
Abstract
Over the course of human history and in most societies, fermented beverages have had a unique economic and cultural importance. Before the arrival of the first Europeans in Australia, Aboriginal people reportedly produced several fermented drinks including mangaitch from flowering cones of Banksia and way-a-linah from Eucalyptus tree sap. In the case of more familiar fermented beverages, numerous microorganisms, including fungi, yeast and bacteria, present on the surface of fruits and grains are responsible for the conversion of the sugars in these materials into ethanol. Here we describe native microbial communities associated with the spontaneous fermentation of sap from the cider gum Eucalyptus gunnii, a Eucalyptus tree native to the remote Central Plateau of Tasmania. Amplicon-based phylotyping showed numerous microbial species in cider gum samples, with fungal species differing greatly to those associated with winemaking. Phylotyping also revealed several fungal sequences which do not match known fungal genomes suggesting novel yeast species. These findings highlight the vast microbial diversity associated with the Australian Eucalyptus gunnii and the native alcoholic beverage way-a-linah.
Similar content being viewed by others


Introduction
Since the establishment of human settlement, alcoholic beverages have played a unique role in most societies, due to their economic and cultural importance1. Archaeological evidence suggests that the oldest fermented beverage was produced in Henan province, Northern China between 7000 and 6650 BCE and which was made of grapes, hawthorn berries, honey, and rice2. Evidence has also dated wine production in Iran as early as 6000 BCE and in Egypt to around 3000 BCE3,4. Since that time, it is believed that the use of fermentation to produce alcoholic beverages expanded from Mesopotamia throughout the world1. It is very likely that the consumption of alcoholic foods and/or beverages precedes modern humans, with frugivorous species, including many primates and hominoid lineages, eating and developing a taste for fermented fruits5,6. Indeed, evidence indicates that the ancestor of humans, chimpanzees, and gorillas adapted to metabolise ethanol long before human-directed fermentation7.
Early communities produced fermented beverages from different substrates, including honey to make mead or honey wine, which was produced in Asia around 1700–1100 BCE; grapes and barley to make wine and beer, respectively, in the Middle east, Rome and China8. In South America grains or fruits were used to produce chicha, while in North America, octli (now known as pulque) was made from agave, a type of cactus8. In Eastern Europe and central Asia, mare’s milk was used to produce a lactic-alcoholic beverage called koumiss9. In Australia before the arrival of the first Europeans, Aboriginal people produced several fermented drinks including mangaitch from flowering cones of Banksia in Western Australia, way-a-linah from Eucalyptus tree sap in Tasmania and kambuda from crushed nuts of the palm-like Pandanus tree in the Northern Territory10. Torres Strait Islanders produced tuba from the fructifying buds of coconut palms, a knowledge gained from Southeast Asian populations and which spread throughout the Pacific11.
In the case of mangaitch, Aboriginal people would dig a trench near a swamp and then line the excavation with a boat-shaped container made of tea-tree bark10. This container was then filled with water and Banksia cones and left to soak to obtain a sugar-rich solution, which was then left to ferment for several days. The drink known as way-a-linah was made by Tasmanian Palawa people from the sap of Eucalyptus gunnii. Stone tools were used to bore a hole in the trunk of the tree and to make a larger depression at the base to collect the flowing sap, which was then left to ferment after covering the collecting hole with a flat stone10. Without human intervention, sap also flows from naturally occurring holes in the trunk and accumulates at the base of the tree. For kambuda, reported from the Borroloola region, only ripe Pandanus nuts were used, which were fire roasted and then crushed with a stone, with the resulting pulp soaked in water for two days in a bark dish before fermentation10. Tuba was made from the syrup that seeps from a cut made in the unopened fructifying bud of Cocos nucifera, which was collected and left to ferment for several days11.
The alcohol present in fermented beverages is the result of yeast metabolism which converts the sugars present in fruits, grains, milk, honey and other carbon sources, into ethanol12. This process was named fermentation, from the Latin word fervere, which means ‘to boil’, probably due to the bubbles of CO2 produced by yeast8. Undoubtedly, the first fermented beverages were the result of spontaneous fermentation by microorganisms (fungi, yeast and bacteria) present on the surface of fruits and grains, inside fermentation pots or inadvertently introduced by human action12.
Spontaneous fermentation, is a complex microbial process involving the action of different species, which not only generate ethanol but also produce numerous secondary metabolites that shape the aroma and flavour of fermented beverages12,13. Although numerous indigenous fermented beverages have been produced worldwide14, only a few have been characterised from a microbiological perspective15. These include: tej, an Ethiopian honey wine16; chicha, a maize fermented drink from Peru17 or a rice-based fermented beverage from Brazil18; pulque, a Mexican fermented beverage produced from the sap of agave plants19,20; and makgeolli, a Korean traditional alcoholic beverage made from rice21, among others. Nowadays, spontaneous fermentation is used in the commercial production of several fermented beverages13,22,23,24. However, the microbial communities associated with the production of certain beer styles (e.g. Belgian lambic beer and American Coolship Ales) and related to wine fermentation are the most thoroughly characterised23,25,26,27,28.
E. gunnii, also known as cider gum, is a tree species endemic to Tasmania which is mainly found in cold, waterlogged habitats, such as lake edges, or poorly drained valley flats29. Factors such as climate change, possum and invertebrate activity, and stock grazing have been linked to a severe E. gunnii decline30. Particularly in the Central Plateau region of Tasmania, increased temperatures and a reduction in summer rainfall are contributing to a significant decrease in the availability of suitable micro‐sites for the successful regeneration of E. gunnii31. In this work, E. gunnii trees from three geographical locations in the Tasmanian Central Plateau in Australia, were sampled in two different dates to collect sap, bark and soil samples. Thus, we report for the first time the native microbial communities associated with E. gunnii and reveal major differences with the microbial composition associated with the production of other fermented beverages.
Results
The Tasmanian Eucalyptus gunnii produces a sweet sap that often flows from naturally occurring holes in the trunk and accumulates at the tree roots and/or on the soil, where it ferments spontaneously (Fig. 1). To evaluate the microbial communities responsible for the fermentation of the cider gum sap we sampled 33 trees from three geographical locations in the Tasmanian Central Plateau in Australia, Trawtha Makuminya, Skullbone Plains and Five Rivers – Serpentine in November 2016 and January 2017 (Fig. 1). In total, 84 different samples, 45 from sap, 29 from bark and 10 from soil, were collected.

The Tasmanian cider gum Eucalyptus gunnii (A). Sap flows from naturally occurring holes in the trunk (B) and accumulates on the tree roots (C) and or on the soil (D). Bark, sap and soil samples were collected on the Tasmanian Central Plateau (E) from three different locations, Trawtha Makuminya (red), Skullbone Plains (yellow) and Five Rivers—Serpentine (blue) (F). Maps were created with ggmap version 3.0.0 in R version 3.6.2.
Chemical composition of Eucalyptus gunnii sap
Sap samples were first analysed by HPLC to evaluate the presence of sugars, organic acids, glycerol and ethanol. A major peak, present in several samples, did not match any of the standards routinely used to quantify fermentation sugars and organic acids. GC/MS analysis on derivatised samples enabled the identification of this unknown compound as maltose. After identification, maltose was included with HPLC standards to obtain the basic chemical composition of E. gunnii sap (Table 1). Three sugars, glucose, fructose and maltose, were present in sap samples with maxima over 150 g/L. Several organic acids were also found with acetic and gluconic acids exceeding 30 g/L in some cases. Ethanol ranged from 0–6.1% (v/v).
Bacterial populations associated with E. gunnii
Sap, bark and soil samples were assessed by 16S phylotyping to identify the bacterial communities associated with E. gunnii. Thirteen different phyla were found across the samples, regardless of geographical location, sample type or sampling date (Table S1), with Proteobacteria (65%), Firmicutes (3%), Actinobacteria (2%) and Bacteroidetes (1%) being the most abundant, while 28% of all OTUs could not be assigned to any phylum (Table S2). Small but significant differences were found between phyla when comparing different ecological niches, with soil samples containing higher proportions of Fibrobacteres, Patescibacteria and Spirochaetes (Table S2). A total of 26 classes, 61 orders, 93 families and 114 unique identified genera were found across all samples (Table S1), with a great diversity of genera for the phylum Proteobacteria (Fig. 2A). The phyla Actinobacteria, Bacteroidetes and Firmicutes also showed a considerable diversity in bacterial genera. Prevalent genera across all samples were Gluconobacter, Acetobacter, Serratia, Dickeya, Komagataeibacter, Zymomonas and Leuconostoc (Fig. 2B), with Gluconobacter, Acetobacter and Zymomonas also being the most abundant (Figure S1). Although small differences were observed when comparing the relative abundance of the same genus by geographical location, ecological niche or sampling date (Figure S2), most differences were not statistically significant. Only some samples from Five Rivers—Serpentine showed significant differences, with the genus Enterococcus exhibiting higher relative abundance in soil samples, whereas the genus Gluconobacter showed lower abundance in sap samples. Similarly, the genus Komagataeibacter showed a higher relative abundance in samples from November 2016 than from January 2017.

Bacterial communities associated with Eucalyptus gunnii. (A) The cladogram shows different taxonomy levels in concentric rings. From the centre, kingdom, phylum, class, order, family and genus are shown. All identified bacterial phyla have been coloured, with taxa abundance proportional to circle size. (B) Most prevalent bacterial genera across samples are shown at different detection thresholds.
Significant differences were also found for diversity indices when samples were analysed by geographical location, ecological niche and sample date (Table 2). These indices included: richness, which indicates the number of species in an ecological community; diversity estimated with the Shannon index, which quantifies the uncertainty in the species identity of a random individual from the dataset; dominance, which indicates the abundance of a particular taxon over others; and divergence, which is calculated as the average dissimilarity of each sample from the group mean 32. Trawtha Makuminya showed higher diversity than other locations and lower dominance than Five Rivers—Serpentine. Soil samples had higher richness than sap samples, whereas bark samples showed lower divergence than sap and soil samples. Increased divergence was also found in samples obtained in January 2017.
Fungal communities associated with E. gunnii
Sap, bark and soil samples were also analysed by ITS phylotyping to identify the fungal communities associated with E. gunnii. Only four fungal phyla were found, comprising 14 classes, 38 orders, 75 families, 136 genera and 204 unique identified species (Table S3). The most abundant classes included Saccharomycetes, Dothideomycetes, Tremellomycetes, Leotiomycetes and Sordariomycetes, while nearly 50% of all OTUs could not be assigned to any class (Table S4). Significative differences in abundance for different classes were found depending on geographical location, ecological niche or sampling date. In particular, Dothideomycetes were more abundant in Five Rivers—Serpentine, while Eurotiomycetes were more abundant in samples from Skullbone Plains. Sap samples showed high abundance in Agaricomycetes, Cystobasidiomycetes, Dothideomycetes, Eurotiomycetes and Sordariomycetes. Only Agaricomycetes showed a significant difference according to sampling date with higher abundance in samples from 2017 (Table S4).
A similar diversity in the number of unique genera was observed for the classes Leotiomycetes, Dothideomycetes, Saccharomycetes and Tremellomycetes (Fig. 3A). The most prevalent genera were Kregervanrija, Hanseniaspora, Lachancea, Zygosaccharomyces, Candida and Pichia (Figure S4A). Diversity indices showed some significant differences when samples were analysed by geographical location and ecological niche (Table 2). Five Rivers—Serpentine showed higher divergence than Skullbone Plains, while sap samples showed increased divergence compared to bark and soil samples. No significant differences were found for sampling date.

Comparison between yeast communities associated with Eucalyptus gunnii (A) and with Vitis vinifera (B). Cladograms show different taxonomy levels in concentric rings. From the centre, kingdom, phylum, class, order, family and genus. The main 8 classes have been coloured, whereas the genus Saccharomyces is indicated in black. Taxa abundance is proportional to circle size. The 15 most abundant yeast genera associated with E. gunnii (C) and with V. vinifera (D). V. vinifera data from Morrison-Whittle and Goddard33,34.
Relative abundances in fungal communities
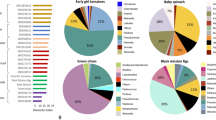
Significative differences in relative abundance for the most abundant fungal genera were only found for geographical location and ecological niche (Fig. 4). The genera Bulleromyces, Cladosporium, Hormonema, Lachancea, Penicillium, Penidiella, Xenophacidiella and Zygosaccharomyces showed different abundances depending on geographical location, while Bulleromyces, Citeromyces, Cladosporium, Cystobasidium, Hormonema, Kregervanrija, Lachancea, Penicillium, Penidiella, Pichia, Schwanniomyces, Torulaspora, Xenophacidiella and Zygosaccharomyces differed according to ecological niche. Interestingly, the genus Lachancea had the lowest abundance in samples from Skullbone Plains and the highest in sap samples. On the other hand, the genus Zygosaccharomyces showed the highest abundance in samples from Skullbone Plains and in bark samples (Fig. 4, Figure S3). The most abundant fungal species were Kregervanrija delftensis, Hanseniaspora valbyensis, Lachancea quebecensis, Citeromyces hawaiiensis, Schwanniomyces pseudopolymorphus, Lachancea cidri and Torulaspora globose (Table S5). When samples were grouped according to geographical location some significative differences were found for sampling date (Figure S5). In Five Rivers – Serpentine, the genera Mortierella and Torulaspora differed in relative abundance according to sampling date, while in Trawtha Makuminya the genera Cladosporium, Hanseniaspora, Lachancea, Metschnikowia and Penidiella showed different relative abundance.

Boxplots showing relative abundance for the 15 most abundant fungal genera associated with Eucalyptus gunnii based on geographical location (A), ecological niche (B) and sampling date (C). Stars indicate statistically significative differences according to the Kruskal–Wallis test (*p < 0.05; **p < 0.01; ***p < 0.001).
Permutational multivariate analysis of variance (PERMANOVA) showed that only ecological niche significantly affected the composition of fungal communities (Table 3). Geographical location, sampling data or the interaction between niche and location did not affect the diversity or composition of fungal populations. Principal Coordinates Analysis (PCoA) enabled to visualise clustering of sap samples according to geographical location for Trawtha Makuminya and Five Rivers—Serpentine (Fig. 5). No clustering was observed for bark or soil samples.

Principal Coordinates Analysis (PCoA) for the top 100 fungal OTUs in Five Rivers—Serpentine (blue), Skullbone Plains (yellow) and Trawtha Makuminya (red) for bark (A), sap (B) and soil (C) samples.
Fungal communities found in cider gum differed to those associated to winemaking
To compare the fungal communities associated with E. gunnii, which we obtained from soil, bark and sap, to those found in winemaking we used the data reported by Morrison-Whittle and Goddard33,34, which described fungal populations found in vineyard soil, on vine bark and grape berries, and during wine fermentation. Although the same main fungal classes were found for both cider gum and winemaking, genus diversity and/or relative abundance differed (Fig. 3A,B). Eurotiomycetes, Leotiomycetes, Microbotryomycetes and Tremellomycetes showed greater genus diversity in populations associated to cider gum, whereas Agaricomycetes, Pezizomycetes and Sordariomycetes had more diversity in communities associated to winemaking. Although genus diversity was similar for Dothideomycetes and Saccharomycetes, genus relative abundance was very different. The most abundant genera for Dothideomycetes associated with E. gunnii included: Hormonema, Xenophacidiella, Penidiella and Cladosporium, while Columnosphaeria, Cladosporium, Alternaria, Davidiella and Keissleriella were the most abundant in winemaking (Fig. 3C,D). For Saccharomycetes, the most abundant genera in cider gum samples were Hanseniaspora, Kregervanrija, Lachancea, Zygosaccharomyces, Citeromyces, Schwanniomyces, Torulaspora, Pichia and Candida, whereas in winemaking samples Saccharomyces, Torulaspora, Hanseniaspora and Wickerhamomyces were the most abundant.
Differences between fungal communities associated with cider gum and with grape growing and winemaking also included the most prevalent genera (Figure S4). Only three of the 16 most prevalent genera, Hanseniaspora, Torulaspora and Cladosporium, were common to both fungal communities. For communities associated with winemaking the most prevalent genera included Cladosporium, Columnosphaeria, Alternaria, Saccharomyces and Davidiella. Interestingly, Saccharomyces, which is the main genus responsible for alcoholic beverage fermentation including beer, cider and wine, was barely found in cider gum samples (Table S3). Species belonging to this genus, S. cerevisiae and S. uvarum, were only observed in some samples. While S. cerevisiae was found in samples from both Trawtha Makuminya and Five Rivers—Serpentine, S. uvarum was only found in the last location.
Discussion
Numerous indigenous fermented beverages have been produced worldwide10,11,14. These beverages are the result of microbial activity, which generally involves the concerted action of numerous bacterial, fungal and yeast species. Here, we report the microbial communities associated with way-a-linah, a mildly alcoholic beverage made from the sap of the cider gum E. gunnii by Aboriginal people in Tasmania.
E. gunnii, which is endemic to Tasmania, shows exceptional cold tolerance for a eucalyptus tree (down to − 20 °C for brief periods) and is now commonly planted as an ornamental tree across the British Isles and some parts of Western Europe35. E. gunnii has been studied as a source of bioenergy36; phytochemicals, including vitamins and fatty acids, given the particularly high concentration of tocopherols compared to other plants37; and essential oils, due to its antioxidant, antimutagenic and antibacterial properties38. E. gunnii sap contained a high concentration of sugars, a trait which is likely associated to its cold tolerance. Indeed, a positive correlation between cold resistance and total soluble carbohydrate content has been reported in E. gunnii cell lines39. While sucrose was the main sugar found in the leaves37, the most abundant soluble sugars found in cells were glucose, fructose, sucrose, stachyose and raffinose39. Here, we found glucose, fructose and maltose to be the main sugars in E. gunnii sap. These sugars are also the main components of honey14, while sucrose, glucose and fructose are the most abundant sugars in maple syrup40.
Several studies have shown biogeographic differentiation between microbial communities with differences between environmental niches and geographical locations, particularly for vineyards and microbial populations associated with winemaking26,33,41,42. Differences among microbial communities have been found at large geographical scales (> 100 km)26,33,34,41, medium scales (> 35 km)43, and small scales (< 2 km)44, indicating that local environmental variables, such as soil type, humidity and nutrient availability among others, can shape microbial populations even within the same vineyard. Microbial communities described here showed significant differences according to geographical location, with sampling sites located within a 20 km radius, suggesting a similar effect of local environmental variables such as those observed for vineyards and winemaking43,44. Interestingly, differences in fungal and bacterial community compositions in vineyards have been described as much clearer in fungi than in bacteria, with leaf and berry fungal community dissimilarities between locations increasing with geographic distance, while bacterial community dissimilarity did not correlate with geographic distance43. Bacterial communities associated with E. gunnii showed only minor differences according to geographical location or ecological niche while fungal populations differed significantly even at genus level, indicating the important role of the environment on shaping fungal communities44,45.
Numerous environmental and anthropogenic factors have been shown to influence the composition of microbial communities found in soil or associated with plant material. These include season for communities in subtropical low mountain forest soils46 or vintage for grape vines26,47; physical characteristics of the soil, including carbon and nitrogen content48; vine vegetative cycle42; topological characteristics of the land49; application of chemical fungicides and fertilizers47,50,51; agricultural management practices in crop plantations and vineyards52,53,54; land-use change55; and erosion in farmlands56. Although none of these factors were investigated here, it is likely that several parameters, including climatic conditions, rainfall and soil nutrient availability, or combinations of these factors, shape the microbial populations associated with E. gunnii. For example, no differences were found for fungal genera according to sampling date when assessing all samples, however significant changes in composition were found when individual geographical locations were compared. This suggests that for E. gunnii, climatic conditions (spring vs summer) only influence fungal communities when combined with other factors.
Considering multiple grape varieties and numerous geographical locations, the main bacterial phyla found associated with vineyards and winemaking are Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes26,42. The same four phyla were also found as the most abundant in cider gum samples. While the genera Klebsiella and Pseudomonas from the phylum Proteobacteria are the most abundant in grape must42, the main bacterial genera found in pulque fermentation include Lactobacillus and Leuconostoc from the Firmicutes phylum, and the Proteobacteria, Zymomonas, Acetobacter and Gluconobacter19. Different lactic acid bacteria (LAB) species have also been shown to dominate the fermentation of tej honey wine, including Lactobacillus, Streptococcus, Leuconostoc, and Pediococcus16; the fermentation of makgeolli, including Enterococcus, Lactobacillus, Leuconostoc, Pediococcus, Weissella and Lactococcus21; and the fermentation of chicha, including Lactobacillus, Bacillus, Leuconostoc and Enterococcus18. LAB species associated with E. gunnii represented < 3% of the total bacterial population, with Leuconostoc as the main genus, whereas the acetic acid bacteria (AAB) Gluconobacter and Acetobacter, were the most abundant bacterial genera. The higher proportion of AAB in cider gum samples highlights the distinctive bacterial communities associated with this Tasmanian drink compared to the bacterial populations involved in the production of other fermented beverages. This enrichment in AAB could be the result of the aerobic conditions in which way-a-linah fermentation occurs.
Although fungal communities associated with E. gunnii and those related to winemaking, including fungal populations found in vineyard soil, on vine bark and grape berries, and during wine fermentation, showed similar diversity, their taxonomic composition and relative abundances differed considerably, particularly for lower taxonomic levels. The most abundant fungal phyla, Ascomycota and Basidiomycota, as well as the most abundant fungal classes, Saccharomycetes, Dothideomycetes and Leotiomycetes were similar for communities associated with winemaking and way-a-linah26,27,34,42. However, the most abundant taxa for order, family and genus diverged for both communities. Interestingly, the main classes found in other Eucalyptus species differed from those associated with E. gunnii, with the most abundant classes found in leaves, twigs and trunks of E. grandis including Dothideomycetes and Sordariomycetes while Saccharomycetes represented only 0.05% of the population57. At family level, the main taxa associated with E. grandis included Mycosphaerellaceae, Botryosphaeriaceae and Teratosphaeriaceae57, whereas those related to E. gunnii were Saccharomycetaceae, Pichiaceae and Saccharomycodaceae. In cider gum Teratosphaeriaceae and Mycosphaerellaceae accounted for less than 3% of the total fungal population, whereas Botryosphaeriaceae was not found. This suggests that at higher taxa levels, fungal populations associated with E. gunnii resembled those found in vineyards and winemaking rather than those of other Eucalyptus species.
The genus Saccharomyces, which is the most abundant in winemaking27,34,42, was practically absent from cider gum samples, in which S. cerevisiae and S. uvarum were found at very low abundance (< 0.03%) and only in a small proportion of samples. Saccharomyces yeasts have a crucial competitive advantage during sugar fermentation due to their ability to grow fast, produce and consume ethanol and their tolerance for various environmental stresses58. The fact that Saccharomyces was not abundant, particularly in fermenting sap samples, suggest that this genus is hardly present in the locations sampled and/or that the conditions in cider gum sap, including the presence of other microorganisms, low alcohol concentration, high organic acid concentration, among others, may make the competition equal to other species.
We found numerous bacterial (close to 30%) and fungal (under 50%) OTUs, which did not match any entries in 16S rRNA and ITS databases, or matched ‘uncultured’ microorganisms providing no taxonomic information for those sequences. This has also been described in other environmental samples, in fact the highest abundances of bacterial OTUs belong to phylogenetically novel uncultured groups in seawater, freshwater, terrestrial subsurface, soil, hypersaline environments, marine sediment, hot springs, hydrothermal vents, nonhuman hosts, snow, and bioreactors59. Thus, not only most bacterial, but also most archaeal taxa remain uncultured and therefore uncharacterised60. Similarly, several studies have shown a significant abundance of unclassified or unassigned fungal OTUs33,50,54,57 suggesting the existence of many novel fungal taxa. Careful examination of culture conditions and meticulous formulation of laboratory culture media have enabled researchers to bring the uncultured into culture and provide essential information about novel species61. Ideally, applying this approach to cider gum samples would make possible the isolation and characterisation of novel bacterial and fungal species.
In conclusion, we have found numerous microbial species associated with the spontaneous fermentation of sap from E. gunnii. While bacterial communities showed small differences between geographical locations, ecological niches or sampling dates, fungal populations showed significant differences. Additionally, fungal communities differed greatly to those associated with winemaking. Phylotyping revealed several bacterial and fungal sequences, which did not match known microbial genomes suggesting potential novel microbial taxa. These findings highlight the vast microbial diversity associated with native Australian plants and beverages.
Materials and methods
Sampling
Bark, sap and soil samples were collected from the cider gum Eucalyptus gunnii on the Tasmanian Central Plateau in Australia. Samples were obtained from three different geographical locations, Trawtha Makuminya (41°59′55.6″S 146°22′58.8″E), Skullbone Plains (42°02′29.2″S 146°19′13.6″E) and Five Rivers—Serpentine (42°05′13.5″S 146°34′07.3″E) at two different dates in November 2016 and January 2017 (Fig. 1).
Chemical analysis
Chemical analysis was performed only on sap samples. Preliminary analysis and compound identification were performed by gas-chromatography mass spectrometry (GC/MS) on an Agilent 7890 gas chromatograph equipped with Gerstel MPS2 multi-purpose sampler and coupled to an Agilent 5975C VL mass selective detector. Instrument control was performed with Agilent G1701A Revision E.02.00 ChemStation software. The gas chromatograph was fitted with a VF-5ms column (30 m × 250 μm × 0.25 μm) fitted with a 10 m guard column and helium (ultra high purity) was used as the carrier gas in constant flow mode at 1 mL/min. Samples were derivatised by adding an aliquot of 22 μL of N-methoxyamine hydrochloride (Sigma-Aldrich) in pyridine (Sigma-Aldrich) and incubating at 37 °C with agitation for 2 h. Following incubation, 4 μL of alkane standard mixture (Cat. No. 68281, Sigma-Aldrich) and 22 μL of N,O-Bis(trimethylsilyl)trifluoroacetamide and trimethylchlorosilane (BSTFA + TMCS, 99:1, Sigma-Aldrich) were added, and incubated at 37 °C with agitation for 60 min. Derivatised samples were incubated at room temperature for 1 h before injection. A volume of 1 μL of derivatized sample was injected into the inlet which was set at 250 °C in pulsed splitless mode. The oven program started at 35 °C for 2 min, then ramped at 10 °C/min to 300 °C for 16 min. Total run time was 46 min. The mass spectrometer quadrupole was set at 150 °C, the source was set at 250 °C while the transfer line was held at 280 °C. Positive ion electron impact spectra at 70 eV were recorded in scan mode with an m/z range of 50–600 and a solvent delay of 9.6 min. After confirming the identity of maltose by GC/MS, quantification of sugars, including glucose, fructose and maltose; organic acids, including gluconic, pyruvic, succinic and acetic acids; glycerol and ethanol was performed by high-performance liquid chromatography (HPLC) using a BioRad HPX87H column at 65 °C, H2SO4 5 mM as mobile phase at 0.5 mL/min, as described previously62.
DNA extraction
Bark samples were incubated at 4 °C for 24 h with 1 mL of PBS (NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, KH2PO4 1.8 mM pH 7.4) before the liquid was used for DNA extraction. Total DNA was isolated from bark and sap samples using the DNeasy PowerFood Microbial kit (Qiagen, Hilden, Germany) as described by the manufacturer. DNA from soil samples was extracted using the DNeasy PowerSoil kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After isolation, DNA was quantified with the Qubit dsDNA HS kit assay (Thermo Fisher Scientific, Massachusetts, USA).
Determination of microbial populations
Microbial populations were assessed using amplicon sequencing. Briefly, 1.0 ng of total DNA from each sample was subjected to a two-step PCR process that amplifies the V3-V4 region of the bacterial 16S rRNA63 or the ITS2 locus from the fungal ribosomal internal transcribed spacer (ITS) region64 while adding both custom in-line barcodes and sequences necessary for Illumina sequencing (including compatible Illumina dual-indexes) according to the process outlined previously65. Briefly, following sequencing, raw reads were quality and adaptor trimmed (Trimmomatic66; Cutadapt67), with paired-end reads overlapped68 to form a single contiguous synthetic read. Reads were then assigned to samples and timepoints using a combination of both the in-line and Illumina barcodes using custom Python scripts. Operational taxonomic units (OTUs) were clustered de novo using Swarm v2.069 and taxonomies assigned using the “assign_taxonomy.py” functionality of QIIME70. Fungal taxonomy was assigned against the QIIME UNITE fungal ITS database (ver7 dynamic 20.11.2016), while bacterial taxonomy used the SILVA 16S database (version 132 QIIME release) as a reference. Sequences and abundances for all bacterial and fungal OTUs identified in this study are listed in Table S1 and Table S3, respectively.
Data and statistical analyses
Data analysis and graphical representation were performed using the R packages phyloseq71, microbiome32, vegan72, microbiomeViz73, ggmap74, ggpubr75 and ggplot276 in R version 3.6.277. OTUs showing less than 0.001% abundance were not considered for analysis. Kruskal tests were used to evaluate statistical differences for relative abundances between species.
References
Legras, J. L., Merdinoglu, D., Cornuet, J. M. & Karst, F. Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Mol. Ecol. 16, 2091–2102 (2007).
McGovern, P. E. et al. Fermented beverages of pre- and proto-historic China. Proc. Natl. Acad. Sci. U.S.A. 101, 17593–17598. https://doi.org/10.1073/pnas.0407921102 (2004).
Cavalieri, D., McGovern, P., Hartl, D., Mortimer, R. & Polsinelli, M. Evidence for S. cerevisiae fermentation in ancient wine. J. Mol. Evol. 57, S226–S232 (2003).
McGovern, P., Hartung, U., Badler, V., Glusker, D. & Exner, L. The beginnings of winemaking and viniculture in the ancient Near East and Egypt. Expedition 39, 3–21 (1997).
Dudley, R. Ethanol, fruit ripening, and the historical origins of human alcoholism in primate frugivory. Integr. Comp. Biol. 44, 315–323 (2004).
Dudley, R. Fermenting fruit and the historical ecology of ethanol ingestion: is alcoholism in modern humans an evolutionary hangover?. Addiction 97, 381–388. https://doi.org/10.1046/j.1360-0443.2002.00002.x (2002).
Carrigan, M. A. et al. Hominids adapted to metabolize ethanol long before human-directed fermentation. Proc. Natl. Acad. Sci. U.S.A. 112, 458–463. https://doi.org/10.1073/pnas.1404167111 (2015).
Alba-Lois, L. & Segal-Kischinevzky, C. Yeast fermentation and the making of beer and wine https://www.nature.com/scitable/topicpage/yeast-fermentation-and-the-making-of-beer-14372813 (2010).
Malacarne, M., Martuzzi, F., Summer, A. & Mariani, P. Protein and fat composition of mare’s milk: some nutritional remarks with reference to human and cow’s milk. Int. Dairy J. 12, 869–877. https://doi.org/10.1016/S0958-6946(02)00120-6 (2002).
Brady, M. First Taste. How Indigenous Australians Learned About Grog (Alcohol Education and Rehabilitation Foundation Ltd, Canberra, 2008).
Brady, M. & McGrath, V. Making Tuba in the Torres Strait islands: the cultural diffusion and geographic mobility of an alcoholic drink. J. Pac. Hist. 45, 315–330. https://doi.org/10.1080/00223344.2010.530811 (2010).
Varela, C. The impact of non-Saccharomyces yeasts in the production of alcoholic beverages. Appl. Microbiol. Biotechnol. 100, 9861–9874. https://doi.org/10.1007/s00253-016-7941-6 (2016).
Jolly, N. P., Varela, C. & Pretorius, I. S. Not your ordinary yeast: non-Saccharomyces yeasts in wine production uncovered. FEMS Yeast Res. 14, 215–237. https://doi.org/10.1111/1567-1364.12111 (2014).
Steinkraus, K. H. Handbook of Indigenous Fermented Foods, Second Edition, Revised and Expanded (Marcel Dekker, New York, 1995).
Tamang, J. P., Watanabe, K. & Holzapfel, W. H. Review: diversity of microorganisms in global fermented foods and beverages. Front. Microbiol. https://doi.org/10.3389/fmicb.2016.00377 (2016).
Bahiru, B., Mehari, T. & Ashenafi, M. Yeast and lactic acid flora of tej, an indigenous Ethiopian honey wine: variations within and between production units. Food Microbiol. 23, 277–282. https://doi.org/10.1016/j.fm.2005.05.007 (2006).
Vallejo, J. A. et al. Atypical yeasts identified as Saccharomyces cerevisiae by MALDI-TOF MS and gene sequencing are the main responsible of fermentation of chicha, a traditional beverage from Peru. Syst. Appl. Microbiol. 36, 560–564. https://doi.org/10.1016/j.syapm.2013.09.002 (2013).
Puerari, C., Magalhães-Guedes, K. T. & Schwan, R. F. Physicochemical and microbiological characterization of chicha, a rice-based fermented beverage produced by Umutina Brazilian Amerindians. Food Microbiol. 46, 210–217. https://doi.org/10.1016/j.fm.2014.08.009 (2015).
Escalante, A. et al. Characterization of bacterial diversity in Pulque, a traditional Mexican alcoholic fermented beverage, as determined by 16S rDNA analysis. FEMS Microbiol. Lett. 235, 273–279. https://doi.org/10.1016/j.femsle.2004.04.045 (2004).
Lappe-Oliveras, P. et al. Yeasts associated with the production of Mexican alcoholic nondistilled and distilled Agave beverages. FEMS Yeast Res. 8, 1037–1052. https://doi.org/10.1111/j.1567-1364.2008.00430.x (2008).
Jung, M. J., Nam, Y. D., Roh, S. W. & Bae, J. W. Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol. 30, 112–123. https://doi.org/10.1016/j.fm.2011.09.008 (2012).
Greppi, A. et al. Determination of yeast diversity in ogi, mawe, gowe and tchoukoutou by using culture-dependent and -independent methods. Int. J. Food Microbiol. 165, 84–88. https://doi.org/10.1016/j.ijfoodmicro.2013.05.005 (2013).
Spitaels, F. et al. The microbial diversity of traditional spontaneously fermented lambic beer. PLoS ONE https://doi.org/10.1371/journal.pone.0095384 (2014).
Tapsoba, F., Legras, J. L., Savadogo, A., Dequin, S. & Traore, A. S. Diversity of Saccharomyces cerevisiae strains isolated from Borassus akeassii palm wines from Burkina Faso in comparison to other African beverages. Int. J. Food Microbiol. 211, 128–133. https://doi.org/10.1016/j.ijfoodmicro.2015.07.010 (2015).
Bokulich, N. A., Bamforth, C. W. & Mills, D. A. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS ONE 7, e35507. https://doi.org/10.1371/journal.pone.0035507 (2012).
Bokulich, N. A., Thorngate, J. H., Richardson, P. M. & Mills, D. A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. U.S.A. 111, E139–E148. https://doi.org/10.1073/pnas.1317377110 (2014).
Siren, K. et al. Taxonomic and functional characterization of the microbial community during spontaneous in vitro fermentation of Riesling must. Front. Microbiol. https://doi.org/10.3389/fmicb.2019.00697 (2019).
Morgan, H. H., du Toit, M. & Setati, M. E. The grapevine and wine microbiome: insights from high-throughput amplicon sequencing. Front. Microbiol. https://doi.org/10.3389/fmicb.2017.00820 (2017).
Williams, K. J. & Potts, B. M. The natural distribution of Eucalyptus species in Tasmania. Tasforests 8, 39–165 (1996).
Calder, J. A. & Kirkpatrick, J. B. Climate change and other factors influencing the decline of the Tasmanian cider gum (Eucalyptus gunnii). Aust. J. Bot. 56, 684–692. https://doi.org/10.1071/BT08105 (2008).
Sanger, J. C., Davidson, N. J., O’Grady, A. P. & Close, D. C. Are the patterns of regeneration in the endangered Eucalyptus gunnii ssp. divaricata shifting in response to climate?. Austral. Ecol. 36, 612–620. https://doi.org/10.1111/j.1442-9993.2010.02194.x (2011).
Lahti, L. & Shetty, S. Tools for microbiome analysis in R. Version 1.9.1 https://microbiome.github.com/microbiome (2017).
Morrison-Whittle, P. & Goddard, M. R. Quantifying the relative roles of selective and neutral processes in defining eukaryotic microbial communities. ISME J. 9, 2003–2011. https://doi.org/10.1038/ismej.2015.18 (2015).
Morrison-Whittle, P. & Goddard, M. R. From vineyard to winery: a source map of microbial diversity driving wine fermentation. Environ. Microbiol. 20, 75–84. https://doi.org/10.1111/1462-2920.13960 (2018).
Brooker, M. I. H. A Key to Eucalypts in Britain and Ireland. (Forestry Commission Booklet 50: The Stationery Office, 1983).
Forrest, M. & Moore, T. Eucalyptus gunnii: a possible source of bioenergy?. Biomass Bioenerg. 32, 978–980. https://doi.org/10.1016/j.biombioe.2008.01.010 (2008).
Guimarães, R. et al. Aromatic plants as a source of important phytochemicals: vitamins, sugars and fatty acids in Cistus ladanifer, Cupressus lusitanica and Eucalyptus gunnii leaves. Ind. Crop Prod. 30, 427–430. https://doi.org/10.1016/j.indcrop.2009.08.002 (2009).
Bugarin, D. et al. Essential oil of Eucalyptus gunnii hook. As a novel source of antioxidant, antimutagenic and antibacterial agents. Molecules 19, 19007–19020. https://doi.org/10.3390/molecules191119007 (2014).
Leborgne, N. et al. Introduction of specific carbohydrates into Eucalyptus gunnii cells increases their freezing tolerance. Eur. J. Biochem. 229, 710–717. https://doi.org/10.1111/j.1432-1033.1995.0710j.x (1995).
Stuckel, J. G. & Low, N. H. The chemical composition of 80 pure maple syrup samples produced in North America. Food Res. Int. 29, 373–379. https://doi.org/10.1016/0963-9969(96)00000-2 (1996).
Taylor, M. W., Tsai, P., Anfang, N., Ross, H. A. & Goddard, M. R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 16, 2848–2858 (2014).
Pinto, C. et al. Wine fermentation microbiome: a landscape from different Portuguese wine appellations. Front. Microbiol. https://doi.org/10.3389/fmicb.2015.00905 (2015).
Miura, T., Sanchez, R., Castaneda, L. E., Godoy, K. & Barbosa, O. Is microbial terroir related to geographic distance between vineyards?. Environ. Microbiol. Rep. 9, 742–749. https://doi.org/10.1111/1758-2229.12589 (2017).
Knight, S. J., Karon, O. & Goddard, M. R. Small scale fungal community differentiation in a vineyard system. Food Microbiol. https://doi.org/10.1016/j.fm.2019.103358 (2019).
Tedersoo, L. et al. Global diversity and geography of soil fungi. Science 346, 1256688. https://doi.org/10.1126/science.1256688 (2014).
Lin, Y. T., Whitman, W. B., Coleman, D. C. & Chiu, C. Y. Effects of reforestation on the structure and diversity of bacterial communities in subtropical low mountain forest soils. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.01968 (2018).
Grangeteau, C. et al. Wine microbiology is driven by vineyard and winery anthropogenic factors. Microb. Biotechnol. 10, 354–370. https://doi.org/10.1111/1751-7915.12428 (2017).
Burns, K. N. et al. Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: differentiation by geographic features. Soil Biol. Biochem. 91, 232–247. https://doi.org/10.1016/j.soilbio.2015.09.002 (2015).
Portillo, M. D. C., Franquès, J., Araque, I., Reguant, C. & Bordons, A. Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food Microbiol. 219, 56–63. https://doi.org/10.1016/j.ijfoodmicro.2015.12.002 (2016).
Castaneda, L. E. & Barbosa, O. Metagenomic analysis exploring taxonomic and functional diversity of soil microbial communities in Chilean vineyards and surrounding native forests. PeerJ 5, e3098. https://doi.org/10.7717/peerj.3098 (2017).
Setati, M. E., Jacobson, D. & Bauer, F. F. Sequence-based analysis of the Vitis vinifera L. cv Cabernet Sauvignon grape must Mycobiome in three South African vineyards employing distinct agronomic systems. Front. Microbiol. https://doi.org/10.3389/fmicb.2015.01358 (2015).
Miura, T. et al. Shifts in the composition and potential functions of soil microbial communities responding to a no-tillage practice and bagasse mulching on a sugarcane plantation. Biol. Fertil. Soils 52, 307–322. https://doi.org/10.1007/s00374-015-1077-1 (2016).
Miura, T., Sanchez, R., Castaneda, L. E., Godoy, K. & Barbosa, O. Shared and unique features of bacterial communities in native forest and vineyard phyllosphere. Ecol. Evol. 9, 3295–3305. https://doi.org/10.1002/ece3.4949 (2019).
Hendgen, M. et al. Effects of different management regimes on microbial biodiversity in vineyard soils. Sci. Rep. https://doi.org/10.1038/s41598-018-27743-0 (2018).
Montecchia, M. S. et al. Pyrosequencing reveals changes in soil bacterial communities after conversion of Yungas forests to agriculture. PLoS ONE 10, 18. https://doi.org/10.1371/journal.pone.0119426 (2015).
Gleeson, D., Mathes, F., Farrell, M. & Leopold, M. Environmental drivers of soil microbial community structure and function at the Avon River Critical Zone Observatory. Sci. Total Environ. 571, 1407–1418. https://doi.org/10.1016/j.scitotenv.2016.05.185 (2016).
Kemler, M. et al. Ion Torrent PGM as tool for fungal community analysis: a case study of Endophytes in Eucalyptus grandis reveals high taxonomic diversity. PLoS ONE https://doi.org/10.1371/journal.pone.0081718 (2013).
Piškur, J., Rozpędowska, E., Polakova, S., Merico, A. & Compagno, C. How did Saccharomyces evolve to become a good brewer?. Trends Genet. 22, 183–186. https://doi.org/10.1016/j.tig.2006.02.002 (2006).
Lloyd, K. G., Steen, A. D., Ladau, J., Yin, J. Q. & Crosby, L. Phylogenetically novel uncultured microbial cells dominate Earth microbiomes. Msystems https://doi.org/10.1128/mSystems.00055-18 (2018).
Steen, A. D. et al. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 13, 3126–3130. https://doi.org/10.1038/s41396-019-0484-y (2019).
Thrash, J. C. Culturing the uncultured: Risk versus reward. Msystems https://doi.org/10.1128/mSystems.00130-19 (2019).
Varela, C., Pizarro, F. & Agosin, E. Biomass content governs fermentation rate in nitrogen-deficient wine musts. Appl. Environ. Microbiol. 70, 3392–3400. https://doi.org/10.1128/Aem.70.6.3392-3400.2004 (2004).
Parker, M. et al. Factors contributing to interindividual variation in retronasal odor perception from aroma glycosides: The tole of odorant sensory detection threshold, oral microbiota, and hydrolysis in saliva. J. Agric. Food Chem. https://doi.org/10.1021/acs.jafc.9b05450 (2019).
Bokulich, N. A. & Mills, D. A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79, 2519–2526. https://doi.org/10.1128/AEM.03870-12 (2013).
Sternes, P. R., Lee, D., Kutyna, D. R. & Borneman, A. R. A combined meta-barcoding and shotgun metagenomic analysis of spontaneous wine fermentation. bioRxiv https://doi.org/10.1101/098061 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. https://doi.org/10.14806/ej.17.1.200 (2011).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. https://doi.org/10.1093/bioinformatics/btr507 (2011).
Mahe, F., Rognes, T., Quince, C., de Vargas, C. & Dunthorn, M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2, e593. https://doi.org/10.7717/peerj.593 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. https://doi.org/10.1038/nmeth.f.303 (2010).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.5.4 https://CRAN.R-project.org/package=vegan (2019).
Li, C., Yu, G. & Zhu, C. microbiomeViz—an R package for visualizing microbiome data https://github.com/lch14forever/microbiomeViz (2018).
Kahle, D. & Wickham, H. ggmap: spatial visualization with ggplot2. R J. 5, 144–161 (2013).
Kassambara, A. ggpubr: 'ggplot2' based publication eady plots. R package version 0.2 https://CRAN.R-project.org/package=ggpubr (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (SpringerVerlag, New York, 2009).
Team, R. C. R: a language and environment for statistical computing https://www.R-project.org/ (2017).
Acknowledgements
The authors would like to thank Steve Cronin, Andry Sculthorpe and the Tasmanian Aboriginal Council for allowing access to Trawtha Makuminya, and Matt Taylor and The Tasmanian Land Conservancy for allowing access to Skullbone Plains and Five Rivers—Serpentine. Special thanks to Michael O’Brian and Associate Professor Maggie Brady for their inputs on the planning stages of this work and to Associate Professor Daniel Durall and Jemma McGilton for their help during sampling. The AWRI and the University of Adelaide are members of the Wine Innovation Cluster in Adelaide. The AWRI is supported by Australia’s grape growers and winemakers through their investment body Wine Australia with matching funds from the Australian Government. University of Adelaide researchers gratefully acknowledge financial support from the offices of the Executive Dean of the Faculty of Science, The Deputy Vice Chancellor (Research) as well as the Yipti Foundation.
Author information
Authors and Affiliations
Contributions
C.V. collected samples, analysed samples and results, drafted the manuscript participated in the conception of the work. J.S. collected and processed samples. K.C. analysed and processed samples. A.B. analysed results and participated in the conception of the work. V.J. collected samples and participated in the conception of the work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Varela, C., Sundstrom, J., Cuijvers, K. et al. Discovering the indigenous microbial communities associated with the natural fermentation of sap from the cider gum Eucalyptus gunnii. Sci Rep 10, 14716 (2020). https://doi.org/10.1038/s41598-020-71663-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-71663-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
