
Abstract
Anaplastic lymphoma kinase (Alk) is a receptor tyrosine kinase of the insulin receptor super-family that functions as oncogenic driver in a range of human cancers such as neuroblastoma. In order to investigate mechanisms underlying Alk oncogenic signaling, we conducted a genetic suppressor screen in Drosophila melanogaster. Our screen identified multiple loci important for Alk signaling, including members of Ras/Raf/ERK-, Pi3K-, and STAT-pathways as well as tailless (tll) and foxo whose orthologues NR2E1/TLX and FOXO3 are transcription factors implicated in human neuroblastoma. Many of the identified suppressors were also able to modulate signaling output from activated oncogenic variants of human ALK, suggesting that our screen identified targets likely relevant in a wide range of contexts. Interestingly, two misexpression alleles of wallenda (wnd, encoding a leucine zipper bearing kinase similar to human DLK and LZK) were among the strongest suppressors. We show that Alk expression leads to a growth advantage and induces cell death in surrounding cells. Our results suggest that Alk activity conveys a competitive advantage to cells, which can be reversed by over-expression of the JNK kinase kinase Wnd.
Similar content being viewed by others


Introduction
The receptor tyrosine kinase anaplastic lymphoma kinase (ALK) was initially described as oncogenic driver in anaplastic large-cell lymphoma and is implicated in a wide range of cancer types including non-small cell lung cancer and neuroblastoma1. During development, the vertebrate ALK receptor is activated by ALKAL (also known as FAM150 or Augmentor) family proteins while LDL-domain protein ligands activate Alk in invertebrates2,3,4,5,6,7. In contrast, cytosolic ALK fusion proteins, such as NPM1-ALK and EML4-ALK, are constitutively active and common drivers of tumor progression in anaplastic large-cell lymphoma and non-small cell lung cancer1,8,9,10. These ALK fusion proteins are ligand-independent and often ectopically expressed in tissues that would normally not express ALK1. In addition to the activation of ALK by fusion events, the full length receptor can be activated by point mutations in its kinase domain which has been described in familial and sporadic neuroblastoma1,11,12,13,14,15,16. Clinical studies suggest that ALK activation drives tumor growth and indeed ALK-driven cancers respond well to initial therapy with ALK tyrosine kinase inhibitors17. However, ALK-driven tumors frequently become resistant to ALK inhibitor treatment, underscoring a need to better understand signaling events that occur during aberrant ALK activity.
To shed light on these signaling events, we and others have used various model organisms, such as Drosophila melanogaster, to complement in vitro cell culture studies. In Drosophila, a single Alk orthologue is required to specify the muscle founder cells of the larval gut musculature2,3,18. In addition, Alk signaling plays a complex role in many aspects of neuronal function19,20,21,22,23,24,25. Interestingly, ectopic expression of Alk in the developing fly eye results in a characteristic phenotype reflecting the activation state of the receptor26,27. Therefore, the fly eye has become a valuable model system to analyze putative activating human ALK mutations identified in neuroblastoma patients28,29,30,31. The sensitivity of the fly eye model to both Drosophila and human ALK activity suggests that a genetic modifier screen in the fly holds the potential to reveal important downstream factors of Alk signaling in vivo. We crossed flies ectopically expressing Alk under control of the sevEP-Gal4 driver to flies carrying genetically mapped deficiencies from the DrosDel collection32 and scored progeny for suppression or modification of the Alk-induced eye phenotype. Among the strongest suppressors, we identified gain of function alleles of the LZK, MAP3K13 (leucine zipper bearing kinase, mitogen-activated protein kinase kinase kinase 13) and DLK, MAP3K12 (dual leucine zipper kinase, mitogen-activated protein kinase kinase kinase 12) homolog Wallenda (wnd). Wnd has previously been shown to regulate JNK-mediated cell death in Drosophila33. JNK signaling in Drosophila tumorigenesis models can promote either cell-invasion or apoptosis dependent on context34,35. Investigating cell death in the context of Alk expression revealed increased cell death in Alk-negative neighboring cells, suggesting that ectopic Alk signaling leads to a competitive advantage that is further strengthened by the finding that Alk-expressing clones have a slight, but significant, growth advantage. This Alk-dependent increased competitiveness can be abrogated by elevated levels of JNK activation, through expression of Wnd or the JNKK Hemipterous (hep) in Alk expressing cells, suggesting that Wnd restrains Alk anti-apoptotic signaling output via modulation of JNK.
Results
Ectopic expression of Alk interferes with Drosophila eye development
We previously showed that the signaling activity of Drosophila and human ALK receptors can be assessed in the fly eye4,26,27,28,29,30,31,36. When Drosophila Alk was ectopically expressed in the developing eye under control of the sevEP-Gal4 driver, we observed severe loss of ommatidia and patches of necrotic tissue in the anterior part of the adult fly eye as well as a “rough-eye–morphology” in the posterior part (Fig. 1A–C). This phenotype was suppressed when co-expressing an RNAi construct (UAS-AlkJF02688) targeting Alk (Fig. 1D). In order to characterize the sevEP-Gal4 driver further, we performed lineage analysis using the G-TRACE system37, which revealed driver activity posterior to the morphogenetic furrow in third instar larval eye discs (Fig. 1E–E’’). The presence of sevEP-Gal4 > UAS-RFP-expressing and EGFP-positive, lineage derived cells suggested that the sevEP reporter resembles the transient expression pattern of sevenless in early photoreceptors and remains active only in a subset of retinal cells38,39,40. This observation was more apparent at later pupal stages when G-TRACE analysis indicated continued RFP expression in the R7 photoreceptor and the cone cells that are recruited later to the ommatidial cluster while other cells of the ommatidium solely expressed EGFP (Fig. 1F). To further analyze the effects of ectopic Alk expression on eye development, we dissected eye discs 50 h after pupa formation (apf) (Fig. 1G–J’). Activity of the sevEP-Gal4 driver revealed by UAS-lacZ (Fig. 1G,G’’’) resembled the previously described pattern of sevenless in R1, R6, R7 photoreceptors and cone cells38,39,40. In control eye discs, DECad staining indicated a regular arrangement of ommatidia upon β-Galactosidase expression (Fig. 1G’’,G’’’). Cut (CT), a lineage marker for cone cells, was expressed in four cells of each ommatidium (Fig. 1G’,G’’’; quantified in Fig. S1A,B). The neuronal marker Elav was expressed in all photoreceptor cells (Fig. 1I’) and Pros was expressed only in R7 cells (Fig. 1I,I’; quantified in Fig. S1C,D). In sevEP-Gal4 > UAS-Alk/ + eye discs, the expression pattern of these markers was highly irregular (Fig. 1H–H’’’,J–J’). A high number of Alk-expressing cells in the anterior region (Fig. 1H,H’’’) correlated with a high degree of ommatidia disorganization as revealed by DECad staining (Fig. 1H’’,H’’’) while, in the posterior region, ommatidia could be identified but were partially fused and irregular (Fig. 1H’’,H’’’). Moreover, the numbers of CT- and Pros-positive cells were significantly higher in sevEP-Gal4 > UAS-Alk/ + eye discs than in controls (Fig. 1G’,H’,I,J; quantified in Fig. S1A–D). This increase was most pronounced in anterior regions of the eye discs (Fig. 1H’,J; quantified in Fig. S1A–D). Taken together, this analysis suggests that ectopic expression of Alk leads to aberrant pattern of expression of lineage markers and interferes with ommatidia differentiation in the developing eye disc.

Ectopic expression of UAS-Alk/ + with the sevEP-Gal4 driver interferes with normal eye development. (A–D) Eyes of adult female flies with the indicated genotypes are shown. n > 200, 100% penetrant (A) “Wild-type” eye morphology of flies with either one copy of the sevEP-Gal4 driver insertion or (B) one copy of the UAS-Alk transgene. (C) Ommatidial disruption and necrotic scars in the anterior eye upon sevEP-Gal4/ + induced UAS-Alk/ + expression. The posterior half exhibits fused ommatidia and missing bristles. (D) Phenotypic rescue by co-expression of an Alk-specific RNAi. (E,F) G-TRACE analysis. Real-time sevEP-Gal4 expression is indicated by RFP (red) while continued expression in the lineage is indicated with GFP (green). (E–E’’) G-TRACE analysis in larval eye discs and in pupal eye discs (F). (G–J’) Immunofluorescence staining of eye discs from pupae (50 h apf) of the indicated genotypes. Antibody staining against Cut (CT) and Drosophila epithelial cadherin (DECad) reveals increased numbers of CT-positive cells and loss of ommatidia organization upon ectopic Alk expression (quantified in Fig. S1). (G,G’’’) βGalactosidase (βGal) reveals driver activity in a control eye disc, (H,H’’’) anti-ALK staining reveals transgene expression. Elav labels all photoreceptor cells and anti-Prospero (Pros) staining was used to identify R7 cells (I–J’). (J,J’) Upon ectopic expression of UAS-Alk the number of R7 photoreceptors is increased (quantified in Fig. S1). Scale bars are 100 µm in (A) and 10 µm in (E’’) and (F,G); anterior is left in all images.
A genetic modifier screen identifies targets of Alk-signaling in Drosophila
We conducted a genetic modifier screen (Fig. 2A) based on the rationale that dose-reduction of factors acting downstream of Alk would modify the observed eye phenotype. We first tested a number of potential suppressors in a validation screen (Fig. S2). These included loss-of-function (lof) alleles of jelly belly (jeb) encoding the Alk-activating ligand in Drosophila2,3,18,41, and RTK-associated signaling molecules such as C3G42,43, Son of sevenless (Sos)44, 14-3-3ε 45, Ras oncogene at 85D (Ras85D)46, kinase suppressor of Ras (ksr)47, rolled (rl) encoding for Drosophila ERK1,248, and pointed (pnt)49. When crossed into the sevEP-Gal4 > UAS-Alk/ + background, single copies of these alleles suppressed the eye phenotype to varying degrees (Fig. S2A-H), indicating that genome-wide screening of deficiency strains should allow us to identify genomic loci implicated in Alk signaling.

A deficiency screen identifies modifiers of ectopic Alk signaling. (A) Layout of the deficiency screen to identify modifiers of the sevEP > , UAS-Alk/ + induced eye phenotype. Scale bar = 100 µm. (B) Schematic representation of chromosome maps including coverage by the DrosDel deficiencies used in the screen (black lines). Suppressor regions appear green; deficiencies that modified the phenotype otherwise are depicted in red.
We screened 388 lines from the DrosDel collection of deficiencies50 covering about 60% of the annotated Drosophila genome and scored 58 modifying genomic regions covered by 133 deficiencies (Fig. 2B, green; Table S1). Ninety-eight deficiencies suppressed the Alk-induced eye phenotype while ten deficiencies led to other modifications such as enhanced necrosis in the eye (Fig. 2B, red; Table S1). We did not obtain adult progeny with 25 deficiencies, of which most were located on the X-chromosome. Further genomic mapping revealed 38 individual loci within these deficiencies that modified the Alk-induced eye phenotype (Fig. 3B, Table S1, see also discussion section). Based on this information, we further tested related factors leading to the identification of 12 additional loci that were not covered by the originally screened DrosDel deficiencies (Table S1). For several modifying deficiencies we were not able to map down individual loci that modified the Alk-induced eye phenotype (Table S1). PANTHER14.0-driven gene ontology (GO) enrichment analysis (https://geneontology.org/, Dec 2019)51 of all Alk modifiers (Fig. 3) revealed a high abundance of factors associated with optic placode development and signal transduction as well as cell dedifferentiation and negative regulation of apoptosis and autophagy (Fig. 3A, Table S1). Among these modifiers, we identified Ras/Raf/ERK network components such as MESR3, cnk and aveugle (ave, also referred to as hyphen) (Fig. 3B). Other identified signaling molecules included PI3K21B, STAT92E, the protein serine/threonine phosphatase alphabet (alph), split ends (spen), Star and moleskin (msk) (Fig. 3B). A number of transcription factors also modified the Alk-induced eye phenotype including the forkhead box, sub-group O transcription factor foxo, tailless (tll), eyeless (eye) and eyes absent (eya). We also identified lilliputian (lilli) and factors involved in regulation of RNA polymerase II-induced transcription (TFIIA-S, TFIIA-L, TFIIEα, TFIIEβ and the TFIID-associated factors Taf4, Taf6, TFIIS, Taf12L) (Fig. 3B).

Candidate genes revealed by the modifier screen. (A) Graphic representation of a PANTHER4.0-driven gene ontology (GO) enrichment analysis of Alk modifying loci. (B) String analysis of the Alk modifier network. For visualization purposes, the line thickness represents the strength of support for an interaction within the network. Suppressors are shown in green; other modifiers are depicted in red. Details of the individual alleles tested are found in Table S1B.
In some cases, more than one gene within a suppressing deficiency was found to suppress Alk-mediated signaling. For example, while mapping down the suppressor contained within deficiency ED6346 we noticed that mutations in both the tailless (tll) transcription factor52 and the warts/lats serine/threonine kinase53,54 suppressed Alk. We also noticed that certain balancer chromosomes modified the Alk-induced phenotype. In case of the CyO chromosome, we identified mutations in the Duox (Curly) gene as moderate suppressors (Table S1).
The activity of human oncogenic ALK is modulated by the identified modifiers
Aberrant ALK activation occurs in a range of cancers55. We therefore tested whether the modifiers found in our screen were also able to modulate signaling output from human ALK. To do this we activated human ALK (Fig. 4A) in three ways by ectopically expressing: (1) human ALK with the ALKAL1 ligand, which leads to high levels of ALK signaling activity4 (Fig. 4A,C), (2) a constitutively active ALK variant (ALK-F1174L) commonly observed in neuroblastoma patients12,13,14,15,16 (Fig. 4A,D), and (3) the chimeric NPM1::ALK oncogene found in anaplastic large cell lymphoma8 (Fig. 4A,E). When expressed in the fly eye under control of the sevEP-Gal4 driver, we observed a rough eye phenotype in all three scenarios but not the severe loss of ommatidia caused by ectopic expression of the Drosophila receptor (Fig. 4C–E, compare to Fig. 1C). Next, we tested the ability of loss of function alleles of 48 candidate genes to modify the human ALK-induced eye phenotype. Many alleles identified in the initial modifier screen also affected the activity of all human ALK variants tested (Fig. 4F), while dose-reduction of ave, cnk, exex, foxo, jeb, Mask, MESR3, Nhe2, Pi3K21B, Ret, spen, Taf12L, and Thor had no effect. Interestingly, P{XP}d00622 and Df(3L)Exel6135 suppressed the phenotype induced by both hALK + ALKAL1 and hALK-F1174L and since neither P{XP}d00622 nor Df(3L)Exel6135 had previously been connected to the function of a particular gene, we decided to analyze these genomic aberrations in more detail.

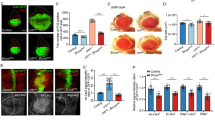
Human ALK variants are suppressed by mutations in the identified candidate genes. (A) Schematic representation of the screening approach, testing (1) hALK with the ALKAL1 ligand, which leads to high levels of ALK signaling activity, (2) a constitutively active hALK-F1174L observed in neuroblastoma and (3) the NPM1::ALK oncogene found in anaplastic large cell lymphoma. Single copies of loss-of-function (lof) mutations in the identified candidate genes were tested for phenotypic modification in flies ectopically expressing one of three oncogenic variants of human ALK (hALK) under sevEP-Gal4 control. (B–E) Eyes of adult female flies expressing either wild-type hALK (B), wild-type hALK activated by ALKAL1 (C), the hALKF1174L activating point mutation (D) or the chimeric hNPM1::hALK protein (E) with sevEP-Gal4. Scale bar = 100 µm. (F) Venn diagram representation of the results of the hALK modifier screens: 28 alleles suppressed (S) the rough-eye phenotype of all hALK variants (corresponding to the 14-3-3-ε, 14-3-3ζ, Asx, Cul2, Dl, Duox, E2f1, eya, eyg, ksr, lilli, Mapmodulin, msk, pll, pnt, Ras85D, rolled, Sos, Star, Stat92E, Taf6, TfIIA-L, TFIIA-S, TFIIEa, tll, and wts loci). One allele affecting two of the predicted five EcR isoforms enhanced (E) the eye phenotype. Modifiers that did not affect all hALK variants are highlighted in the diagram.
Misexpression alleles of the DLK/LZK homolog wallenda (wnd) suppress the Alk-induced eye phenotype
P{XP}d00622 and Df(3L)Exel6135 were identified while mapping the suppressor activity within two partially overlapping deficiencies (Df(3L)ED228 and Df(3L)ED229, Table S1) that also contained the strong suppressor candidate Taf656. P{XP}d00622 is a P-element57 inserted in the 5´UTR region of wallenda (wnd), the homolog of human LZK and DLK58 (Fig. 5A,C; control shown in 5B), while Df(3L)Exel6135 is a small deficiency with a proximal breakpoint mapped 48 bp upstream of wnd (Fig. 5A,D). Surprisingly, three previously characterized wnd loss of function alleles58 were unable to suppress the sevEP-Gal4 > UAS-Alk/ + eye phenotype (Figs. 5A,E, S3C,D), prompting us to reevaluate the P{XP}d00622 and Df(3L)Exel6135 suppressors. We noted that the P{XP}d00622 insertion contains UAS-sequences that allow Gal4-driven miss-expression of nearby genes57. UAS-sequences are also present on the Df(3L)Exel6135 chromosome that harbors a hybrid P{XP-U}Exel6135-element immediately upstream of the wnd transcription start site57. We therefore reasoned that the sevEP-Gal4 driver in our screen could have induced ectopic expression of Wnd in eye discs of sevEP-Gal4 > UAS-Alk/+ ; P{XP}d00622/+ and sevEP-Gal4 > UAS-Alk/+ ; P{XP-U}Exel6135/+ animals. To test the possibility that Wnd overexpression was suppressing the Alk-induced eye phenotype, we expressed an independent UAS-wnd transgene58 in sevEP-Gal4/+ control and sevEP-Gal4 > UAS-Alk/+ eye discs (Fig. 5F-I, Fig. S3E, F). While expression of Wnd alone (Figs. 5G, S4B) only had a subtle effect on eye morphology leading to occasionally reduced rhabdomere numbers per ommatidia (Fig. 5G, circle) and more frequent absence of single photoreceptor cells (Fig. 5G, arrows), it rescued the sevEP-Gal4 > UAS-Alk/ + phenotype (Figs. 5H,I, S4B). However, fused ommatidia (Fig. 5I, circle) and ommatidia with a reduced number of photoreceptor cells (Fig. 5I, arrow) could still be observed in the eye discs of these animals (Fig. 5I). Notably, UAS-wnd transgene expression was also able to rescue the rough-eye phenotype observed in all three activating conditions of human ALK, likely reflecting higher levels of expression of the UAS-wnd transgene compared to P{XP}d00622 and Df(3L)Exel6135 (Table S1). Wnd has been described as a cell death regulator in the JNK pathway, and over-expression of UAS-wnd with GMR-Gal4 results in cell death and a characteristic “small-eye” phenotype33. In line with these findings, we observed a “small-eye” phenotype when we combined GMR-Gal4 with either P{XP}d00622 or P{XP-U}Exel6135 (Fig. 5J,N, controls shown in Fig. S3). This “small-eye” phenotype was suppressed by wnd-specific RNAi supporting that both P{XP}d00622 or P{XP-U}Exel6135 result in ectopic expression of Wnd when combined with a Gal4-driver (Fig. 5K,O). In agreement with the proposed role of Wnd in the JNK-pathway, co-expression of either a dominant-negative variant of the Jun kinase Basket (UAS-bskDN) or the JNK-antagonist protein phosphatase Puckered (UAS-puc) suppressed the “small-eye” phenotype (Fig. 5L,M,P,Q). Taken together, these experiments suggest that P{XP}d00622 and P{XP-U}Exel6135 are inducible gain of function (gof) alleles of wnd and are henceforth referred to as wndd00622 and wndExel6135.

Inducible wnd gain-of-function alleles antagonize the effects of ectopic Alk expression. (A) Schematic representation of the wnd gene structure according to FlyBase (version FB2019_05). Black boxes represent protein-coding exons, grey boxes UTRs; lines indicate intronic regions of the four predicted wnd-transcripts. The positions of characterized point mutations and P-element insertions are highlighted. P{XP}d00622 and P{XP-U}Exel6135 (wndd00622 and wndExel6135) contain UAS-elements (direction indicated by arrows) and are inserted in or immediately upstream of the wnd 5′UTR. P{XP-U}Exel6135 is a hybrid P-element flanking the chromosomal deficiency Df(3L)Exel6135. Scale bar = 1 kb. (B–E, J–Q) Eyes of adult female flies with the indicated genotypes are shown. n > 100 animals, 100% penetrance. Scale bar is 100 µm. (F–I) To visualize ommatidia patterns in adult female Drosophila eyes, eye sections were stained with toluidine blue. (F,G) sevEP-Gal4/ + and sevEP-Gal4 > Wnd/ + controls. (H) Visible necrotic scars and absence of ommatidia in the anterior part of an eye upon sevEP-Gal4/ + induced UAS-Alk/ + expression. (I) Overexpression of Wnd/ + rescues ommatidia patterns in sevEP > UAS-Alk flies (H). Scale bar = 20 µm.
JNK pathway activation suppresses the effect of ectopic Alk signaling
We next investigated the role of JNK signaling in our sevEP-Gal4 > UAS-Alk model. First, we tested whether Wnd kinase activity was required to suppress the sevEP-Gal4 > UAS-Alk phenotype. Expression of a kinase-dead Wnd variant (UAS-wndK185A)58 did not rescue the rough eye phenotype, indicating that indeed Wnd kinase activity is important for the suppression (Figs. 6C,D,S4). We then addressed the effect of modulating individual components of the JNK signaling cascade on the sevEP-Gal4 > UAS-Alk/ + eye phenotype (Fig. 6A,B). While co-expression of the JNKKK Tak1 had a weak rescuing effect, expression of a constitutively activated variant of the JNKK Hemipterous (hep; UAS-hepCA) strongly suppressed the sevEP-Gal4 > UAS-Alk/ + eye phenotype (Figs. 6E,F, S4), suggesting that JNK activity is required for suppression. In agreement with this, blocking JNK signaling by expressing either dominant negative basket (UAS-bskDN), Jura (UAS-JraDN), or kayak (UAS-kayDN) in sevEP-Gal4 expressing cells failed to suppress the Alk-driven eye phenotype (Fig. 6G,I,J; controls in Fig. S4), as did expression of puckered (UAS-puc) (Fig. 6H), a negative regulator of JNK.

Ectopic JNK pathway activation rescues the sevEP-Gal4 > UAS-Alk eye phenotype. (A) Schematic representation of the JNK signaling pathway and the suggested role for Alk signaling in repressing Wnd-mediated apoptosis. (B–J) Eyes of adult female flies ectopically expressing UAS-Alk and JNK pathway components as indicated under the control of the sevEP-Gal4 driver. n > 50, 100% penetrance. Scale bar = 100 µm.
Alk expression induces non-cell autonomous apoptosis
Our previous experiments indicated that accumulation of Alk expressing cells in the anterior part of the fly eye correlated with the loss of ommatidia organization (Fig. 1). Therefore, it could be possible that Alk-negative neighboring cells play an important role in restricting the expansion of Alk-positive cells, eventually suppressing the phenotype in the posterior region of the eye. Supporting this assumption, broad expression of the cell death inhibitors Diap1 and p35 under direct control of the GMR promoter but not under sevEP-Gal4 control, strongly suppressed the Alk-induced eye phenotype (Fig. 7A–E, controls in Fig. S5). Moreover, reducing the dose of hid, grim and rpr using the deficiency Df(3L)H99 resulted in strong suppression of the Alk-induced eye phenotype (Fig. 7F), further suggesting that protecting Alk-negative neighbors from cell death enhances their ability to resist the Alk expressing cells.

Ectopic wnd expression attenuates Alk-induced, JNK-mediated cell competition. (A–E) Eyes of adult female flies expressing the indicated transgene(s) under the control of the binary Gal/UAS- system or the GMR promotor. n > 75, 100% penetrance. (F) Phenotypic rescue of a sevEP-Gal4 > UAS-Alk fly eye by one copy of Df(3L)H99, a deficiency for the pro-apoptotic genes hid, grim, and rpr. n > 40, 100% penetrance. Scale bar = 100 µm. (G–I’’) Eye discs from pupae (50 apf) expressing sevEP-driven UAS-YFP and the JNK activity reporter TRE-RFP. TRE-activity is not observed in controls (G-G’’) but upon ectopic Alk expression (H–H’’). (I–I’’) TRE-activity in sevEP-Gal4 > UAS-Alk, UAS-wnd/ + eye discs co-expressing Wnd and Alk.
To analyze this further, we used the TRE-JNK activity reporter system59 in sevEP-Gal4 > UAS-Alk/ + eye discs. No TRE-JNK activity was observed in control 50 h hpf eye discs expressing UAS-YFP under the control of the sevEP-Gal4 driver (Fig. 7G–G’’). In Alk-expressing discs however, the TRE-DsRed reporter was activated in both YFP-negative and YFP-positive cells (Fig. 7H–H’’). Co-expression of Wnd in sevEP-Gal4 > UAS-Alk, UAS-wnd/ + eye discs resulted in strong TRE-DsRed reporter activity in Alk-expressing cells (Fig. 7I–I’’). One explanation for these observations could be that sevEP-Gal4 > UAS-Alk expressing cells acquire a competitive advantage that is abrogated by Wnd-induced JNK activity. The role of JNK in cell competition involves both pro- and anti-tumorigenic responses and depends on the duration of JNK activation as well as on the genetic context60,61. For instance, JNK activation has been observed in invasive and migrating cells in Drosophila tumor models62,63, but also efficiently triggers death of cells with reduced fitness during cell competition64,65,66. To discriminate between these effects we analyzed cell death in sevEP-Gal4 > UAS-Alk expressing eye discs with antibodies against cleaved Dcp-1, an effector caspase downstream of Dronc67. Increased Dcp-1 staining could already be observed in third instar larval eye discs, when compared to the controls (Fig. 8A–B’). G-TRACE analysis further suggested that many Dcp-1 positive cells were in close vicinity to, but not derived from, the sevEP-Gal4 lineage (Fig. 8B,D). Increased Dcp-1 staining could still be observed in sevEP-Gal4 > UAS-Alk (Fig. 8D,D’) but not in control (Fig. 8C–C’) eye discs at pupal stages (50 h apf; quantified in Fig. 8E), suggesting that Alk-expressing cells induce cell death in neighboring cells from early stages of ectopic Alk expression. To further test the hypothesis that ectopic Alk-expression provides a competitive advantage at the expense of cell death in neighboring cells we used the hsFLP/Ay-Gal4 system68 to induce Alk-expressing clones in developing wing discs. Wing discs with Alk-expressing clones displayed significantly increased numbers of Dcp-1-labelled cells when compared to control discs with only GFP-expressing clones (Fig. 8F–G’’’, quantified in 8H). In addition, the number of Dcp-1-positive cells within a range of 8–10 cells around Alk-expressing clones was also significantly increased compared to GFP-clone controls (Fig. 8I). Moreover, Alk-expressing clones were slightly increased in size relative to GFP controls, which further supports a competitive advantage of Alk-expressing cells (Fig. 8J). Taken together, these results show that expression of Alk results in cells with enhanced fitness and suggest a role for Alk in non-cell autonomous induction of cell death in surrounding cells.

Alk-expressing cells exhibit features of super-competitors. (A–D’) Anti cleaved Drosophila Death caspase-1 (Dcp-1) staining of sevEP-Gal4 > UAS-G-TRACE (A,A’,C,C’) and sevEP-Gal4 > UAS-G-TRACE/UAS-Alk (B,B’,D,D’) eye discs at the wandering 3rd instar larval stage and 50 h apf. Scale bars = 10 µm. (E) Quantification of Dcp-1 positive cells using a bee-swarm boxplot representation. Mann Whitney Test reveals statistical significance, ***p < 0.001 (control n = 10, UAS-Alk n = 9). (F–G’’’) Immunofluorescence staining of Alk-expressing clones in wing imaginal discs dissected from wandering 3rd instar larvae. Discs were stained for Dcp-1, GFP and Alk. Scale bar = 50 µm in F. Scale bar = 10 µm in G’’. (H) Quantification of Dcp-1 positive cells in GFP control and Alk overexpressing wing discs. Bars depict means. Error bars represent S.D.; ***p < 0.001 (n = 7 wing discs). An unpaired, two-tailed t-test was applied to reveal significant differences. (I) Quantification of Dcp-1 positive cells in an 8–10 cell diameter surrounding GFP-control (n = 110) and Alk-overexpressing (n = 113) clones in the L3 wing discs. Means and SD (error bars) are depicted. Mann Whitney test was applied to reveal significant differences, ***p < 0.001. (J) Bee-swarm boxplot representation of the quantification of clone size of age-matched GFP-control (n = 16) and Alk-overexpressing (n = 19) clones in L3 wing discs. Mann Whitney test was applied to reveal significant differences, *p < 0.05.
Discussion
Malignant transformation by ALK involves several mechanisms including hyper-activation of Ras/MAPK-, PI3K- and STAT3 signaling pathways as well as increased expression of transcriptional regulators and stem-cell factors, which shifts the affected cell to a more undifferentiated state69,70,71,72. In this study, we conducted a genome-wide deficiency screen in Drosophila to identify modifiers of Alk signaling, resulting in the identification of many known and anticipated signaling molecules, such as Ras/Raf/ERK network components (MESR3, Cnk, Aveugle), PI3K21B, STAT92E, and the protein serine/threonine phosphatase Alphabet which is in agreement with previous screening efforts73. Amongst the signaling molecules we also found split ends (spen)74 and Star75,76, consistent with their known roles in RTK signaling73. Another gene identified as a suppressor of the Alk-induced eye phenotype was moleskin, which is a homologue of the vertebrate Importin-7, and has been proposed to be important for the nuclear import of activated ERK77. Among the identified transcription factors, Foxo and the orphan nuclear receptor transcription factor Tailless (Tll) were modifiers of Alk signaling activity. Nuclear localization of Foxo is regulated by Alk-driven AKT/PI3K signaling in insulin-producing cells in the Drosophila brain23 and, similarly, oncogenic ALK variants affect FOXO3a localization in human anaplastic large-cell lymphoma (ALCL) and neuroblastoma cell culture models71,78. In addition to its function in terminal patterning in the Drosophila embryo52,79, tll is expressed in the optic lobe primordium80, where it controls cell-fate decisions during Bolwig’s organ and optic lobe precursor development81. Interestingly, the mammalian Tll homolog TLX/NR2E1 acts as self-renewal factor in neural stem cells and neuroblastoma tumor spheres, and elevated expression of TLX/NR2E1 is correlated with an unfavorable prognosis in neuroblastoma patients82,83. We also identified tissue-specific transcription factors, like the PAX6 homolog Eyeless and Eyes absent (EYA) which have been linked to human neuroblastoma84,85. Moreover, eight factors involved in initiation of transcription from RNA polymerase II regulated promoters (TFIIA-S, TFIIA-L, TFIIEα, TFIIEβ and the TFIID-associated factors Taf4 (TafII110), Taf6 (TafII60), TFIIS (TCEA1), and Taf12L (TafII30α-2)), were revealed as downstream effectors of Alk signaling. However, mutations in core TFII-complex members could lead to a general reduction of transcriptional activation that would also affect Alk transgene expression. Similarly, mutations of the AF4/FMR2 family (AFF) homolog lilliputian (lilli) were strong suppressors of the Alk-induced eye phenotype86,87. As for the identified TFII-complex members, suppression by lilli alleles should be interpreted with caution as they affect hsp70- and sE-promotor-dependent transgene expression and are therefore frequently found as suppressors in genetic overexpression screens87.
Most strikingly, inducible gain of function alleles of the MAP3K Wallenda (Wnd) that were serendipitously identified during the candidate mapping process were amongst the strongest suppressors. Wnd is one of several JNKKKs in Drosophila and has previously been shown to induce JNK-mediated cell death33,34,88. Testing other components of the JNK signaling pathway, we noted that only activators of the pathway, such as the Wnd JNKKK and an activated version of the Hep JNKK, were able to suppress the Alk-induced eye phenotype, while transgenes that inhibit JNK signaling had no effect, suggesting that activation of the JNK pathway leads to suppression of the phenotype. Interestingly, Wnd was more effective than Tak1 in suppressing the Alk-induced eye phenotype, which may reflect earlier observations that the JNKKKs provide context-dependent output, in contrast to Hep, which is part of the core JNK signaling module33,89. These observations led us to hypothesize that Wnd overexpression in our sevEP-Gal4 > UAS-Alk model might have shifted the cells into a state where competitive advantages gained by ectopic Alk signaling are abrogated upon high levels of JNK pathway activity. A possible mechanism how Alk-expressing cells acquire this advantage could be the repression of pro-apoptotic factors like Hid upon increased Ras/ERK and PI3K/Akt signaling which not only involves direct phosphorylation of Hid by MAPK/ERK but also transcriptional regulation by Foxo and E2F1 which were also found as modifiers in our screen90. Alk-expressing cells activate multiple signaling pathways that result in promotion of the pro-tumorigenic response, and we know from previous work that Alk overexpression leads to activation of MAPK signaling in both eye discs and clones of imaginal discs26.
Cell death is a central mechanism in the process of cell competition and the JNK signaling pathway is thought to play an important role64,91. Work from many investigators has identified numerous context-dependent effects of JNK signaling in this process, including scenarios where cells with higher fitness (“winners”) eliminate their less-competitive neighbors by activating the JNK-pathway in these “loser-cells”34,35,65,66,91. Conversely, JNK activation can lead to increased cellular fitness if the apoptotic pathway downstream of JNK is blocked60,92. A number of genes with oncogenic potential have been reported to confer a competitive advantage on Drosophila cells. One of the best studied examples is Myc that transforms cells into super-competitors at the expense of surrounding cells that are induced to undergo apoptosis66,93, which has been reported to be mediated by both JNK-dependent66 and JNK-independent mechanisms93. A number of observations support a role for Alk in conveying a competitive advantage. Firstly, inhibiting cell death in all cells of the eye disc substantially rescued eye morphology, while blocking cell death in Alk-expressing cells alone had no effect. Secondly, expression of Alk led to an increased TRE-DsRed signal in eye discs. Thirdly, Alk-negative neighbors appear to be sensitized to undergo cell death as reflected in the increased Dcp-1 staining of wing discs containing Alk-expressing clones. Intriguingly, apoptosis is increased in the Drosophila eye when EGFR-triggered cell differentiation is blocked94,95, suggesting that undifferentiated cells involved in cell competition may be more sensitive to JNK-pathway activation, and we noted more Dcp-1 staining in the third instar larval eye disc in the region that is closest to the morphogenetic furrow which may reflect this. Finally, clones expressing Alk were slightly, but significantly increased in size compared with controls. We also noted TRE-activity in Alk-expressing cells of the sevEP lineage as well as YFP-negative cells which could resemble different contexts of JNK activation in the two cell populations. In that case, it would be plausible to speculate that Alk signaling could protect cells by activating the Ras/Raf/ERK pathway which in turn could inhibit pro-apoptotic effector genes. Repression of apoptosis by EGFR signaling has indeed been observed in the developing eye disc and revealed the pro-apoptotic gene hid as direct target of the Ras/MAPK pathway96,97,98. In addition, suppression of apoptosis by activated ALK in a ALK-F1174L/MYCN-driven zebrafish neuroblastoma model99, and pro-apoptotic responses in human cell cultures after ALK inhibitor treatment100,101 have been reported further strengthening a connection between Alk-signaling and cell-intrinsic inhibition of apoptosis.
These findings shed light on a previously unappreciated potential of Alk to induce cell death in the surrounding environment that may ultimately promote growth of Alk-expressing cells. Whether this is also true in human ALK-driven cancer remains to be elucidated, although our initial analysis with ALK-F1174L and the NPM1::ALK fusion oncogenes that are known oncogenic variants in human cancer would suggest that this may be the case. In mammalian cells, ALK signaling leads to activation of multiple signaling pathways, including anti-apoptotic responses55 that may act in concert to convey a competitive advantage. Moreover, as mentioned above, Drosophila Myc transforms cells into super-competitors66, which may be relevant in human cancers such as neuroblastoma where ALK is known to cooperate with MYCN to promote tumorigenesis11,55. In conclusion, while our screening efforts identified many genes that will require further analysis to define their role in Alk signaling, it also allowed us to identify a role for Alk in cell fitness and competition that may be of relevance in human ALK-driven cancer.
Methods
Drosophila melanogaster stocks and genetics
Standard Drosophila husbandry procedures were followed102. Stocks were maintained on a potato-mash-based diet at room temperature and crosses were performed in constant climate chambers at controlled 25 °C and 60% humidity levels, 12:12 h day and night cycle. Fly stocks were obtained from the BDSC (NIH P40OD018537), the KYOTO Stock Center (DGRC) at Kyoto Institute of Technology and the Vienna Drosophila Resource Center (VDRC) at the Campus Science Support Facilities GmbH (CSF). Other lines used were: aveCC9-36B 36, C3GMS 43, cnkΔY2H 36, jeb™.3xOLLAS (this study), gzlΔ2 103, UAS-Alk.L 26, UAS-hALKAL1.G and UAS-hALKAL2.G 4, UAS-hALKF1174L 104, UAS-hNPM1::hALK (this study), UAS-Jra.DN, UAS-kay.DN 105, UAS-puc.M 106, tllg 107, and wnd2 58.
Generation of jeb™.3xOLLAS flies
We used CRISPR/Cas9 genome editing to induce homology-directed repair (HDR) in the Drosophila jeb locus108,109. The pBluescriptII KS[-] HDR donor construct (GenScript) contained homology arms (corresponding to 357 bp immediately upstream and 406 bp downstream of the jeb stop codon) that flanked a central segment encoding for a six amino acid linker (Gly-Ser-Gly-Gly-Ser-Ala), the transmembrane domain of Drosophila Neurotactin (Ala325-His347), a triple OLLAS-tag and a TAA stop codon. Two CRISPR target sites (AGCGGATGCGTTACTACGCTGGG and TGTGTGTGCTGTGTATTAGACGG) in close proximity to the endogenous jeb stop codon were identified using the flyCRISPR optimal target finder tool109. Targeting gRNAs were cloned into pU6-BbsI-chiRNA (Addgene) as described109. Donor and U6-chiRNA plasmids were injected into y1 M{vas-Cas9}ZH-2A embryos (BestGene Inc.). HDR-events were identified by single fly PCR screening using a primer combination specific to the knocked-in sequence (5′-GTTCCTCTTGCAGAGTGTCCT-3′ and 5′-CTAATTGTTTGTCA-ATGTGAGTTCGCT-3′). Candidates were further analyzed by Sanger sequencing (GATC Services—Eurofins Genomics) and tested for non-complementation with the jebweli null allele18,36.
Construction of the UAS-hNPM1::hALK transgene
We employed the deduced amino acid sequence of the human nucleophosmin 1 (hNPM1)-human anaplastic lymphoma kinase (hALK) chimera reported by Morris et al. (8, GenBank: AAA58698.1) as template to generate the UAS-hNPM1::hALK transgene. gBlock fragments (Integrated DNA Technologies) of the hNPM1 n-terminus and the hALK kinase domain were assembled into EcoRI/XbaI-cut pUASTattB vector using the NEBuilder HiFi assembly cloning kit (NEB). The construct was verified by Sanger sequencing (GATC Services—Eurofins Genomics) and injected in y1 w67c23; P{y+t7.7 = CaryP}attP1 embryos (BestGene Inc.).
Genetic screening
Transgenic flies ectopically expressing the Alk receptor in a subset of cells of the developing ommatidia (sevEP-Gal4, UAS-Alk/CyO) exhibit a temperature-sensitive phenotype. Stocks reared at 29 °C exhibited pupal lethality while a characteristic eye phenotype (Fig. 1C) is observed at 25 °C. Screening was performed under controlled conditions (constant 25 °C and 60% humidity levels, 12:12 h day and night cycle). sevEP-Gal4, UAS-Alk/CyO flies were crossed to stocks of the DrosDel deficiency collection kit32 which offers the advantage of an isogenic background and high-percentage coverage of the Drosophila genome. More than 20 F1 female flies per cross that did not exhibit balancer chromosome-associated markers were scored for modifications of the eye phenotype. Suppression was defined as reduction or absence of the area of “undifferentiated cells” in the anterior part of the eye. Expansion of this area, as well as dark spots and excessive necrotic tissue were scored as “other modifications” (detailed in Table S1). All scored flies had to exhibit a similar effect on the sevEP-Gal4, UAS-Alk phenotype although slight variations in the phenotypic strength of the observed effect were tolerated. Crosses that did not yield viable offspring of the desired genotype were performed in triplicate before assigning them to the “lethal/no progeny” group. Non-curly winged female offspring of sevEP-Gal4, UAS-Alk/CyO and the DrosDel project starter stock (#5905) were used as controls. Deficiencies that modified the eye phenotype in this initial screen were tested again to ensure that the results were reproducible. In order to map down the various modifier regions, we scored additional stocks carrying either smaller chromosomal deficiencies, or available mutations in loci located within the suppressing region, or Gal4/UAS-based transgenic constructs of the associated genes. Alleles used to reveal the modifying effect of the identified candidates are listed in Table S1. More than 20 non-balanced F1 female flies per cross were scored, crosses with each allele were performed at least twice. We did not score for partial suppression/modification but slight variations in phenotypic strength were tolerated. For the human ALK modifier experiment, more than 15 non-balanced F1 female flies per cross were scored for modifications of the induced “rough-eye” phenotype. Only complete penetrance was scored, and crosses were performed twice.
Antibody staining and clonal induction
Dissection and antibody staining of imaginal discs was carried out according to110. Alk- or GFP-expressing clones were generated with hsFLP/Ay-Gal4 system68. Briefly, freshly hatched first larvae were picked at 24–26 h after egg laying (AEL), prior to 30 min heat shock at 54–56 h AEL to induce overexpression clones in imaginal discs. Animals were dissected at wandering third instar larval stage (104–106 AEL) and analyzed. The following antibodies were purchased from the Developmental Studies Hybridoma Bank (DSHB, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242) and used in the specified dilutions: mouse anti-Cut (CT; 2B10; 1:500), mouse anti-Prospero (PROS; MR1A; 1:100), rat anti-Drosophila epithelial cadherin (DECad; DCAD2; 1:100), rat anti-embryonic lethal abnormal vision (Elav; 7E8A10; 1:500). Other antibodies used: guinea pig anti-Drosophila Alk (Alk, 1:1,000, 2), rabbit anti β-Galactosidase pre-absorbed on w1118 embryos (β-Gal, Cappel #0855976, 1:1,500), rabbit anti-cleaved Drosophila Death caspase-1 (Dcp-1; Asp216; 1:200, Cell Signaling #9578), chicken anti-GFP (1:500; Abcam ab13970), rabbit anti-RFP (1:1,000; Abcam ab62341). Secondary antibodies and animal sera were purchased from Jackson ImmunoResearch. Stained samples were embedded in Fluoromount-G (SouthernBiotech, #0100-01) and analyzed with a ZEISS Axio Imager.Z2 microscope. Images were acquired with a ZEISS LSM800 confocal microscope using the ZEN Blue edition software.
Preparation of adult Drosophila eyes for thin sectioning and microscopic analysis
To visualize ommatidia patterns, heads from adult flies were dissected and fixed in a mix of 2% glutaraldehyde, and 4% formaldehyde in 50 mM sodium cacodylate buffer (pH 7.4) and post-fixed in 4% Osmium and 2% KFeCN in 200 mM sodium cacodylate buffer (pH 7.4) followed by stepwise dehydration in 30%, 50%, 70%, 85%, 95%, 100% ethanol. Samples were embedded in Hard Plus Resin 812 (Electron Microscopy Sciences) for 48 h at 60 °C. Eye Section (1 µm) were obtained using a Reichert-Jung Ultracut E ultramicrotome with Ultra 450 diamond knife (Electron Microscopy Sciences) on glass slides and stained with a 1% toluidine-blue solution. Images were obtained using a ZEISS Axio Imager.Z2 microscope equipped with a ZEISS Axiocam 503 color camera and the ZEN Blue edition software.
Imaging of adult Drosophila eyes
Image stacks of adult fly eyes were acquired with a ZEISS AxioZoom.V16 stereo zoom microscope equipped with LED ring light and an Axiocam 503 color camera. The enhanced depth of focus (EDF) module of the ZEN Blue edition software was used for further image processing.
Databases, data analysis and statistics
Genomic coordinates refer to the Dmel_Release_6 sequence assembly. Detailed information about Drosophila stocks and genetics is available on FlyBase (https://flybase.org/), the Database of Drosophila Genes & Genomes (Thurmond et al., 2019). Modifying loci (as listed in Table S1) were analyzed by PANTHER14.0-driven gene ontology (GO) enrichment analysis (https://geneontology.org/)51). Interaction analysis of Alk modifying loci (Fig. 3B, listed in Table S1) was analyzed with STRING version 11 (https://string-db.org)111 employing a medium confidence score of 0.4 for interactions. GraphPad Prism 7 software was used for all statistical analyses.
References
Hallberg, B. & Palmer, R. H. The role of the ALK receptor in cancer biology. Ann. Oncol. 27(Suppl 3), iii4–iii15. https://doi.org/10.1093/annonc/mdw301 (2016).
Englund, C. et al. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 425, 512–516. https://doi.org/10.1038/nature01950 (2003).
Lee, H. H., Norris, A., Weiss, J. B. & Frasch, M. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature 425, 507–512 (2003).
Guan, J. et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4, e09811. https://doi.org/10.7554/eLife.09811 (2015).
Reshetnyak, A. V. et al. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. U. S. A. 112, 15862–15867. https://doi.org/10.1073/pnas.1520099112 (2015).
Fadeev, A. et al. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.1719137115 (2018).
Mo, E. S., Cheng, Q. N., Reshetnyak, A. V., Schlessinger, J. & Nicoli, S. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc. Natl. Acad. Sci. U. S. A. 114, 12027–12032. https://doi.org/10.1073/pnas.1710254114 (2017).
Morris, S. W. et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263, 1281–1284 (1994).
Soda, M. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007).
Rikova, K. et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 (2007).
Matthay, K. K. et al. Neuroblastoma. Nat. Rev. Dis. Primers 2, 16078. https://doi.org/10.1038/nrdp.2016.78 (2016).
Janoueix-Lerosey, I. et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970. https://doi.org/10.1038/nature07398 (2008).
Mosse, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935 (2008).
Chen, Y. et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974 (2008).
George, R. E. et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978 (2008).
Caren, H., Abel, F., Kogner, P. & Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 416, 153–159 (2008).
Lin, J. J., Riely, G. J. & Shaw, A. T. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov. 7, 137–155. https://doi.org/10.1158/2159-8290.CD-16-1123 (2017).
Stute, C., Schimmelpfeng, K., Renkawitz-Pohl, R., Palmer, R. H. & Holz, A. Myoblast determination in the somatic and visceral mesoderm depends on Notch signalling as well as on milliways(mili(Alk)) as receptor for Jeb signalling. Development 131, 743–754 (2004).
Bazigou, E. et al. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell 128, 961–975. https://doi.org/10.1016/j.cell.2007.02.024 (2007).
Cheng, L. Y. et al. Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell 146, 435–447. https://doi.org/10.1016/j.cell.2011.06.040 (2011).
Gouzi, J. Y. et al. The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet. 7, e1002281 (2011).
Lasek, A. W. et al. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS ONE 6, e22636. https://doi.org/10.1371/journal.pone.0022636PONE-D-10-06421[pii] (2011).
Okamoto, N. & Nishimura, T. Signaling from Glia and Cholinergic neurons controls nutrient-dependent production of an insulin-like peptide for drosophila body growth. Dev. Cell 35, 295–310. https://doi.org/10.1016/j.devcel.2015.10.003 (2015).
Rohrbough, J. & Broadie, K. Anterograde Jelly belly ligand to Alk receptor signaling at developing synapses is regulated by Mind the gap. Development 137, 3523–3533. https://doi.org/10.1242/dev.047878 (2010).
Pecot, M. Y. et al. Sequential axon-derived signals couple target survival and layer specificity in the Drosophila visual system. Neuron 82, 320–333. https://doi.org/10.1016/j.neuron.2014.02.045 (2014).
Loren, C. E. et al. Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes Cells 6, 531–544 (2001).
Hugosson, F. et al. The Drosophila midkine/pleiotrophin homologues Miple1 and Miple2 affect adult lifespan but are dispensable for alk signaling during embryonic gut formation. PLoS ONE 9, e112250. https://doi.org/10.1371/journal.pone.0112250 (2014).
Chand, D. et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis. Model Mech. 6, 373–382. https://doi.org/10.1242/dmm.010348 (2013).
Martinsson, T. et al. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res. 71, 98–105. https://doi.org/10.1158/0008-5472.CAN-10-2366 (2011).
Guan, J. et al. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb. Mol. Case Stud. https://doi.org/10.1101/mcs.a002550 (2018).
Guan, J. et al. Novel mechanisms of ALK activation revealed by analysis of the Y1278S neuroblastoma mutation. Cancers (Basel) https://doi.org/10.3390/cancers9110149 (2017).
Ryder, E. et al. The DrosDel collection: a set of P-element insertions for generating custom chromosomal aberrations in Drosophila melanogaster. Genetics 167, 797–813 (2004).
Ma, X. et al. Wallenda regulates JNK-mediated cell death in Drosophila. Cell Death Dis. 6, e1737. https://doi.org/10.1038/cddis.2015.111 (2015).
LaMarca, J. E. & Richardson, H. E. Two-faced: roles of JNK signalling during tumourigenesis in the Drosophila model. Front. Cell Dev. Biol. https://doi.org/10.3389/fcell.2020.00042 (2020).
Bowling, S., Lawlor, K. & Rodriguez, T. A. Cell competition: the winners and losers of fitness selection. Development https://doi.org/10.1242/dev.167486 (2019).
Wolfstetter, G. et al. The scaffolding protein Cnk binds to the receptor tyrosine kinase Alk to promote visceral founder cell specification in Drosophila. Sci. Signal. https://doi.org/10.1126/scisignal.aan0804 (2017).
Evans, C. J. et al. G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila. Nat. Methods 6, 603–605. https://doi.org/10.1038/nmeth.1356nmeth.1356[pii] (2009).
Hafen, E., Basler, K., Edstroem, J. E. & Rubin, G. M. Sevenless, a cell-specific homeotic gene of Drosophila, encodes a putative transmembrane receptor with a tyrosine kinase domain. Science 236, 55–63. https://doi.org/10.1126/science.2882603 (1987).
Banerjee, U., Renfranz, P. J., Pollock, J. A. & Benzer, S. Molecular characterization and expression of sevenless, a gene involved in neuronal pattern formation in the Drosophila eye. Cell 49, 281–291. https://doi.org/10.1016/0092-8674(87)90569-1 (1987).
Tomlinson, A., Bowtell, D. D., Hafen, E. & Rubin, G. M. Localization of the sevenless protein, a putative receptor for positional information, in the eye imaginal disc of Drosophila. Cell 51, 143–150. https://doi.org/10.1016/0092-8674(87)90019-5 (1987).
Weiss, J. B., Suyama, K. L., Lee, H. H. & Scott, M. P. Jelly belly: a Drosophila LDL receptor repeat-containing signal required for mesoderm migration and differentiation. Cell 107, 387–398 (2001).
Schonherr, C., Yang, H. L., Vigny, M., Palmer, R. H. & Hallberg, B. Anaplastic lymphoma kinase activates the small GTPase Rap1 via the Rap1-specific GEF C3G in both neuroblastoma and PC12 cells. Oncogene 29, 2817–2830. https://doi.org/10.1038/onc.2010.27onc201027 (2010).
Shirinian, M. et al. The Rap1 guanine nucleotide exchange factor C3G is required for preservation of larval muscle integrity in Drosophila melanogaster. PLoS ONE 5, e9403. https://doi.org/10.1371/journal.pone.0009403 (2010).
Rogge, R. D., Karlovich, C. A. & Banerjee, U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 64, 39–48 (1991).
Chang, H. C. & Rubin, G. M. 14-3-3 epsilon positively regulates Ras-mediated signaling in Drosophila. Genes Dev. 11, 1132–1139 (1997).
Simon, M. A., Bowtell, D. D., Dodson, G. S., Laverty, T. R. & Rubin, G. M. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701–716 (1991).
Therrien, M. et al. KSR, a novel protein kinase required for RAS signal transduction. Cell 83, 879–888 (1995).
Biggs, W. H. 3rd. et al. The Drosophila rolled locus encodes a MAP kinase required in the sevenless signal transduction pathway. EMBO J. 13, 1628–1635 (1994).
O’Neill, E. M., Rebay, I., Tjian, R. & Rubin, G. M. The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137–147 (1994).
Ryder, E. et al. The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics 177, 615–629 (2007).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189. https://doi.org/10.1093/nar/gkw1138 (2017).
Pignoni, F. et al. The Drosophila gene tailless is expressed at the embryonic termini and is a member of the steroid receptor superfamily. Cell 62, 151–163. https://doi.org/10.1016/0092-8674(90)90249-e (1990).
Justice, R. W., Zilian, O., Woods, D. F., Noll, M. & Bryant, P. J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546 (1995).
Xu, T., Wang, W., Zhang, S., Stewart, R. A. & Yu, W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121, 1053–1063 (1995).
Hallberg, B. & Palmer, R. H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 13, 685–700 (2013).
Aoyagi, N. & Wassarman, D. A. Developmental and transcriptional consequences of mutations in Drosophila TAF(II)60. Mol. Cell Biol. 21, 6808–6819. https://doi.org/10.1128/MCB.21.20.6808-6819.2001 (2001).
Parks, A. L. et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292 (2004).
Collins, C. A., Wairkar, Y. P., Johnson, S. L. & DiAntonio, A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69 (2006).
Chatterjee, N. & Bohmann, D. A versatile PhiC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS ONE 7, e34063. https://doi.org/10.1371/journal.pone.0034063 (2012).
Pinal, N., Martin, M., Medina, I. & Morata, G. Short-term activation of the Jun N-terminal kinase pathway in apoptosis-deficient cells of Drosophila induces tumorigenesis. Nat. Commun. 9, 1541. https://doi.org/10.1038/s41467-018-04000-6 (2018).
Uhlirova, M., Jasper, H. & Bohmann, D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl. Acad. Sci. U. S. A. 102, 13123–13128 (2005).
Uhlirova, M. & Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. Embo J. 25, 5294–5304 (2006).
Wu, M., Pastor-Pareja, J. C. & Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 463, 545–548. https://doi.org/10.1038/nature08702 (2010).
Moreno, E., Basler, K. & Morata, G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416, 755–759. https://doi.org/10.1038/416755a (2002).
Pinal, N., Calleja, M. & Morata, G. Pro-apoptotic and pro-proliferation functions of the JNK pathway of Drosophila: roles in cell competition, tumorigenesis and regeneration. Open Biol. 9, 180256. https://doi.org/10.1098/rsob.180256 (2019).
Moreno, E. & Basler, K. dMyc transforms cells into super-competitors. Cell 117, 117–129. https://doi.org/10.1016/s0092-8674(04)00262-4 (2004).
Song, Z., McCall, K. & Steller, H. DCP-1, a Drosophila cell death protease essential for development. Science 275, 536–540. https://doi.org/10.1126/science.275.5299.536 (1997).
Ito, K., Awano, W., Suzuki, K., Hiromi, Y. & Yamamoto, D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development 124, 761–771 (1997).
Werner, M. T., Zhao, C., Zhang, Q. & Wasik, M. A. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood 129, 823–831. https://doi.org/10.1182/blood-2016-05-717793 (2017).
Montes-Mojarro, I. A., Steinhilber, J., Bonzheim, I., Quintanilla-Martinez, L. & Fend, F. The pathological spectrum of systemic anaplastic large cell lymphoma (ALCL). Cancers (Basel) https://doi.org/10.3390/cancers10040107 (2018).
Van den Eynden, J. et al. Phosphoproteome and gene expression profiling of ALK inhibition in neuroblastoma cell lines reveals conserved oncogenic pathways. Sci. Signal. https://doi.org/10.1126/scisignal.aar5680 (2018).
Sattu, K. et al. Phosphoproteomic analysis of ALK downstream signaling pathways identifies STAT3 as a functional target of activated ALK in neuroblastoma cells. Febs J https://doi.org/10.1111/febs.12453 (2013).
Therrien, M., Morrison, D. K., Wong, A. M. & Rubin, G. M. A genetic screen for modifiers of a kinase suppressor of Ras-dependent rough eye phenotype in Drosophila. Genetics 156, 1231–1242 (2000).
Rebay, I. et al. A genetic screen for novel components of the Ras/Mitogen-activated protein kinase signaling pathway that interact with the yan gene of Drosophila identifies split ends, a new RNA recognition motif-containing protein. Genetics 154, 695–712 (2000).
Heberlein, U., Hariharan, I. K. & Rubin, G. M. Star is required for neuronal differentiation in the Drosophila retina and displays dosage-sensitive interactions with Ras1. Dev. Biol. 160, 51–63. https://doi.org/10.1006/dbio.1993.1285 (1993).
Kolodkin, A. L., Pickup, A. T., Lin, D. M., Goodman, C. S. & Banerjee, U. Characterization of Star and its interactions with sevenless and EGF receptor during photoreceptor cell development in Drosophila. Development 120, 1731–1745 (1994).
Lorenzen, J. A. et al. Nuclear import of activated D-ERK by DIM-7, an importin family member encoded by the gene moleskin. Development 128, 1403–1414 (2001).
Gu, T. L. et al. NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood 103, 4622–4629 (2004).
Jurgens, G., Wieschaus, E., Nusslein-Volhard, C. & Kluding, H. Mutations affecting the pattern of the larval cuticle inDrosophila melanogaster : II. Zygotic loci on the third chromosome. Wilehm Roux Arch. Dev. Biol. 193, 283–295. https://doi.org/10.1007/BF00848157 (1984).
Gui, H., Li, M. L. & Tsai, C. C. A tale of tailless. Dev. Neurosci. 33, 1–13. https://doi.org/10.1159/000321585 (2011).
Daniel, A., Dumstrei, K., Lengyel, J. A. & Hartenstein, V. The control of cell fate in the embryonic visual system by atonal, tailless and EGFR signaling. Development 126, 2945–2954 (1999).
Sobhan, P. K. & Funa, K. TLX-its emerging role for neurogenesis in health and disease. Mol. Neurobiol. 54, 272–280. https://doi.org/10.1007/s12035-015-9608-1 (2017).
Chavali, P. L. et al. TLX activates MMP-2, promotes self-renewal of tumor spheres in neuroblastoma and correlates with poor patient survival. Cell Death Dis. 5, e1502. https://doi.org/10.1038/cddis.2014.449 (2014).
Hansen, J. N., Lotta, L. T. Jr., Eberhardt, A., Schor, N. F. & Li, X. EYA1 expression and subcellular localization in neuroblastoma and its association with prognostic markers. J. Cancer Res. Ther. (Manch) 4, 11–18. https://doi.org/10.14312/2052-4994.2016-3 (2016).
Pinson, J., Simpson, T. I., Mason, J. O. & Price, D. J. Positive autoregulation of the transcription factor Pax6 in response to increased levels of either of its major isoforms, Pax6 or Pax6(5a), in cultured cells. BMC Dev. Biol. 6, 25. https://doi.org/10.1186/1471-213X-6-25 (2006).
Wittwer, F., van der Straten, A., Keleman, K., Dickson, B. J. & Hafen, E. Lilliputian: an AF4/FMR2-related protein that controls cell identity and cell growth. Development 128, 791–800 (2001).
Tang, A. H., Neufeld, T. P., Rubin, G. M. & Muller, H. A. Transcriptional regulation of cytoskeletal functions and segmentation by a novel maternal pair-rule gene, lilliputian. Development 128, 801–813 (2001).
Ma, X. et al. Rho1-Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene 35, 846–855. https://doi.org/10.1038/onc.2015.137 (2016).
Muzzopappa, M., Murcia, L. & Milan, M. Feedback amplification loop drives malignant growth in epithelial tissues. Proc. Natl. Acad. Sci. U. S. A. 114, E7291–E7300. https://doi.org/10.1073/pnas.1701791114 (2017).
Bilak, A. & Su, T. T. Regulation of Drosophila melanogaster pro-apoptotic gene hid. Apoptosis 14, 943–949. https://doi.org/10.1007/s10495-009-0374-2 (2009).
Madan, E., Gogna, R. & Moreno, E. Cell competition in development: information from flies and vertebrates. Curr. Opin. Cell Biol. 55, 150–157. https://doi.org/10.1016/j.ceb.2018.08.002 (2018).
Kucinski, I., Dinan, M., Kolahgar, G. & Piddini, E. Chronic activation of JNK JAK/STAT and oxidative stress signalling causes the loser cell status. Nat. Commun. 8, 136. https://doi.org/10.1038/s41467-017-00145-y (2017).
de la Cova, C., Abril, M., Bellosta, P., Gallant, P. & Johnston, L. A. Drosophila myc regulates organ size by inducing cell competition. Cell 117, 107–116. https://doi.org/10.1016/s0092-8674(04)00214-4 (2004).
Sawamoto, K. et al. Argos induces programmed cell death in the developing Drosophila eye by inhibition of the Ras pathway. Cell Death Differ. 5, 262–270. https://doi.org/10.1038/sj.cdd.4400342 (1998).
Freeman, M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 87, 651–660. https://doi.org/10.1016/s0092-8674(00)81385-9 (1996).
Kurada, P. & White, K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95, 319–329. https://doi.org/10.1016/s0092-8674(00)81764-x (1998).
Bergmann, A., Agapite, J., McCall, K. & Steller, H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95, 331–341. https://doi.org/10.1016/s0092-8674(00)81765-1 (1998).
Bergmann, A., Tugentman, M., Shilo, B. Z. & Steller, H. Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Dev. Cell 2, 159–170 (2002).
Zhu, S. & Thomas Look, A. Neuroblastoma and its zebrafish model. Adv. Exp. Med. Biol. 916, 451–478. https://doi.org/10.1007/978-3-319-30654-4_20 (2016).
McDermott, U. et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 68, 3389–3395 (2008).
Christensen, J. G. et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 6, 3314–3322 (2007).
Ashburner, M. Drosophila: A Laboratory Handbook and Manual. Two volumes. (1989).
Yamazaki, Y. et al. Goliath family E3 ligases regulate the recycling endosome pathway via VAMP3 ubiquitylation. EMBO J. 32, 524–537. https://doi.org/10.1038/emboj.2013.1 (2013).
Schonherr, C. et al. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem. J. 440, 405–413 (2011).
Balakireva, M. et al. The Ral/exocyst effector complex counters c-Jun N-terminal kinase-dependent apoptosis in Drosophila melanogaster. Mol. Cell Biol. 26, 8953–8963. https://doi.org/10.1128/MCB.00506-06 (2006).
Martin-Blanco, E. et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557–570 (1998).
Pankratz, M. J., Hoch, M., Seifert, E. & Jackle, H. Kruppel requirement for knirps enhancement reflects overlapping gap gene activities in the Drosophila embryo. Nature 341, 337–340. https://doi.org/10.1038/341337a0 (1989).
Ren, X. et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc. Natl. Acad. Sci. U. S. A. 110, 19012–19017. https://doi.org/10.1073/pnas.1318481110 (2013).
Gratz, S. J., Rubinstein, C. D., Harrison, M. M., Wildonger, J. & O’Connor-Giles, K. M. CRISPR-Cas9 genome editing in Drosophila. Curr. Protoc. Mol. Biol. 111(31), 20. https://doi.org/10.1002/0471142727.mb3102s111 (2015).
Tea, J. S., Cespedes, A., Dawson, D., Banerjee, U. & Call, G. B. Dissection and mounting of Drosophila pupal eye discs. J. Vis. Exp. https://doi.org/10.3791/52315 (2014).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. https://doi.org/10.1093/nar/gky1131 (2019).
Acknowledgments
The authors would like to thank K.V. Anderson, C. Collins, B. Dickson, A. Holz, P. Ingham, C. Klämbt, E. Knust, A. Müller, C. Nowell, T. Schupbach, M. Therrien, C. Thummel, S. Wasserman, T. Wolff, T. Xu., and C. Zuker for generous gifts of fly stocks and reagents during the course of this work. We are also grateful to the stock centers at Bloomington, Szeged, Harvard and Tübingen for providing stocks. This work has been supported by grants from the Swedish Cancer Society (RHP, CAN18/729), the Swedish Childhood Cancer Foundation (RHP, 2015-96), the Swedish Research Council (RHP, 2015-04466), the Swedish Foundation for Strategic Research (RHP, RB13-0204, www.nnbcr.se), the Knut och Alice Wallenbergs Stiftelse (RHP, KAW 2018.0057), the Carl Trygger Foundation, Västra Götalandsregionen ALFGBG-238561 and the Göran Gustafsson Foundation (RHP, 2016).
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
G.W. contributed to conceptualization, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, original draft preparation, review & editing. K.P. contributed to formal analysis, investigation, methodology, visualization, original draft preparation, writing/review & editing. M.B. contributed to conceptualization, formal analysis, investigation, methodology, visualization, original draft preparation, review & editing. T.A.M. contributed to investigation, visualization, original draft preparation, review & editing. P.M-G. contributed to formal analysis, investigation, visualization, original draft preparation, review & editing. S.C. contributed to investigation, visualization, review & editing. H.S. contributed to investigation, visualization, original draft preparation, review & editing. S.K.K. contributed to investigation, visualization, review & editing. E.U. contributed to investigation, visualization, original draft preparation, review & editing. G.V. contributed to investigation, review & editing. A.U. contributed to project administration, resources, review & editing. R.H.P. contributed to conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, original draft preparation, review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wolfstetter, G., Pfeifer, K., Backman, M. et al. Identification of the Wallenda JNKKK as an Alk suppressor reveals increased competitiveness of Alk-expressing cells. Sci Rep 10, 14954 (2020). https://doi.org/10.1038/s41598-020-70890-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70890-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
