
Abstract
Freshwater mussels belonging to the genus Nodularia (Family Unionidae) are known to be widely distributed in East Asia. Although phylogenetic and population genetic studies have been performed for these species, there still remain unresolved questions in their taxonomic status and biogeographic distribution pathways. Here, the nucleotide sequences of CO1 and 16S rRNA were newly determined from 86 N. douglasiae and 83 N. breviconcha individuals collected on the Korean Peninsula. Based on these data, we revealed the following results: (1) N. douglasiae can be divided into the three genetic clades of A (only found in Korean Peninsula), B (widely distributed in East Asia), and C (only found in the west of China and Russia), (2) the clade A is not an independent species but a concrete member of N. douglasiae given the lack of genetic differences between the clades A and B, and (3) N. breviconcha is not a subspecies of N. douglasiae but an independent species apart from N. douglasiae. In addition, we suggested the plausible scenarios of biogeographic distribution events and demographic history of Nodularia species.
Similar content being viewed by others

Introduction
Freshwater mussels belonging to the family Unionidae (Unionida, Bivalvia) consist of 753 species distributed over a wide area of the world encompassing Africa, Asia, Australia, Central and North America1, 2. The unionid mussels are known to play key roles in maintaining aquatic ecosystems such as nutrient circulation, water purification, and habitat supply3. Over the recent years, populations of unionid mussel species have rapidly been decreased due to global anthropogenic disturbances including desertification, water pollution, and river fragmentation4, 5. A total of 247 unionid species are now identified from the IUCN Red list of Threatened species6, 7. Interest in the taxonomic system and evolutionary relationships of unionids began with increasing reports about the fauna, ecological significance, and endangered status of freshwater mussels in North America and Europe since the late of twentieth century century5, 8, 9. In East Asia, ecological and evolutionary studies of unionids are still scarce though numerous species are currently at the blink of extinction in this taxon10, 11.
Unionid species survive parasitically on the scales, gills and fins of various fish species passing by in the first larval stage for several weeks or a month, fall off as juveniles, and grow burying themselves in the sediments or sticking to the crevice of rocks12, 13. Given the unique life history of the parasitic larval and adherent adult periods of this taxon, the biogeographic distribution patterns and genetic diversity of current species can reflect the history of past freshwater system formation and geological fluctuations12, 13. The Korean Peninsula is located at the end of East Asia. The seas between China and Japanese Archipelago were the lakes during the last glacial maximum which later served as the origin of the freshwater systems on the Korean Peninsula14,15,16. A tremendous endemism is observed on this peninsula, regardless of taxa, because many organisms that escaped glaciers could settle in countless refugia that had been formed due to complex terrain on this peninsula and have been speciated in the processes of isolation and adaptation16,17,18,19. Despite the biogeographical importance of the Korean Peninsula, the researches on the biogeographic origins and genetic diversity of unionids have been conducted insufficiently5, 10.
The Nodularia species are widely distributed throughout East Asia including the Korean Peninsula, China and Japanese Archipelago as well as Russia and Vietnam9, 20. N. douglasiae and N. breviconcha are only known species on the Korean Peninsula (Supplementary Fig. S1), and these two species were classified based on the morphological characteristics such as the size and inner epidermis color of the shell21. N. douglasiae has a bigger shell size than N. breviconcha. N. douglasiae is widely distributed throughout East Asia, while N. breviconcha is known to be a native species inhabiting only the Korean Peninsula11, 22. N. breviconcha had previously been treated as a subspecies of N. douglasiae21, 23, but it was recently revealed that these two species may be genetically and phylogenetically separated from each other10, 12, 20, 24. However, the studies conducted so far may not be enough to address the taxonomic status of N. breviconcha because they only used a small number of individuals from very limited sampling areas10, 12, 20. Further intensive studies are still required with more extensive sampling to clarify the taxonomic boundaries of this species.
Regarding the taxonomic status of N. douglasiae, Lopes-Lima et al.10 recently suggested that South Korean N. douglasiae should be divided into two species of N. douglasiae and Nodularia sp.1 based on CO1 sequence difference. Although they attempted a sophisticated phylogeographic analysis on Nodularia, their study was not based on extensive sampling and population structure of N. douglasiae inhabiting the Korean Peninsula. The detailed population genetic structure of N. douglasiae in the Korean Peninsula should be uncovered with abundant sample collections, given that this peninsula would have been the most important intermediate link in shaping the contemporary distribution of this genus around the East Asia.
In this study, the phylogenetic relationships and population genetic structures of the East Asian Nodularia species were examined with mitochondrial CO1 and 16S rRNA data newly obtained from 86 N. douglasiae and 83 N. breviconcha individuals inhabiting the freshwater systems on the Korean Peninsula. The present analysis covers most of the areas inhabited by the two species on the Korean Peninsula and is based on extensive sampling to accurately determine the distribution of genetic variation, which was not expected from the previous studies4, 10, 12, 20. The obtained data here were applied to estimate and discuss the historical origin and distribution processes of Nodularia across East Asia by integrating with all previously known data. The present study can address the following four questions. First, are N. douglasiae and N. breviconcha distinctly separated to the extent of species level? Second, in N. douglasiae inhabiting the Korean Peninsula, is there a genetic lineage that is distinct enough to be considered a new Nodularia species? Third, what biogeographic pathways did Nodularia species inhabiting the Korean Peninsula come through? Finally, is there any pattern of genetic differentiation among the populations of N. douglasiae and N. breviconcha reflecting the known historical events of freshwater system formation and fluctuations?
Results
Sequence analysis of CO1 and 16S rRNA
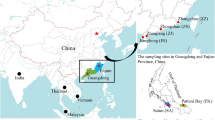
The mitochondrial CO1, which is 524 bp in length, was amplified and sequenced from 86 individuals of N. douglasiae and 83 of N. breviconcha collected from seven and six rivers, respectively, on the Korean Peninsula (Table 1, Fig. 1). Extensive sampling was performed in populations BH, GM, and ND of N. douglasiae, and in populations BH and NH of N. breviconcha. We had trouble collecting individuals from the other populations, due to the scarcity, and ended up sampling only one to five individuals per population. In our analysis, 25 and 16 CO1 haplotypes were detected from N. douglasiae and N. breviconcha, respectively (Supplementary Tables S1 and S2). No haplotype was interspecifically shared. A total of 30 and 21 polymorphic sites of N. douglasiae and N. breviconcha were identified, respectively, of which eight and nine were singleton variable sites, and 22 and 12 were parsimoniously informative sites. As shown in Table 1, in N. douglasiae, the number of CO1 haplotypes per population varied from one (SJ, TJ, and YS) to 13 (ND). Haplotype diversity was highest in the population ND (h = 0.878). The population ND only showed the statistically significant negative Fu's Fs value, which could be a signature of a sudden historical population expansion. In N. breviconcha, the number of CO1 haplotypes per population was higher in BH (6) and NH (7) than the others. Haplotype diversity was relatively high in BH (h = 0.695) and SJ (h = 1; but estimated from only two individuals). N. breviconcha showed statistically significant negative Fu's Fs value (− 5.203) when considering all observed haplotypes.

Sampling sites for 86 Nodularia douglaisae and 83 Nodularia breviconcha in the Korean Peninsula. The sampling locations of the seven N. douglasiae populations (BH, GM, MG, ND, SJ, YS, and TJ) and the six N. breviconcha populations (BH, NH, GM, SJ, YS, and TJ) were depicted on the map. For the comparison Nodularia species inhabiting in East Asian countries, we used the data from Japan, China, Russia, and Vietnam as well as the Korean Peninsula. The maps were generated by Adobe Illustrator CC 2020 using a GIS shape file retrived from administrative areas database in DIVA GIS (https://www.diva-gis.org/).
The mitochondrial 16S rRNA (357 bp long) was successfully amplified and sequenced from 79 out of 86 individuals of N. douglasiae and 79 out of 83 individuals of N. breviconcha (Fig. 1, Table 1), which yielded 16 haplotypes for N. douglasiae and five for N. breviconcha (Supplementary Tables S3 and S4). No haplotype was interspecifically shared in 16S rRNA. A total of 18 and five polymorphic sites from N. douglasiae and N. breviconcha were identified, respectively, of which ten and two were singleton variable sites, and 8 and 3 parsimoniously informative sites. In N. douglasiae, relatively high haplotype diversities were found in populations BH, GM, and ND because of limited sampling in the other populations (Table 1). The highest haplotype diversity (h = 0.794) and nucleotide diversity were observed in population GM. Only population ND showed significant negative Tajima's D and Fu's Fs, indicating a drastic population expansion. In N. breviconcha, only population NH showed significant negative Tajima’s D and Fu’s Fs, which is also the signature of demographic expansion. Populations BH (though statistically insignificant) and NH of N. breviconcha consistently showed negative Tajima’s D and Fu’s Fs like in CO1, strongly implying historical expansions of these populations.
Phylogenetic and population analyses
For the present phylogenetic and population analyses based on CO1 haplotypes, we retrieved and aligned additional genetic information from NCBI GenBank database including 42 CO1 haplotypes from 108 sequences for N. douglasiae, one haplotype from 2 sequences for Nodularia sp.1, 14 haplotypes from 26 sequences for Nodularia sp.2, 7 haplotypes from 9 sequences for N. nipponensis, and one haplotype from one sequence of N. nuxpersicae (Supplementary Table S5). Five haplotypes for five outgroup species were also retrieved. With 25 haplotypes for N. douglasiae and 16 haplotypes for N. breviconcha newly obtained in this study, a total of 106 Nodularia CO1 haplotypes were used for the phylogenetic and population genetic analyses. According to the ML and BI phylogenetic trees reconstructed with 106 Nodularia CO1 haplotypes (Fig. 2a), N. douglasiae (BP 99% and BPP 1.00) and N. breviconcha (BP 96% and BPP 1.00) formed independent monophyletic groups within the clade of the genus Nodularia with high node confidence values, respectively. These two species were clearly separated by other intrageneric species, N. nipponensis, N. nuxpersicae, N. sp.1, and N. sp.2. N. douglasiae was directly clustered with N. nipponensis and N. nuxpersicae, with N. breviconcha being placed at the basal node. Within the group of N. douglasiae, there were three genetic lineages of the clades A, B, and C, though the clade A was paraphyletic to the clade B. In the results of the PCoA and TCS network analyses (Fig. 2; Supplementary Fig. S2a), N. douglasiae was evidently separated from N. breviconcha as shown in the phylogenetic analyses (Fig. 2a). In the PCoA (Fig. 2a), clades B and C of N. douglasiae were almost overlapped with each other, though clade A was slightly distant from clades B and C. In the TCS network (Fig. 2b), the three clades were also closely related with small number of mutation steps.

modified by Adobe Illustrator CC 2020.
(a) Phylogenetic tree reconstructed using 106 CO1 haplotypes of six Nodularia species based on Bayesian inference (BI) and maximum likelihood (ML) methods. The tree was reconstructed using 67 CO1 haplotypes of N. douglasiae, 16 of N. breviconcha, one of N. sp.1, 14 of N. sp.2, seven of N. nipponensis, and one of N. nuxpersicae. Five species were used as outgroups. The species used in this phylogenetic analyses were listed in Supplementary Tables S5 and S7. The numbers on branches indicate bootstrap values (ML) and posterior probabilities (BI) being separated by slash. The PCoA graph was generated based on the CO1 haplotypes from six Nodularia species. (b) The unrooted TCS haplotype network was constructed based on the CO1 haplotypes from the six Nodularia species. Haplotype frequency is related to the size of the circle. Different colors within the nodes refer to different sampling sites shown in Fig. 1. TCS network was generated using PopART (https://popart.otago.ac.nz/index.shtml) and then
As shown in Fig. 2, the clade A consisted of the haplotypes of N. douglasiae only from BH, ND, YS, and GM populations in the Korean Peninsula, the clade B included the haplotypes from relatively wide distribution range including northeast China, west Japan, Russia, Vietnam, as well as South Korea (GM, MG, SJ and TJ). On the other hand, the haplotypes of the clade C were only from west China and Russia. Thus, CO1 haplotypes of N. douglasiae collected from the Korean Peninsula could be allocated into clades A and B; no clade C was not found in this peninsula. Also, in population GM, haplotypes from the clades A and B coexisted.
Based on the haplotypes of 16S rRNA, the ML and BI trees (Fig. 3a) showed that N. breviconcha (BP 94% and BPP 1.00) and N. nipponensis (BP 97% and BPP 0.99) formed strong monophyletic groups, though the monophyly of N. douglasiae (BP 69% in ML) was supported with relatively lower node confidence value. N. nipponensis appeared to be a sister species to N. douglasiae. Overall, tree topology inferred from 16S rRNA was consistent with that reconstructed based on CO1 data (Fig. 2a). N. douglasiae and N. breviconcha formed independent monophyletic groups. Due to the missing of Russian samples and the limited number of Chinese haplotypes, the clade C shown in CO1 data was not observed in N. douglasiae. The monophyly of clade A was weakly supported, and the clade B was paraphyletic to the clade A. According to the results of the PCoA and TCS network analyses based on 16S rRNA (Fig. 3; Supplementary Fig. S2b), N. douglasiae was evidently separated from N. breviconcha, just as observed in the CO1 results (Fig. 2; Supplementary Fig. S2a). The clades A and B of N. breviconcha appeared to be almost completely overlapped with each other in the PCoA (Fig. 3a; Supplementary Fig. S2b), with these two being separated by only a single mutation step in TCS network (Fig. 3b).

modified by Adobe Illustrator CC 2020.
(a) Phylogenetic tree reconstructed based on the maximum likelihood (ML) and Bayesian inference (BI) methods using 16S rRNA haplotypes of the three Nodularia species. The tree is constructed using 16S rRNA haplotypes of 20 N. douglasiae and five of N. breviconcha and seven of N. nipponensis. Two species were used as outgroups. The species used in this phylogenetic analysis were listed in Supplementary Tables S6 and S8. The numbers on branches indicate the bootstrap values (ML) and posterior probabilities (BI) being separated by slash. The PCoA graph was generated based on the 16S rRNA haplotypes from the three Nodularia species showed on phylogenetic tree. (b) The unrooted TCS haplotype networks was constructed based on the haplotypes of 16S rRNA haplotypes from the three Nodularia species. Haplotype frequency is related to the size of the circle. Different colors within the nodes refer to different sampling sites shown in Fig. 1. TCS network was generated using PopART (https://popart.otago.ac.nz/index.shtml) and then
Demographic analysis
The neutrality tests were performed with CO1 and 16S rRNA haplotypes of N. douglasiae and N. breviconcha (Table 2). All the three clades of N. douglasiae in the CO1 data showed significant negative values in Fu’s Fs, respectively, but not in Tajima’s D. When measured on the species itself, the same pattern was appeared. In N. breviconcha, both Tajima’s D and Fu’s Fs values were significantly negative. On the other hand, based on the 16S rRNA data, both N. douglasiae and N. breviconcha showed significant negative values only in Fu’s Fs, respectively, but not in Tajima’s Ds. When a mismatch distribution analyses (MDA) based on only CO1 were performed for each species, both N. douglasiae and N. breviconcha (Fig. 4a) showed multimodal curves. When analyzed for each clade of N. douglasiae (Fig. 4a) a unimodal curve was observed.

(a) The mismatch distribution analysis (MDA) and (b) Bayesian skyline plot (BSP) analysis estimated based on the CO1 haplotypes of Nodularia douglasiae, the three N. douglasiae clades of A, B, and C, and Nodularia breviconcha. The 67 CO1 haplotypes for N. douglasiae (Supplementary Table S7), and 16 haplotypes for N. breviconcha (Supplementary Table S2) were employed. The number of haplotypes for the clades of N. douglasiae refers to Table 2.
Bayesian skyline plot (BSP) analyses based on the CO1 data were performed to examine the pattern of fluctuation in effective population size for N. douglasiae, and N. breviconcha (Fig. 4b), and each of the three clades of N. douglasiae (Fig. 4b). The effective population size of N. douglasiae was dramatically increased from ca. 100 kya but was ceased at around ca. 50 kya. N. breviconcha began to gradually grow from ca. 100 kya, which was ceased at round ca. 50 kya. Among the three clades within N. douglasiae, a noticeable growth event was observed only in A and C at a certain period. Demographic expansion occurred slightly earlier in the clade C (ca. 100 – 50 kya) than A (ca. 130 –70 kya).
Divergence time estimation
According to the molecular clock analysis by the BEAST program (Supplementary Fig. S3), it is estimated that N. breviconcha and the other examined Nodularia species shared a common ancestor at about 28.21 mya. The divergence time of N. douglasiae and N. nipponensis was estimated to be around 20.57 mya. Within N. douglasiae, clade A was first diverged off about 12.26 mya, and then the clades B and C were diverged from each other at around 8.88 mya. The S-DIVA analysis under a Bayesian Binary Markov Chain Monte-Carlo (BBM) model (Fig. 5) indicated that a hypothetical common ancestor of the six examined Nodularia species originated from around Chinese Yangtze River where the clade B haplotypes are dominantly observed. This result may also indicate that the region may be regarded as a plausible origin of all examined Nodularia species in East Asia, with raising the possibility of the clade B as ancestral haplotypes (Fig. 6; Supplementary Fig. S4).

Time-calibrated Bayesian tree reconstructed with 106 CO1 haplotypes of six Nodularia species using the BEAST 2.6.0. program with the inference of ancestral areas under Bayesian binary MCMC (BBM) model implemented in the RASP 3.2 program. Ancestral areas were hypothesized based on the distribution ranges of each Nodularia species identified in the phylogenetic tree, which were shown as the format of pie chart on the nodes. See Fig. 1 for the details about how to generate the map.

The plausible scenario of changes of biogeographical distribution of the six Nodularia species in East Asia, focusing on Nodularia douglasiae and Nodularia breviconcha in the Korean Peninsula. See Fig. 1 for the details about how to generate the map.
Discussion
This study was designed to provide a taxonomic and phylogenetic revision of the species in the genus Nodularia distributed throughout northeastern Asia (Beringia, Amur-Korea, Japan-Skalin, and Eastern China) with a focus on N. douglasiae and N. breviconcha. Although N. breviconcha is now known as a native species endemic to South Korea, it has often been treated as a subspecies of N. douglasiae, even after the start of extensive taxonomic study for Nodularia species20, 21. It has been consistently pointed out that these two species have distinct morphological differences21. Recent studies based on CO1 sequences10, 12, 20 have insisted that N. breviconcha should be considered as an independent species apart from N. douglasiae on the basis of the remotely related phylogenetic relationships between the two species within the genus Nodularia. Lopes-Lima et al.10 gave a new scientific name to this species as N. breviconcha. However, recent taxonomic and phylogenetic studies on the genus Nodularia have an obvious drawback, limited sampling for N. breviconcha. For examples, Klishko et al.20 used only a single individual, and Lopes-Lima et al.10 considered 6 haplotypes for the analysis (individual information not shown). Therefore, more extensive sampling was needed to address the distribution of its intraspecific variation of N. breviconcha. Thereby, a total of 83 N. breviconcha and 86 N. douglasiae individuals were collected from the six and seven different drainages in South Korea, respectively, in the present study. With the collected samples, the nucleotide sequences of CO1 and 16S rRNA were determined, which were subjected to the subsequent population genetic and phylogenetic analyses.
Summarizing all the present results including the phylogenetic trees, PCoA, and TCS networks, it was rather ironic that there have been debates over the taxonomic status of N. douglasiae and N. breviconcha. Their genetic differentiation was highly evident. Thus, we here confirmed N. breviconcha to be an independent species. In the phylogenetic trees reconstructed using CO1 and 16S rRNA, N. breviconcha occupied a basal position in the clade of the genus Nodularia without exception. In addition, N. nipponensis, N. nuxpersicae, and Nodularia sp.1 and sp.2 were even more closely related to N. douglasiae than N. breviconcha. According to the BEAST analysis, N. breviconcha was first branched out and colonized in the freshwater systems of the Korean Peninsula during the late Paleogene (ca. 28 mya). The appearance of this new endemic species may be due to the adaptation to new habitats of the Korean Peninsula, markedly different from other parts of the East Asia. Given a much smaller shell size than N. douglasiae21, N. breviconcha may have occupied different types of niche while avoiding competition with N. douglasiae. No ecological studies have yet been conducted on the adaptation of these two species to their microhabitats on the Korean Peninsula so far.
In N. breviconcha, a star-like pattern appeared in the haplotype network without any strong genetic structure among populations, indicating that there was little spatial isolation process after geographical dispersion. However, the genetic structure was slightly formed in the CO1 haplotype network. Moreover, the mismatch analyses (MDA) with CO1 showed a multi-modal curve, suggesting the probability that two slightly different genetic groups existed within N. breviconcha. The results of examining both the Tajima’s D and Fu’s Fs using CO1 and 16S rRNA at the species level clearly support the sudden demographic expansion of this species following colonization on the Korean Peninsula.
In the phylogenetic trees, PCoA, and TCS networks obtained from the CO1 sequences, N. douglasiae haplotypes were assigned to the three clades of A, B, and C. With 16S rRNA sequences, we could also reconfirm the structure of the clades A and B, though the existence of clade C was not confirmed as NCBI GenBank lacked 16S rRNA information from Chinese and Russian specimens. In CO1 sequences, clades B and C showed a star-like haplotype network pattern, unlike clade A, showing the signature of population expansion during the formation of frequent confluences in the geological period. The individuals belonging to the clade A are dominantly distributed in the Nakdong River (ND) and the upper reaches of the Han River (BH). Probably, clade A may not have the opportunity of demographic expansion in the process of dispersing along the limited habitat areas on the narrow peninsula. The MDA analysis based on CO1 showed a more complicated multi-modal curve than that of N. breviconcha, but each of N. douglasiae clades showed a unimodal curve. Such pattern suggests the probability that no genetic structure existed within each clade, despite a definite genetic structure within the whole N. douglasiae clades. However, we could not find a possible signature of population expansion within whole N. douglasiae population, considering that statistically significant values were not observed in the Tajima’s D tests for CO1 and 16S rRNA at the species level and the clade level, though the Fu’s Fs values were significant in both loci.
Lopes-Lima et al.10 recently suggested that the clade A inhabiting the Korean Peninsula should be assigned as Nodularia sp.1 with the possibility of being an independent species apart from N. douglasiae. However, this suggestion was made based on three different haplotypes. Our robust phylogenetic results reconstructed based on CO1 and 16S rRNA do not support their view of considering N. douglasiae as a species complex with multiple species candidates including the clade A. The clade A was not monophyletic but paraphyletic to the monoclade of B and C in our phylogenetic results. In addition, the degree of genetic differences between clades A and B was not enough to consider the level of interspecific difference in our haplotype network analyses. Clade A was overlapped with clade B in PCoA results obtained based on both CO1 and 16S rRNA.
According to the results of the molecular clock analysis using the BEAST program, clade A was first branched off from the ancestral N. douglasiae and colonized the Korean Peninsula around mid-Neogene (ca. 12 mya). Afterwards, it seems that clades B and C seem to be born around late Neogene (ca. 9 mya). The clade B of N. douglasiae consisted of the haplotypes from east China, west Japan, east Russia and Vietnam, as well as South Korea, whereas the clade C haplotypes were obtained from the samples of central China and Russian Far East. Through the S-DIVA analysis under BBM model, geographic locations of the most common ancestors were examined at each node of speciation of Nodularia and clade formation within N. douglasiae. The clade B haplotypes (= a plausible common ancestor of Nodularia) were dominantly located at the Yangtze River in China, and the clade A was diverged on the way of the eastward inflow into the Korean Peninsula. In addition, it is thought that the Korean Peninsula probably served as a bridgehead for the dispersal of clade B to west Japan.
As summarized in Fig. 6, we attempted to reconstruct the historical pathway of the two South Korean Nodularia species, N. douglasiae and N. breviconcha, to colonize the Korean Peninsula. Clade B haplotypes can be regarded as the potential origin of N. douglasiae, as shown in the S-DIVA results (B-like ancestor; Fig. 6a). The distribution of the clade B is concentrated in the eastern part of China and the western part of the Korean Peninsula, centered on the Yellow Sea10, 12. In particular, the clade B populations are abundantly found mainly in Hongze Lake, Taihu Lake and the surrounding freshwater systems10 that are thought to have been frequently connected to the Yangtze River, a long river system running through south China, along geological times10. The Yangtze River in China is the hotspot with the largest number of endemic freshwater mussel species in East Asia and is believed to have been the origin of the contemporary species diversity11. Taken together, it is likely that the present data supported the demographical history of Nodularia species that they were originated from the Yangtze River in China and spread to the East Asian freshwater systems including the Korean Peninsula and Japan (around 28 mya during the late Paleogene). After wide distribution of the clade B of N. douglasiae, it is likely that N. breviconcha endemic to South Korea was first raised possibly at the Han River including BH and NH during the late Paleogene (around 28–25 mya; Fig. 6b). The speciation events of N. sp.1 (east China), N. sp.2 (east China), N. nipponensis (west Japan), and N. nuxpersicae (north Vietnam) might occur from the late Paleogene to the middle Neogene (around 25–12 mya; Fig. 6c). In the middle Neogene around 12–9 mya (Fig. 6d), it is likely that the clade A of N. douglasiae might have colonized the middle area (i.e. GM) of South Korea, while N. breviconcha seemed to expand its habitats into the southward of South Korea. From late Neogene (ca. 9 mya), it is thought that the clade C of N. douglasiae was raised in the west China or Russia and the clade A of N. douglasiae expanded to the eastward of South Korea (Fig. 6e). As shown in Fig. 6e, it is interesting that the distribution of N. breviconcha were overlapped with clade A (ⓐ; BH) and clade B (ⓑ; SJ, YS, and TJ). The distribution of clades A and B were also overlapped with each other (ⓒ; GM).
According to the present results, there still remain two big questions as follows. First, why is the clade A of N. douglasiae relatively rare in the west coastal rivers but overwhelming in the east areas of the Korean Peninsula, mainly in the Nakdong River and the upper reaches of Han River? Although the Han River including BH has likely been colonized through the paleo-Yellow River confluence, it is difficult to infer the biogeographic pathways that formed the Nakdong River population. Since continental shelf is not well-developed around the mouth of the Nakdong River, estuary coalescence with adjacent rivers would not have been possible25,26,27,28. If so, it is somewhat surprising that the genetic diversity of the Nakdong River population was relatively and significantly higher than the others in the present results, considering that this population would have flourished via the small-scale migration likely with watershed capture26. Given the high genetic diversity, there is no other way than to assume that there has been a persistent gene flow elsewhere, such as the Han River. However, no empirical evidence exists to support this assumption yet. Second, the investigation of genetic admixture between clades A and B was not conducted in this study because the mitochondrial loci were only used. Perhaps, genotyping using nuclear loci is necessary through further studies.
N. breviconcha and N. douglasiae clade A, which have been found to inhabit only the Korean Peninsula, are believed to have very important values in elucidating the evolutionary and demographic history of Nodularia species in East Asia. It is quite sure that these species are very useful for tracking the structure and changes of past freshwater systems that have formed on the Korean Peninsula. Given that most studies regarding the historical changes of freshwater systems have been centered on fish, comparative studies using these freshwater mussel species would be very effective and provide new perspectives on it. Considering their ecological and phylogeographical importance, concrete and sustainable management plans for N. breviconcha and N. douglasiae clade A should be established to preserve their historical imprints on the Korean Peninsula in the near future.
Methods
Sample collection and DNA extraction
A total of 86 N. douglasiae and 83 N. breviconcha individuals were collected from the seven (Bukhan, Geum, Nakdong, Seomjin, Tamjin, Yeongsan, and Mangyeong) and six (Bukhan, Geum, Namhan, Seomjin, Tamjin, and Yeongsan) rivers in South Korea between 2016 and 2017, respectively (Fig. 1; Supplementary Fig. S1). The collected individuals were fixed in 95% alcohol in the field, and were taken to the laboratory. Species identification was performed based on the shell morphology. The genomic DNA was isolated from muscle tissues (foot) using a DNeasy Blood and Tissue Kit (QIAGEN, Valencia, California, USA) following the manufacturer’s protocol. The concentration of extracted DNA was evaluated using NanoDrop 2000 (Thermo Fisher Scientific Co, USA) and 1% agarose gel electrophoresis.
PCR amplification and sequencing
To amplify partial mitochondrial DNA fragments of CO1 and 16S rRNA, and PCR was carried out using the previously known universal primers, LCO1490/HCO219829 and LCO22me2/HCO700dy230 for CO1 and 16Sar-L-myt/16Sbr-H-myt31, 16SarL/16SbrH32, and 16Sar/16Sbr33 for 16S rRNA (Supplementary Table S1). The information of the primers are shown in Supplementary Table S9. The thermal cycling profile consisted of a denaturation at 95 °C for 2 min, 35 cycles of 95 °C for 20 s, 48–50 °C for 40 s, and 72 °C for 1 min, and a final extension at 72 °C for 5 min. One microliter of each PCR product was electrophoresed on 1% agarose gel including eco-dye and observed under UV light. When the PCR bands were detected, the PCR products were purified using a QIAquick PCR Purification Kit (QIAGEN Co, USA) and directly sequenced with an ABI Prism 3730 DNA sequencer (PerkinElmer Inc, USA) using a Big Dye Termination Sequencing Kit (PerkinElmer Inc, USA). All the novel sequences of CO1 and 16S rRNA discovered in this study were deposited under the GenBank accession numbers (Supplementary Tables S5 and S6).
Population genetic analyses
The nucleotide sequences of mitochondrial CO1 and 16S rRNA obtained from N. douglasiae and N. breviconcha were aligned using Clustal X234 and BioEdit 7.2.535. The identification of variable and parsimoniously informative sites and the number of haplotypes (h) were estimated using DnaSP 6.1136. A mismatch distribution analysis (MDA)37 was also conducted to infer demographic stability of phylogenetic clades and species using DnaSP 6.11. Based on the haplotype list generated from DnaSP, the number of private haplotypes unique to each population was determined (Supplementary Tables S1–S4, and S7, S8). The Arlequin 3.5 program38 was used to examine population demographic history and evolutionary neutrality of N. douglasiae and N. breviconcha based on the tests of Tajima’s D39 and Fu’s Fs40. A haplotype network was constructed to estimate gene genealogies using the statistical parsimony approach at the population level using PopART41. To further evaluate and visualize the geographic genetic structure among the populations, a Principal Coordinates Analysis (PCoA) was conducted using the DARwin 6.0.9 program42. A Bayesian skyline plot (BSP) was computed in the BEAST 2.6.0 program43, 44 to examine the historical demographic fluctuation since the time of the most recent common ancestor. We used the HKY substitution model and mutation rates of 2.0 × 10–8 under a strict molecular clock used by Liu et al.12. Markov chain Monte Carlo was run for 5 million steps, with sampling every 1,000 generations, and the TRACER 1.5 program45 was used to construct the BSP46.
Phylogenetic analyses
For phylogenetic analyses, a Bayesian topology was inferred under the GTR + I + G model using MrBayes 3.2.247, and a maximum likelihood tree was reconstructed under the GTR + I + G model in IQtree online site (https://iqtree.cibiv.univie.ac.at) for the CO1 and 16S rRNA haplotype datasets. The 86 CO1 and 83 16S rRNA nucleotide sequences newly obtained from N. douglasiae and N. breviconcha inhabiting the Korean Peninsula were employed for the phylogenetic analyses. As listed in Supplementary Tables S5 and S6, previously reported data were retrieved from the NCBI GenBank and added to our final nucleotide sequence alignment sets. The added sequences were the haplotypes of the samples obtained from South Korea10, China4, 12, Japan10, Russia20, and Vietnam10 for CO1 of N. douglasiae, and from Japan (N. nipponensis and N. biwae) and South Korea (N. breviconcha)48 for 16S rRNA. The outgroup species used were Unio pictorum, Cuneopsis heudei, Cuneopsis rufescens, Cuneopsis pisciculus, and Schistodesmus lampreyanus for the CO1 dataset and Unio pictorum and Cuneopsis heudei for the 16S rRNA dataset.
Divergence time estimation
Divergence time estimation of the nodes on the Nodularia phylogeny was conducted in the BEAST 2.6.0. program43, 44 based on CO1 sequences. The BEAST analysis was conducted based on fossil calibration points using a lognormal relaxed molecular clock algorithm49 under the calibrated-Yule model. A HKY model was applied with correlations for gamma distribution, and we designated priors for outgroup taxa using a “Monophyly” option of the BEAUti 2 program as (Parreysiinae, (Nodularia)). The fossil calibrations adapted to the analysis were estimated from the outgroup taxa50, since fossil records of the genus Nodularia have not known yet. The absolute age of outgroup taxa was referred in Bolotov et al.50. Posterior distributions of parameter were estimated using 1,000,000 MCMC generations with sampled every 1,000 generations. In the TreeAnnotator 2.6.0 program51, the initial 20% of generations were removed as burn-in, and resultant 1,001 trees were combined to a maximum clade credibility tree. The consensus tree was visualized in the FigTree 1.4.2 program52. To estimate the distribution of a hypothetical common ancestor, a Bayesian binary MCMC method (BBM)53, which is implemented in the RASP 3.2 program54, was adapted to the BEAST tree. When running the program, three possible distributed areas were coded for the ingroup taxa as follows: A) the distribution range of the clade A of N. douglasiae including Bukhan River and Nakdong River in South Korea; B) the distribution range of the clade B of N. douglasiae including the rest of rivers in South Korea except Bukhan River and Nakdong River, Japan, Vietnam, Hongze lake and Taihu lake in China, and Amur River and Ussury River in Russia; C) The distribution range of the clade C including Poyang lake, Duguan lake, Qinglan lake, Gan River, Liangzi lake, Dongting lake, and Xiannv lake in China, and Onon River in Russia.
References
Bolotov, I. N. et al. Ancient river inference explains exceptional Oriental freshwater mussel radiations. Sci. Rep. 7, 2135. https://doi.org/10.1038/s41598-017-02312-z (2017).
Lopes-Lima, M. et al. Phylogeny of the most species-rich freshwater bivalve family (Bivalvia: Unionida: Unionidae): defining modern subfamilies and tribes. Mol Phylogenet. Evol. 106, 174–191. https://doi.org/10.1016/j.ympev.2016.08.021 (2017).
Vaughn, C. C. Ecosystem services provided by freshwater mussels. Hydrobiologia https://doi.org/10.1007/s10750-017-3139-x (2017).
Liu, X. J. et al. Effects of dams and their environmental impacts on the genetic diversity and connectivity of freshwater mussel populations in Poyang Lake Basin China. Freshw. Biol. 65, 264–277 (2019).
Zieritz, A. et al. Diversity, biogeography and conservation of freshwater mussels (Bivalvia: Unionida) in East and Southeast Asia. Hydrobiologia 1, 1–16. https://doi.org/10.1007/s10750-017-3104-8 (2017).
IUCN. Guidelines for Application of IUCN Red List Criteria at Regional and National Levels: Version 2016–3 (IUCN, 2017).
Lee, J. H., Choi, E. H., Kim, S. K., Ryu, S. H. & Hwang, U. W. Mitochondrial genome of the cockscomb pearl mussel Cristaria plicata (Bivalvia, Unionoida, Unionidae). Mitochondrial DNA 23, 39–41 (2012).
Lopes-Lima, M. et al. Biology and conservation of freshwater bivalves: past, present and future perspectives. Hydrobiologia 735, 1–13. https://doi.org/10.1007/s10750-014-1902-9 (2014).
Lopes-Lima, M. et al. Conservation status of freshwater mussels in Europe: state of the art and future challenges. Biol. Rev. 92, 572–607. https://doi.org/10.1111/brv.12244 (2017).
Lopes-Lima, M. et al. Freshwater mussels (Bivalvia: Unionidae) from the Rising Sun (Far East Asia): Phylogeny, systematics and distribution. Mol. Phylogenet. Evol. https://doi.org/10.1016/j.ympev.2020.106755 (2020).
Huang, X. C. et al. Towards a global phylogeny of freshwater mussels (Bivalvia: Unionida): species delimitation of Chinese taxa, mitochondrial phylogenomics, and diversification patterns. Mol. Phylogenet. Evol. 130, 45–59. https://doi.org/10.1016/j.ympev.2018.09.019 (2019).
Liu, X. et al. Genetic structure and diversity of Nodularia douglasiae (Bivalva: Unionida) from the middle and lower Yantze River drainage. PLoS One, 12 (2017).
Graf, D. L., Jones, H., Geneva, A. J., Pfeiffer, J. M. & Klunzinger, M. W. Molecular phylogenetic analysis supports a Gondwanan origin of the Hyriidae (Mollusca: Bivalvia: Unionida) and the paraphyly of Australasian taxa. Mol. Phylogenet. Evol. 85, 1–9. https://doi.org/10.1016/j.ympev.2015.01.012 (2015).
Tominaga, K., Nakajima, J. & Watanabe, K. Cryptic divergence and phylogeography of the pike gudgeon Pseudogobio esocinus (Teleostei: Cyprinidae): a comprehensive case of freshwater phylogeography in Japan. Ichthyol. Res. https://doi.org/10.1007/s10228-015-0478-3 (2015).
Lee, E. & Nam, S. Freshwater supply by Korean rivers to the East Sea during the last glacial maximum: a review and new evidence from the Korean Strait region. Geo Mar. Lett. 23, 1–6 (2003).
Song, K. H., Jung, J. & Kim, W. Genetic variation in a freshwater prawn species, Palaemon paucidens South Korea. Biochem. Syst. Ecol. 65, 23–32 (2016).
Kaizuka, S. Late cenozoic paleogeography. GeoJournal 4, 101–109 (1980).
Kimura, M. Paleogeography of the ryukyu islands. Tropics 10, 5–24 (2000).
Lee, J. H., Kim, S. K. & Hwang, U. W. Complete mitochondrial genome of Lamprotula coreana (Unionidae, Unionoida, Bivalvia). Mitochondrial DNA 27, 1026–1027 (2016).
Klishko, O. K., Lopes-Lima, M., Froufe, E., Bogan, A. E. & Abakumova, V. Y. Unravelling the systematics of Nodularia (Bivalvia, Unionidae) species from eastern Russia. Syst. Biodivers. 16, 287–301. https://doi.org/10.1080/14772000.2017.1383527 (2018).
Kwon, O. K. Ilustrated encyclopedia of fauna and flora of Korea. Mollusca (I) (ed. Kwon, O. K.) 32, 153–159 (Ministry of Education, 1990).
Cha, S. H. et al. The complete mitochondrial genome of a freshwater mussel Nodularia douglasiae (Bivalvia : Unionidae). Mitochondrial DNA. 3, 945–946 (2018).
Kwon, O. K. & Habe, T. A list of non-marine moluscan fauna of Korea. Korean Journal of Limnology 12, 25–33 (1979).
Klishko, O. K., Lopes-Lima, M., Froufe, E. & Bogan, A. E. Solution of taxonomic status of Unio mongolicus Middendorff, 1851 (Bivalvia: Unionidae) from the type locality in Transbaikalia and history of its taxonomy. Ruthenica. 29, 55–70 (2019).
Jeon, H., Kim, D., Lee, Y. J., Bae, H. & Suk, H. Y. The genetic structure of Squalidus multimaculatus revealing the historical pattern of serial colonization on the tip of East Asian continent. Sci. Rep. 8, 10629. https://doi.org/10.1038/s41598-018-28340-x (2018).
Won, H., Jeon, H. & Suk, H. Y. Evidence of an ancient connectivity and biogeodispersal of a bitterling species, Rhodeus notatus, across the Korean Peninsula. Sci. Rep. 10, 1011. https://doi.org/10.1038/s41598-020-57265-3 (2018).
Unmack, P. J., Hammer, M. P., Adams, M., Johnson, J. B. & Dowling, T. E. The role of continental shelf width in determining freshwater phylogeographic patterns in south-eastern Australian pygmy perches (Teleostei: Percichthyidae). Mol. Ecol. 22, 1683–1699 (2013).
Zúñiga-Vega, J. J., Ingley, S. J., Unmack, P. J. & Johnson, J. B. Do freshwater ecoregions and continental shelf width predict patterns of historical gene flow in the freshwater fish Poecilia butleri?. Biol. J. Linn. Soc. 112, 399–416 (2014).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molec. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Walker, J. M. et al. Taxonomic distribution and phylogenetic utility of genderassociated mitochondrial genomes in the Unionoida (Bivalvia). Malacologia. 48, 265–282 (2006).
Lydeard, C., Mulvey, M. & Davis, G. M. Molecular systematics and evolution of reproductive traits of North American freshwater unionacean mussels (Mollusca: Bivalvia) as inferred from 16S rRNA gene sequences. Philos. Trans. R. Soc. Lond. B 351, 1593–1603 (1996).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Ent. Soc. Am. 87, 651–701 (1994).
Palumbi, S. R. Nucleic acids II: the polymerase chain reaction. (ed. Hillis, D. M., Moritz, C. & Mable, B. K.) 205–248. (Molecular Systematics, 1996).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948 (2007).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 3299–3302 (2017).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol 9, 552–569 (1992).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Leigh, J. W. & Bryant, D. popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Perrier, X. & Jacquemoud-Collet, J. P. DARwin software, available at, https://darwin.cirad.fr/Darwin (2006).
Bouckaert, R. et al. BEAST2: a software platform for Bayesian evolutionary analysis (PLoS Comput, Biol, 2014).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2010).
Rambaut, A. & Drummond, A. J. Tracer v1.6. Available at https://beast.bio.ed.ac.uk/tracer (2014).
Heled, J. & Drummond, A. J. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8, 289 (2008).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Sano, I., Shirai, A., Kondo, T. & Miyazaki, J. I. Phylogenetic relationships of Japanese Unionoida (Mollusca: Bivalvia) based on mitochondrial 16S rDNA sequences. J. Water Resour. Prot. 9, 493–509 (2017).
Drummond, A. J., Ho, S. Y., Philips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, 669 (2006).
Bolotov, I. N. et al. New taxa of freshwater mussels (Unionidae) from a species-rich but overlooked evolutionary hotspot in Southeast Asia. Sci. Rep. 7, 11573 (2017).
Rambaut, A. & Drummond, A. J. TreeAnnotator v.2.3.0. Available at https://beast2.org (2014).
Rambaut, A. FigTree v.1.4., available at, https://tree.bio.ed.ac.uk/sofware/fgtree/ (2012).
Nylander, J. A. A., Olsson, U., Alström, P. & Sanmartín, I. Accounting for phylogenetic uncertainty in biogeography: a Bayesian approach to dispersal-vicariance analysis of the thrushes (Aves: Turdus). Syst. Biol. 57, 257–268 (2008).
Yu, Y., Harris, A. J., Blair, C. & He, X. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49 (2015).
Acknowledgements
Special thanks should be given to Dr. Jin-Hee Lee from Daegu Science High School (Daegu, South Korea), and Dr. Sang-Ki Kim from Animal & Plant Research Team, Nakdonggang National Institute of Biological Resources (Sangju, South Korea). They partly dedicated to collect some Nodularia samples and to produce their nucleotide sequence data employed in this study. This study was financially supported by a year-2017 grants of Nakdonggang National Institute of Biological Resources, “Genetic diversity research of freshwater biological resources”.
Author information
Authors and Affiliations
Contributions
E.H.C., S.H.R., Y.S.L., and U.W.H. designed this study. S.H.C. and E.H.C. did the experimental works. E.H.C., G.K., S.Y.B., H.Y.S., and U.W.H. wrote the manuscript. G.K. and H.Y.S. were mainly responsible for data analysis. J.S.L. collected and identified some samples used in this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, E.H., Kim, G., Cha, S.H. et al. Molecular phylogenetic, population genetic and demographic studies of Nodularia douglasiae and Nodularia breviconcha based on CO1 and 16S rRNA. Sci Rep 10, 16572 (2020). https://doi.org/10.1038/s41598-020-72015-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72015-5
This article is cited by
-
Possible species discrimination of a blotched nerite Nerita albicilla with their distribution pattern and demographic history in the Indo-Pacific
Scientific Reports (2023)
-
Liolophura species discrimination with geographical distribution patterns and their divergence and expansion history on the northwestern Pacific coast
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
