (nt 308,541–309,308), TvRIP2 (nt 315,008–315,853) and TvRIP3 (nt 327,149–327,907). The amino acid identity among TvRIPs ranged from 38 to 72%. These proteins showed 30–35% identity when compared to BtRIPs.
Whitefly RIP genes are phylogenetically closer to plant than to mosquito homologues
Next, we performed phylogenetic inferences using the recently discovered whitefly RIPs along with a representative dataset of RIP sequences from our previous works13,14. The phylogenetic relationships among whitefly RIPs and previously characterized homologues are expected to shed light on the evolutionary origin and history of these genes and their possible relationship with homologous genes from dipterans. Notably, as can be seen in Fig. 1B, whitefly RIPs are embedded into a clade of plant sequences (Transfer Boostrap Expectation; TBE = 0.94). These RIPs are distantly related to mosquito RIPs, which on the contrary are gathered in a clade of bacterial homologues (TBE = 0.85). B. tabaci and T. vaporariorum genes form sister clades, revealing that the duplication events occurred after divergence of these species yielding two and three different paralogs, respectively (Supplementary Figure S4).
Whitefly RIPs are derived from a plant genome via a single HGT event
To determine the presence/absence of RIPs in other insects, we performed homology searches using whitefly RIPs as queries in complete genomes of insects other than B. tabaci and T. vaporariorum (797 assemblies with full representation). No hits were retrieved even in other hemiptera species. This fact, along with the phylogenetic inferences, indicates that these genes are not derived from vertical inheritance through the insect lineage. Instead, they originate from an independent HGT event of a plant RIP gene to an insect genome. Figure 1C shows the phylogenetic relationships among selected Neoptera species. As it can be observed only two clades harbor RIP genes: the Diptera lineage including Culicini and Aedini tribes and, the Hemiptera lineage that includes B. tabaci and T. vaporariorum species.
As previously reported, Diptera RIP genes are derived from a single HGT event which took place between the divergence of Anopheles and Culex/Aedes lineages (around 190 MYA) and before the separation of Aedes and Culex genus (150 MYA) (Fig. 1C). In a similar way, whitefly RIP genes seem to have been also acquired by HGT but from an eukaryotic, plant genome source. The absence of RIP genes in other hemiptera species suggests that HGT took place in the lineage of suborder Sternorrhyncha before the divergence between B. tabaci and T. vaporariorum species (in the range of 300–80 MYA).
We have previously postulated that HGT from bacteria to mosquitoes could have been facilitated by the weakness of the Weisman barrier (i.e. the physical separation between somatic and germinal cells in multicellular organisms) at early developmental stages. Feeding mosquito larva on bacterial species in their ecological niches is consistent with the acquisition of a RIP gene from prokaryotes14. The findings presented in this work seem to support this hypothesis, since early development stages whiteflies feed on plant sap. Therefore, our previous and current findings pinpoint that in addition to phylogenetic inconsistencies and taxonomic distribution, developmental and ecological features of animal species should be carefully analyzed when investigating and testing the plausibility of HGT events.
Whitefly RIP genes have evolved under purifying selection
After the integration in the genome of ancestral whitefly, RIP genes have not been lost by genetic drift over 80 million years of evolution. This strongly suggests that these genes are playing a functional role in their hosts. We tested this hypothesis by analyzing the nonsynonymous/synonymous substitution rates ω(dN/dS). As expected, this analysis yielded a global ω value of 0.34 consistent with evolution under purifying selection.
We have previously shown that almost all sequenced metazoan genomes are devoid of RIP genes13,14, the only two exceptions are insect lineages of Culicidae14 and Aleyrodidae (reported in this work) families. Both of these lineages have incorporated RIP encoding genes via HGT. These genes are actively transcribed and evolve under purifying selection. Based on this evidence we propose that after integration in their host genomes, RIP genes have a positive impact on species fitness. The defensive role of Spiroplasma encoded RIPs reported in Drosophila16 suggests that exogenous insect RIPs might play a similar protective role.
Methods
Homology searches and sequence analyses
BLASTp and tBLASTn homology searches were performed under default parameters on metazoan databases, using as queries a previously reported set of RIP sequences14. Two automatically annotated protein sequences (GenBank: XP_018902206, XP_018902288) were retrieved from the B. tabaci MEAM 1 genome database. Pfam analysis was performed to confirm the presence of RIP domain (PF00161)22. Homology models were generated using Swiss-Model server23 and visualized in PyMOL. Active site residues24 were detected by sequence alignment to momordin (PDB 3MY6). The T. vaporariorum non-annotated draft genome (GenBank: VJOP01000000) was downloaded and standalone tBLASTn searches performed using B. tabaci RIPs (GenBank: XP_018902206, XP_018902288) as queries.
Synteny analysis
The contig region containing RIP genes from B. tabaci MEAM 1 (NW017547285)21 was compared employing Mauve software25 (version 2.4.0, available at https://darlinglab.org/mauve) to the corresponding genomic fragments of other members of the B. tabaci complex; namely B. tabaci SSA-ECA (PGTP01000858)19 and B. tabaci MED/Q (ML134445)20.
Transcriptomic data analysis
The full RNA-seq dataset expressed as Reads Per Kilobase of transcript per Million mapped reads (RPKM) of B. tabaci MEAM 1 was kindly provided by Dr. Zhangjun Fei21. The logarithm of the average level (among different experimental conditions) of gene expression for BtRIP1 (Gene ID: Bta13094) and BtRIP2 (Gene ID: Bta13103) were plotted along with box and whisker graph showing quartiles for expression level of the whole set of B. tabaci genes using GraphPad Prism version 5.00 for Windows.
Multiple sequence alignment and phylogenetic inferences
The new B. tabaci and T. vaporariorum RIP protein sequences were incorporated to our previous dataset13. The conserved region in the RIP domain (from Y14 to S196 residues according to trichosanthin (GenBank: AAT91090) was selected for alignment as previously reported13,14,26. Multiple sequence alignment (MSA) was constructed using MAFFT 7 server27 employing BLOSUM 30 as scoring matrix. Poorly aligned regions were trimmed as blocks. This MSA containing 125 sequences and 199 sites was used to perform phylogenetic analysis by Maximum Likelihood in RAxML (version 8.2.10, available at https://github.com/stamatak/standard-RAxML)28. The WAG substitution matrix was selected using ProtTest 3.429. To estimate the robustness of the phylogenetic inference 500 rapid bootstrap (BS) were selected. Transfer bootstrap expectation was calculated in BOOSTER30. Phylogenetic relationships and divergence times among species were obtained from TimeTree knowledge-base31. FigTree (version 1.4.2, available at https://tree.bio.ed.ac.uk/software/figtree) was used to visualize and edit the trees.
Selection pressure analysis
The omega (ω) ratio between nonsynonymous/synonymous substitution rates was calculated with codeml in PAML 4.932. This analysis was performed under the one ratio model (M0) with κ = 2.096, α = 5.234 under 4 gamma categories (parameters calculated in jModelTest 2.1.10). The codon alignment used as input was created in PAL2NAL and the protein based tree in MAFFT by (Neighbor Joining) NJ method.
References
Dagan, T., Artzy-Randrup, Y. & Martin, W. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc. Natl. Acad. Sci. USA 105, 10039–10044. https://doi.org/10.1073/pnas.0800679105 (2008).
Gogarten, J. P., Doolittle, W. F. & Lawrence, J. G. Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238 (2002).
Treangen, T. J. & Rocha, E. P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 7, e1001284 (2011).
Ku, C. & Martin, W. F. A natural barrier to lateral gene transfer from prokaryotes to eukaryotes revealed from genomes: The 70 % rule. BMC Biol. 14, 89. https://doi.org/10.1186/s12915-016-0315-9 (2016).
Zhao, C. & Nabity, P. D. Phylloxerids share ancestral carotenoid biosynthesis genes of fungal origin with aphids and adelgids. PLoS One 12, e0185484. https://doi.org/10.1371/journal.pone.0185484 (2017).
Hotopp, J. C. D. Horizontal gene transfer between bacteria and animals. Trends Genet. 27, 157–163 (2011).
Wybouw, N., Pauchet, Y., Heckel, D. G. & Van Leeuwen, T. Horizontal gene transfer contributes to the evolution of arthropod herbivory. Genome Boil. Evol. https://doi.org/10.1093/gbe/evw119 (2016).
Brown, A. M. V. Endosymbionts of plant-parasitic nematodes. Annu. Rev. Phytopathol. 56, 225–242. https://doi.org/10.1146/annurev-phyto-080417-045824 (2018).
Endo, Y., Mitsui, K., Motizuki, M. & Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 262, 5908–5912 (1987).
Hudak, K. A., Dinman, J. D. & Tumer, N. E. Pokeweed antiviral protein accesses ribosomes by binding to L3. J. Biol. Chem. 274, 3859–3864 (1999).
Rajamohan, F., Ozer, Z., Mao, C. & Uckun, F. M. Active center cleft residues of pokeweed antiviral protein mediate its high-affinity binding to the ribosomal protein L3. Biochemistry 40, 9104–9114 (2001).
Lapadula, W. J. & Ayub, M. J. Ribosome Inactivating Proteins from an evolutionary perspective. Toxicon 136, 6–14. https://doi.org/10.1016/j.toxicon.2017.06.012 (2017).
Lapadula, W. J., Sanchez Puerta, M. V. & Juri Ayub, M. Revising the taxonomic distribution, origin and evolution of ribosome inactivating protein genes. PLoS One 8, e72825. https://doi.org/10.1371/journal.pone.0072825 (2013).
Lapadula, W. J., Marcet, P. L., Mascotti, M. L., Sanchez-Puerta, M. V. & Juri Ayub, M. Metazoan ribosome inactivating protein encoding genes acquired by horizontal gene transfer. Sci Rep. 7, 1863. https://doi.org/10.1038/s41598-017-01859-1 (2017).
Lapadula, W. J., Marcet, P. L., Taracena, M. L., Lenhart, A. & Juri Ayub, M. Characterization of horizontally acquired ribotoxin encoding genes and their transcripts in Aedes aegypti. Gene 754, 144857. https://doi.org/10.1016/j.gene.2020.144857 (2020).
Ballinger, M. J. & Perlman, S. J. The defensive Spiroplasma. Curr. Opin. Insect Sci. 32, 36–41. https://doi.org/10.1016/j.cois.2018.10.004 (2019).
Lusk, R. W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data. PLoS One 9, e110808. https://doi.org/10.1371/journal.pone.0110808 (2014).
Francois, C. M., Durand, F., Figuet, E. & Galtier, N. Prevalence and implications of contamination in public genomic resources: A case study of 43 reference arthropod assemblies. G3 10, 721–730. https://doi.org/10.1534/g3.119.400758 (2020).
Chen, W. et al. Genome of the African cassava whitefly Bemisia tabaci and distribution and genetic diversity of cassava-colonizing whiteflies in Africa. Insect Biochem. Mol. Biol. 110, 112–120. https://doi.org/10.1016/j.ibmb.2019.05.003 (2019).
Xie, W. et al. Genome sequencing of the sweetpotato whitefly Bemisia tabaci MED/Q. GigaScience 6, 1–7. https://doi.org/10.1093/gigascience/gix018 (2017).
Chen, W. et al. The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol. 14, 110. https://doi.org/10.1186/s12915-016-0321-y (2016).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. https://doi.org/10.1093/nar/gky995 (2019).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. https://doi.org/10.1093/nar/gky427 (2018).
Shi, W. W., Mak, A. N., Wong, K. B. & Shaw, P. C. Structures and ribosomal interaction of ribosome-inactivating proteins. Molecules 21, 20. https://doi.org/10.3390/molecules21111588 (2016).
Darling, A. C., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14, 1394–1403. https://doi.org/10.1101/gr.2289704 (2004).
Lapadula, W. J., Sanchez-Puerta, M. V. & Ayub, M. J. Convergent evolution led ribosome inactivating proteins to interact with ribosomal stalk. Toxicon 59, 427–432. https://doi.org/10.1016/j.toxicon.2011.12.014 (2012).
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M. & Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10. https://doi.org/10.1093/nar/gkz342 (2019).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. https://doi.org/10.1093/bioinformatics/btr088 (2011).
Lemoine, F. et al. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556, 452–456. https://doi.org/10.1038/s41586-018-0043-0 (2018).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. https://doi.org/10.1093/molbev/msm088 (2007).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. Timetree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. https://doi.org/10.1093/molbev/msx116 (2017).
Acknowledgements
W.J.L, M.L.M and M.J.A. are members of the scientific career of CONICET. This work has been funded by grants from ANPCyT (Agencia Nacional de Promoción Científica y Tecnológica) PICT 2016-1446 to W.J.L. The authors are deeply grateful to Dr. Zhangjun Fei for kindly sharing B. tabaci transcriptomic data.
Author information
Authors and Affiliations
Contributions
W.J.L. performed sequence alignment, phylogenetic inferences and synteny analysis. M.L.M. performed 3D modeling and selection analysis. W.J.L. and M.L.M. prepared the figures. M.J.A. conceived the original idea and conducted initial database searches. All authors wrote, corrected and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lapadula, W.J., Mascotti, M.L. & Juri Ayub, M. Whitefly genomes contain ribotoxin coding genes acquired from plants. Sci Rep 10, 15503 (2020). https://doi.org/10.1038/s41598-020-72267-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72267-1
This article is cited by
-
Evidence of venom factor-like in crocodilians
Organisms Diversity & Evolution (2023)
-
Contribution of cryptochromes and photolyases for insect life under sunlight
Journal of Comparative Physiology A (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
