
Abstract
Hepatitis C virus (HCV) infection remains a global health problem. Previously, the prevalence of NS5A resistance-associated substitutions (RASs) to elbasvir, a new direct-acting antiviral (DAA) against the NS5A viral protein was assessed by our group before its introduction into clinical use in Spain. However, the origin, epidemic history, transmission dynamics, diversity and baseline RASs to NS5A direct-acting agents of HCV-GT1a in Spain remain unknown. A nationwide cross-sectional survey of individuals chronically-infected with HCV-G1a and DAAs-naïve was performed. HCV population sequencing, phylogenetic analysis and Bayesian methods were used. GT1a clade II was more prevalent than clade I (82.3% vs. 17.7%; P < 0.001) and older (estimated origin in 1912 vs. 1952). Clade II epidemic is currently declining whereas clade I epidemic has reached equilibrium. A total of 58 single RASs were identified, which account for the moderate level (10%) of baseline resistance observed. When considering the regional data, marked differences were observed, with thirteen regions showing an intermediate level (5–15%) and one a high level (20%) of resistance. Current HCV-GT1a epidemic in Spain is driven by clade I which seem to have different dissemination routes relative to clade II. A moderate level of baseline RASs to NS5A-DAAs with marked differences among regions was observed. Close surveillance of response to treatment with DAAs will be important.
Similar content being viewed by others
Introduction
Hepatitis C virus (HCV) infection is a global health problem with latest data estimates 71.1 million viraemic chronic infections (1.1% of world population)1. HCV displays high genetic heterogeneity and is classified into eight major genotypes (1–8)2 and 86 subtypes3. Worldwide there are significant differences in epidemic history among the HCV genotypes (GT) which may differ in response to treatment, with GT1 being the most represented4. Among HCV-infected patients also coinfected with human immunodeficiency virus (HIV) subtype 1a (GT1a) prevails5,6,7.
Despite the high rates of cure with the new direct-acting antivirals (DAAs), the presence of NS5A resistance-associated substitutions (RASs) may compromise the efficacy of NS5A inhibitors. Recently, Zeuzem and colleagues (2017) observed that pretreatment ledipasvir-specific RASs (identified in 8–16% of patients) may compromise treatment outcome particularly in treatment-experienced patients with GT1a, as they found a sustained virological response (SVR) rate of only 76% in patients with pretreatment RASs8.
The seroprevalence of HCV infection in Spain is 1.1%9 and hepatitis C leads the list of infectious disease related mortality10. GT1 accounts for 67% of all HCV infections and GT1a causes 40% of all GT1 infections11. Until October 2018 117,452 patients had access to DAAs and 95.5% had SVR in the frame of the national plan of universal access to treatment12 while 1.1% of those cured are reinfected7.
Our group previously evaluated the prevalence of NS5A RASs to elbasvir before its introduction into clinical use in Spain13. However, the HCV-GT1a spatiotemporal distribution, epidemic history and resistance to all NS5A inhibitors in Spain remain unknown. In this study we aimed to make the first description of the origin, epidemic history, transmission dynamics and diversity of HCV-GT1a in Spain.
Methods
Study design and patients
Overall, 588 patients harboring HCV-GT1a were included. We used the STROBE checklist14 to design and carry out a cross-sectional survey of individuals chronically-infected with HCV-G1a and naïve to NS5A inhibitors who were attended in 84 health centers distributed throughout the national territory (Supplementary Information SI1). The samples were collected prior to anti-NS5A therapy initiation, between October 2014 and October 2015. Genotyping testing was performed using the Real-Time HCV genotype II assay (Abbott Laboratories, Illinois, USA)15. The identification of NS5A RASs was performed using plasma specimens at the National Center of Microbiology (Instituto de Salud Carlos III [ISCIII]). Anonymized samples and a minimum data set were then transferred to the ISCIII National Biobank (REF: 0000984).
Amplification and direct sequencing of NS5A gene
Amplification and direct sequencing of complete NS5A gene (1,343 nt) was performed as described elsewhere13. Standard Sanger sequencing of the PCR product was carried out using an ABI PRISM 377 DNA sequencer (AppIied Biosystems, USA), with a threshold of sensitivity of 15%16.
HCV subtyping, phylogenetic tree reconstruction and transmission clusters analysis
HCV subtyping was performed using COMET HCV subtyping tool17, and HCV-GT1a lineages (clade I and clade II) were confirmed by geno2pheno[HCV] (Bonn, Germany; https://hcv.geno2pheno.org/). The sequences were aligned with the MAFFT algorithm18 and manually edited in MEGA v7.0.2619. Reference HCV Genotype 1 sequences were retrieved from the Los Alamos HCV database and added to the final alignment (Table SI3). Using the RaxMLGUI v1.5 software, a maximum likelihood tree (ML) was then reconstructed under the GTR + Gamma evolutionary model with 1,000 bootstrap replicates as implemented in RaxMLGUI v1.520. Final visualization and annotation of the ML tree was implemented in iTOL v4.0.321. Transmission clusters including two or more individuals (TC ≥ 2) were identified on the ML tree using ClusterPickerGUI v1.2.322 with bootstrap and genetic distance thresholds set at 70% and 0.045 respectively.
Bayesian evolutionary analysis
To improve the convergence of MCMC chains during the Bayesian phylogenetic analysis in the BEAST v1.10 package23 sequences with < 50% sequence length coverage (n = 14) were excluded from the Spanish HCV-GT1a sequences and the remaining sequences split into two clades, clade I (n = 100) and clade II (n = 474) and analyzed separately. Next, a maximum likelihood tree was built using IqTree24 and a temporal signal analysis was performed using Tempest25. The Bayesian analysis was carried out as described formerly26. Briefly, the SRD06 model of nucleotide substitution27, and an uncorrelated lognormal relaxed molecular clock (UCLD) with a Bayesian skyline plot (BSP) coalescent prior were specified in BEAUti in the BEAST package. Because of lack of sufficient temporal signal in our dataset which is often encountered with HCV dataset, and as this can result in unreliable and inaccurate estimates, we relied on previously published data with a strong temporal signal to calibrate the molecular clock28,29,30. Hence, we specified a Normal prior distribution on the clock rate (0.001 ± 0.0005 substitutions/site/year)31. For the selection of appropriate molecular clock model, Bayes factor support was calculated by specifying the path-sampling and stepping-stone marginal likelihood estimation (Table SI4)32. Three independent Markov Chain Monte Carlo (MCMC) chains were run for 1–2 × 108 generations for clades I and II respectively and combined after discarding 10% burn-in. The convergence of MCMC chains were monitored in Tracer v1.7 (https://tree.bio.ed.ac.uk/software/tracer/). Maximum clade credibility (MCC) trees were summarized using tree annotator and tree visualization was implemented in Figtree v1.4.3 (https://tree.bio.ed.ac.uk/software/figtree/).
Phylogeography analysis
Discrete phylogenetic analyses of the HCV-GT1a clades I and II epidemic circulating in the autonomous regions in Spain were analyzed using the Bayesian stochastic search variable selection (BSSVS) procedure with an asymmetric model of among-location transition for the determination of the HCV lineage migration rates as implemented in BEAST v1.10 (Table SI4). The MCMC chains were run as described above and the spatiotemporal visualization and estimation of the Bayes factor (BF) support (BF support ≥ 3 was assumed to be relevant33 for all the geographical location transitions were performed in the “Spatial Phylogenetic Reconstruction of Evolutionary Dynamics” software using Data-Driven Documents (D3) (spreaD3 v0.9.6)34. The georeferencing of the RASs with demonstrated clinical relevance among the Spanish Autonomous Communities (CCAA) was performed by using the GPS coordinates in decimal degrees and the prevalence maps were elaborated using the data visualization tool Tableau 9.3, (Tableau Software, Inc., USA)35.
Resistance-associated substitutions
NS5A-specific clinically relevant RASs analyzed were the following according to the latest European Guidelines: K24G/N/R, K26E, M28A/G/T/V, Q30C/D/E/G/H/I/K/L/N/R/S/T/Y, L31I/F/M/P/V, P32L/S, S38F, H58D/L/R, A92K/T, Y93C/F/H/L/N/R/S/T/W36. Fold-change was evaluated according to the European and American Guidelines36,37 and additional references listed in Table SI5. RASs were categorized as follow: RASs with a high fold-change (> 100×, > 1,000×, > 10,000× with a probable clinical impact; RASs that have an intermediate impact on efficacy (fold-change < 100×); RASs with a low fold-change (< 20×) with no clinical significant impact38. The prevalence of RASs observed regionally was classified as follows: low level (< 5%), intermediate level (5–15%) and high level (> 15%). Codon sites corresponding to these RASs were removed from the HCV NS5A sequence alignment for phylogenetic analysis.
Statistical analysis
The Pearson chi-square test or Fisher’s exact test were used to analyze categorical variables while continuous variables were compared by Mann–Whitney U test. P-values were 2-tailed and statistical significance α was 5%. The SPSS software v.25 (SPSS Inc., Chicago, IL) was used to perform analyses.
Ethics statement
The study was approved by the Institutional Review Board and the Research Ethic Committee of ISCIII (Nº CEI PI 43_2015) and was conducted in accordance with the Declaration of Helsinki.
Results
Overall, 588 patients harboring HCV-GT1a were included. The 80.8% of subjects were men and were 50 years old (IQR: 47–53). HCV-monoinfected and HIV/HCV-coinfected patients were equally represented. Clade II was much more prevalent than clade I (82.7% vs. 17.3%; P < 0.0001) and clade II was more represented in HCV-monoinfected than in HIV/HCV-coinfected patients (87.0% vs. 78.1%; P = 0.004) (Table 1; Figure SI2). Importantly, consistent GT1a lineages results were obtained when comparing the clade assignment by phylogenetic method with the geno2pheno[HCV] algorithm, with only 4 sequences having a different assignment (Table SI6).
Male-dominated transmission pairs and clade II viruses predominate the Spanish HCV-GT1a population
Twenty-six transmission pairs and three transmission clusters (TC including more than 2 individuals) were identified (70% bootstrap support; genetic distance threshold < 0.045) compared to ten transmission pairs that were identified with a more restrictive transmission cluster criteria (90% bootstrap support; genetic distance threshold < 0.015) (Fig. 1, Table 2), representing 10.5% versus 3.4% (n = 62/588 vs 20/588) of the study population. The transmission pairs and clusters predominantly comprised clade II viruses (79.3%, n = 23/29) and occurred intra-regionally (55.2%, n = 16/29). Fourteen transmission pairs and three clusters (58.6%, n = 17/29) were made up of men only. We further investigated the influence of RASs on the transmission clusters. Identical transmission clusters but short of three transmission pairs (n = 23 vs n = 26) were identified when the RASs sites were not excluded from the alignment; of these, only two clusters, cluster 7 and 8, had transmissible M28V and Q30R + Y93H RASs, respectively (Table 2).

Phylogenetic tree reconstruction of GT1a strains circulating in Spain. A maximum likelihood tree (ML) produced under the GTR + Gamma substitution model with 1,000 bootstrap replicates segregates the GT1a into two clades, clade I (blue) and clade II (red). Bootstrap supports ≥ 70% are depicted as purple filled circles at the nodes and transmission clusters (TC ≥ 2) are highlighted in grey colour.
The largest transmission cluster, cluster 29 (TC = 4 patients) also had the most diverse population encompassing patients from three different autonomous regions, namely, Cantabria, Castille and Leon and Basque Country. The highest number of transmission pairs and clusters were found in Galicia and Cantabria autonomous regions. Overall these results are consistent with a longstanding HCV epidemic in Spain that is driven by all modes of transmission.
Origin and spatial-temporal dynamics of GT1a dispersion in Spain
Clade II was introduced in Spain in the beginning of the twentieth century [median estimate of the date of the most recent common ancestor (MRCA), 1912, 95% highest posterior density (95% HPD) interval 1822–1964] whereas clade I was introduced several decades later in 1952 (95% HPD, 1905–1980) (Fig. 2A, B). Both viruses were unnoticed during several decades before going through exponential epidemic growth, clade II starting in the 1950s and clade I twenty years later in the 1970’s (Fig. 2C, D). Clade II epidemic dissemination was more successful than clade I and leveled off in the early 1990’s with Ne ≈ 2 × 105 which was almost 20-fold higher than clade I epidemic at its peak (Ne ≈ 1.2 × 104 in mid-1990s). However, clade II epidemic is now waning whereas clade I epidemic is still ongoing.

Bayesian estimation of the epidemic history of NS5A GT1a clades in Spain. Maximum clade credibility trees for each of the clades, clade I (A) and clade II (B) are presented with the mean tMRCA (95% HPD interval) estimates in calendar years annotated at the root nodes for each of the GT1a clades. Posterior probability cut-off value ≥ 0.9 are annotated at the nodes. The Bayesian skyline plots (BSP) for GT1a clade I (C) and GT1a clade II (D) showing the epidemic growth over time are presented. The solid blue line represents the changes in the median effective population size through time on a log10 scale, with the grey shaded area corresponding to the 95% highest posterior density (95% HPD) interval.
The earliest migration of HCV-GT1a strains in Spain most likely occurred from the Basque country and involved very strongly supported dispersal of clade II viruses from the Basque country to Andalusia and Madrid (BF support = 192.13 and 170.24 respectively) (Table 3). Subsequently, these clade II viruses were successfully propagated from Andalusia and Madrid (BF support = 181.91 and 42.61 respectively). Moreover, Andalusia and Madrid have continued to be the important sources for the dispersal of clade I viruses in contrast to the Basque country which had more support for being a sink (Andalusia-to-Basque country and Galicia-to-Basque country, BF support: > 20 and > 3 respectively) rather than a source of viral dispersion. Thus, the migration pattern of the HCV-GT1a strains in Spain, together with the paraphyly of clade I relative to clade II and its later origin compared to clade II strongly suggest that clade I has evolved from clade II (Fig. 1) and is a driver of the ongoing HCV-GT1a epidemic in Spain.
Baseline polymorphisms and RASs at the NS5A domain
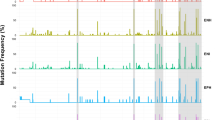
Overall, almost 13,000 baseline mutations were detected, of which 8,602 were considered polymorphisms, being found in > 2% of the samples (Supplementary Dataset and Table 4). Viruses bearing RASs were present in 50 individuals (8.5%), 22/300 (7.3%) HCV-monoinfected and 28/288 (9.7%) HCV/HIV-coinfected and were similarly observed in those infected with clade 1 (8.8%; n = 9/102) and clade 2 (8.4%; n = 41/486). Six of those subjects harbored viruses with double RASs and one had triple RASs (14.0%, n = 7/50). In total 58 single RASs were identified in the study population being the most common M28A/T/V (37.9%; n = 22/58), Y93C/F/H/N (24.1%; n = 14/58) and Q30E/H/R (20.7%; n = 12/58). The double mutations 30H + 93H (n = 2), 28V + 30R (n = 2), 30R + 93H (n = 2) and triple mutations 30H + 93H + 24R (n = 1) were also observed (Table 1). The prevalence of RASs observed regionally was as follow: low level (< 5%) in four regions, intermediate (5–15%) in thirteen regions, high (20%) in Cantabria (Fig. 3 and Table 5). Among patients harboring RASs, those with mutations which confer high resistance were: 62.0% (n = 31/50) to daclatasvir, 52.0% (n = 26/50) to ledipasvir, 50.0% (n = 25/50) to ombitasvir, 16.0% (n = 8/50) to elbasvir, 16.0% (n = 8/50) to velpatasvir and 4.0% (n = 2/50) to pibrentasvir; 64.0% (n = 32/50) of patients harbored RASs conferring resistance to more than 1 DAA. Eleven subjects had RASs to velpatasvir and one to pibrentasvir that were not likely to confer a clinically significant impact.

Spatial epidemiology of natural polymorphisms at the HCV NS5A gene associated with resistance to NS5A inhibitors among patients infected with HCV genotype 1a in Spain, (A) total prevalence of the five resistance mutations under analysis in the 17 Spanish Autonomous Communities and in the city of Ceuta; (B) prevalence of K24R (C) prevalence of M28A/T/V; (D) prevalence of Q30E/H/R; (E) prevalence of L31M; (F) prevalence of H58D; (G) prevalence of Y93C/F/H/N.
Discussion
In this study we performed the first nationwide survey assessing the origin, diversity and spatiotemporal transmission dynamics of GT1a using sequences derived from DAA-treatment naïve patients. Phylogenetic analysis confirmed the presence of two divergent GT1a clades, clades I and II39. Consistent with our previous observations, clade II showed a prevalence more than fourfold higher than clade I13,26. These findings illustrate the distinct geographic distributions of these GT1a clades, since clade I is more common in the United States and both clades are identically represented in other European countries40,41. Clade distribution differed among groups of patients, with clade II being more prevalent among HCV-monoinfected patients than among those co-infected with HIV. These differences confirm those reported in our previous nationwide cross-sectional surveys of NS5A RASs to elbasvir13 and of NS3 RASs in Spain (2014–2015)42, and may be related to differences in mode of transmission of HIV and HCV, such as risky sexual behaviors or parental use of drugs43,44.
There were major differences in the origin and spatiotemporal distribution of both clades. The origin of clade II in Spain dates back to the beginning of the twentieth century, preceding clade I by at least 40 years. Clade II transitioned from endemic in the Basque Country to epidemic in Spain. Importantly, the dispersion of clade II from the Basque Country to Andalusia and Madrid coincides with the propagation and spread of clade I from these autonomous communities to the rest of Spain. Overall, Andalusia and Madrid were the most important sources of clade I dissemination in Spain while the Basque Country was the major source of clade II dissemination. De Luca et al.40 reported an earlier origin of clade I, 1966 (95% HPD, 1952–1972) compared with clade II, 1975 (95% HPD, 1961–1989) using time-scaled phylogeny applied to NS3 sequences from Italian and German patients and additional sequences from the Los Alamos National Laboratory HCV sequence database with sampling dates from 1977 to 2013 and originated mainly from US but also from other countries such as France and Spain. Overall, these results illustrate fundamental differences in the epidemic behavior of these two GT1a clades both in Spain and in the rest of Europe.
Importantly, our data also shows that clade II epidemic is now declining whereas clade I epidemic has reached equilibrium. The impact of this new clade I epidemic in Spain may be seen on the level of NS3 Q80K prevalence that was reported to be mainly associated with clade I40,41,42,45 and that is associated with reduced treatment response to the macrocyclic protease inhibitor simeprevir46,47.
HCV and HIV share common transmission risk behaviors48. In Spain, HCV/HIV coinfection has been historically associated with parenteral drug use. Data from the Spanish National Epidemiological Bulleting show that HIV was mainly acquired through parental drug use since the first AIDS cases were reported in 198249,50, while men having sex with men have accounted for most incident cases since the year 200051. Spain is among the eight European countries with high (> 15%) estimates of HCV/HIV coinfection52. The most prevalent HCV genotypes associated with parenteral drug use in Europe are GT1a and GT3a. GT1a dominates in Spain among people who inject drugs53,54 and HIV/HCV coinfected7,55. According to a recent observational analysis, from 1988 to 2015 in North-Eastern Spain there has been an increase in the prevalence of GT1a, GT3 and GT4 associated with male gender and parental use of drugs, currently the most prevalent route of infection, and a concomitant decline of GT1b, associated with transfusion or parenteral/nosocomial transmission56. In this context, it is likely that the initial spread of GT1a clade II during the first half of the twentieth century in Spain occurred when HIV had still not found its way to this country, which may clarify the higher prevalence of clade II among the HCV-monoinfected individuals of our cohort.
Regarding the baseline polymorphisms and RAS at the NS5A domain, in a combined analysis carried out in 22 countries of baseline samples from Phase II/III HCV trials, baseline NS5A RASs were present in 13% of 3,501 GT1a samples8. Reports of the frequency of naturally occurring NS5A RASs are still scarce in the clinical settings. A cross-sectional cohort of ~ 130 GT1a subjects enrolled in São Paulo (Brazil), reported a prevalence of RASs of 14.6% in HCV-monoinfected (M28V and Q30H/R) and 3.9% in HIV/HCV-coinfected subjects (M28T and Q30H/R)57.
A low prevalence of NS5A RASs to the six DAAs evaluated was observed in the present study. RASs were similarly distributed between monoinfected and coinfected patients and between the two GT1a clades in each group. However, when considering the regional data, RASs prevalence level showed a marked geographical variability across regions. In Spain few observational regional or sub-regional studies have been carried out, as resistance testing is only recommended for subjects who are being considered for therapy with grazoprevir/elbasvir. A cohort study including 53 G1a-patients from a main hospital in Madrid (Spain) found that 18.9% (n = 10) harbored at least one RAS58. Another study including 166 G1a-patients from a tertiary hospital in A Coruña (Galicia, Spain), found a low (5.5%) prevalence of NS5A RASs using population-based sequencing59. Differently from our analyses, this study performed in Galicia did not consider clinically relevant the RASs at position K24.
Thirteen regions showed an intermediate level (5–15%) of baseline RASs to DAAs and Cantabria showed a high level (20%). This suggests that the proportion of patients who will benefit from these treatments may vary according to the Spanish regions. In this sense, it will be important to monitor carefully the response to treatment with NS5A-specific DAAs in patients from these regions and in particular in patients bearing the RASs. The results from these studies will determine whether baseline testing is appropriate for candidates to these new drugs. Nevertheless, results from the observational HCV-TARGET cohort, in USA, indicate that longer treatments may surmount the negative impact of baseline RASs on SVR12 in the clinical setting60.
Effective treatment for HCV is currently available in Spain. However, there are several barriers left to disease eradication, including deficiencies in screening and diagnosis. Indeed, almost 50,000 of those living with HCV are still unaware of their infection61. Understanding the structural barriers that undermine the implementation of screening programs and the scale-up of diagnosis coverage is fundamental to assure linkage to care and implementation of the HCV care cascade62,63.
This study presents some limitations. A more comprehensive dataset of the clinical and epidemiological characteristics of enrolled patient may allow a deeper understanding of the results. Furthermore, the lack of follow-up limited the evaluation of the possible impact of NS5A RASs on the treatment efficacy. Finally, RASs were assessed by population sequencing instead of deep sequencing which may slightly underestimate RASs prevalence8.
In conclusion, current HCV-GT1a epidemic in Spain is mainly driven by clade I viruses which seem to have different spreading routes relative to clade II viruses. With the exception of Cantabria, viruses bearing RASs to NS5A-DAAs were present at low to intermediate level in HCV infected patients at baseline. Close surveillance of response to treatment with DAAs will be important.
References
World Health Organization. Global hepatitis report, 2017. http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1 (2017).
Borgia, S. M. et al. Identification of a novel hepatitis C virus genotype from Punjab, India: expanding classification of hepatitis C virus into 8 genotypes. J. Infect. Dis. 218, 1722–1729 (2018).
Smith, D. B. et al. International Committee on Taxonomy of Viruses (ICTV). HCV classification. A web resource to manage the classification and genotype and subtype assignments of hepatitis C virus. https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/56/hcv-classification.
Blach, S. et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol. Hepatol. 2, 161–176 (2017).
McNaughton, A. L. et al. Spatiotemporal reconstruction of the introduction of hepatitis C virus into Scotland and its subsequent regional transmission. J. Virol. 89, 11223–11232 (2015).
Acero Fernández, D. et al. Changes in the epidemiology and distribution of the hepatitis C virus genotypes in North-Eastern Spain over the last 35 years. Gastroenterol. Hepatol. 41, 2–11 (2018).
Berenguer, J. et al. Human immunodeficiency virus/hepatits C virus coinfection in Spain: elimination is feasible, but the burden of residual cirrhosis will be significant. Open Forum Infect. Dis. 5, 1–8 (2018).
Zeuzem, S. et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J. Hepatol. 66, 910–918 (2017).
Lavin, A. C. et al. Prevalence of hepatitis C in the Spanish population: the prevhep study (ethon cohort). J. Hepatol. 66, S272 (2017).
García-Fulgueiras, A. et al. Hepatitis C and hepatitis B-related mortality in Spain. Eur. J. Gastroenterol. Hepatol. 21, 895–901 (2009).
Aguilera, A. et al. Prevalence and distribution of hepatitis C virus genotypes in Spain during the 2000–2015 period (the GEHEP 005 study). J. Viral Hepat. 24, 725–732 (2017).
Secretaría General de Sanidad y Consumo Ministerio de Sanidad, Consumo y Bienestar Social. Plan Estratégico para el Abordaje de la Hepatitis C en el Sistema Nacional de Salud (PEAHC) (2018). https://doi.org/10.1016/j.ceramint.2013.10.089.
Palladino, C. et al. Low frequency of NS5A relevant resistance-associated substitutions to Elbasvir among hepatitis C virus genotype 1a in Spain: a cross-sectional study. Sci. Rep. 7, 3–8 (2017).
von Elm, E. et al. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Int. J. Surg. 61, 344–349 (2014).
Abbott Molecular Inc. Abbott RealTime HCV Genotype II (2013).
Leitner, T. et al. Analysis of heterogeneous viral populations by direct DNA sequencing. Biotechniques 15, 120–127 (1993).
Struck, D., Lawyer, G., Ternes, A. M., Schmit, J. C. & Bercoff, D. P. COMET: adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 42, 1–11 (2014).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Ragonnet-Cronin, M. et al. Automated analysis of phylogenetic clusters. BMC Bioinform. 14, 317 (2013).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Rambaut, A., Lam, T. T., Carvalho, L. M. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, 1–7 (2016).
Palladino, C. et al. Epidemic history of hepatitis C virus genotypes and subtypes in Portugal. Sci. Rep. 8, 12266 (2018).
Shapiro, B., Rambaut, A. & Drummond, A. J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23, 7–9 (2006).
Iles, J. C. et al. Phylogeography and epidemic history of hepatitis C virus genotype 4 in Africa. Virology 464–465, 233–243 (2014).
Pybus, O. G. et al. Genetic history of hepatitis C virus in East Asia. J. Virol. 83, 1071–1082 (2009).
Pybus, O. G., Drummond, A. J., Nakano, T., Robertson, B. H. & Rambaut, A. The epidemiology and iatrogenic transmission of hepatitis C virus in egypt: A Bayesian coalescent approach. Mol. Biol. Evol. 20, 381–387 (2003).
Gray, R. R. et al. The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol. Biol. 11, 131 (2011).
Xie, W., Lewis, P. O., Fan, Y., Kuo, L. & Chen, M.-H. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst. Biol. 60, 150–160 (2011).
Kass, R. E. & Raftery, A. E. Bayes factors. J. Am. Stat. Assoc. 90, 773–795 (1995).
Bielejec, F. et al. SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169 (2016).
Tableau. https://www.tableau.com/. https://www.webcitation.org/6w4yCTPjb. Accessed 1 Oct 2018.
Pawlotsky, J.-M. et al. EASL recommendations on treatment of hepatitis C 2018. J. Hepatol. 69, 461–511 (2018).
AASLD-IDSA HCV Guidance Panel. Hepatitis C guidance 2018 update: AASLD-IDSA recommendations for testing, managing, and treating hepatitis C virus infection. Clin. Infect. Dis. 67, 1477–1492 (2018).
Wyles, D. L. & Luetkemeyer, A. F. Understanding hepatitis C virus drug resistance: clinical implications for current and future regimens. Top Antivir. Med. 25, 103 (2017).
Pickett, B. E., Striker, R. & Lefkowitz, E. J. Evidence for separation of HCV subtype 1a into two distinct clades. J. Viral Hepat. 18, 608–618 (2011).
De Luca, A. et al. Two distinct hepatitis C virus genotype 1a clades have different geographical distribution and association with natural resistance to NS3 protease inhibitors. Open Forum Infect. Dis. 2, ofv043 (2015).
Id, G. S. et al. Hepatitis C virus genetic diversity by geographic region within genotype 1–6 subtypes among patients treated with glecaprevir and pibrentasvir. PLoS ONE 13, e0205186 (2018).
Jimenez-Sousa, M. A. et al. NS3 Resistance-Associated Variants (RAVs) in patients infected with HCV genotype 1a in Spain. PLoS ONE 11, e0163197 (2016).
Bradshaw, D., Matthews, G. & Danta, M. Sexually transmitted hepatitis C infection: the new epidemic in MSM?. Curr. Opin. Infect. Dis. 26, 66–72 (2013).
Parikh, N. et al. Substance Abuse, HIV-1 and Hepatitis. Curr. HIV Res. 10, 557–571 (2012).
Bagaglio, S. et al. Distribution of natural resistance to NS3 protease inhibitors in hepatitis C genotype 1a separated into clades 1 and 2 and in genotype 1b of HIV-infected patients. Clin. Microbiol. Infect. 22(386), e1-386.e3 (2016).
Bae, A. et al. Susceptibility of treatment-naive hepatitis C virus (HCV) clinical isolates to HCV protease inhibitors. Antimicrob. Agents Chemother. 54, 5288–5297 (2010).
Asselah, T. & Marcellin, P. Second-wave IFN-based triple therapy for HCV genotype 1 infection: simeprevir, faldaprevir and sofosbuvir. Liver Int. 34, 60–68 (2014).
Wiktor, S., Ford, N., Ball, A. & Hirnschall, G. HIV and HCV: distinct infections with important overlapping challenges. J. Int. AIDS Soc. 17, 1–2 (2014).
Lissen, E., Wichmann, I., Jimenez, J. M. & Andreu-Kern, F. AIDS in haemophilia patients in Spain. Lancet 321, 992–993 (1983).
Estruch, R. et al. Acquired immunodeficiency syndrome in a male residing in Barcelona. Med Clin 81, 645 (1983).
Sistema de información sobre nuevos diagnósticos de VIH. Registro nacional de casos de SIDA. Ministerio de Sanidad Servicios Sociales e Igualdad. Vigilancia Epidemiológica del VIH y SIDA en España 2017. Actualización 30 de junio de 2018. 1–43 (2018).
Wiessing, L. et al. Hepatitis C virus infection epidemiology among people who inject drugs in europe: a systematic review of data for scaling up treatment and prevention. PLoS ONE 9, e103345 (2014).
Serra, M. A., Rodríguez, F., Del Olmo, J. A., Escudero, A. & Rodrigo, J. M. Influence of age and date of infection on distribution of hepatitis C virus genotypes and fibrosis stage. J. Viral Hepat. 10, 183–188 (2003).
Touceda, S., Pereira, M. & Agulla, A. Prevalence of hepatitis C virus genotypes in the area of El Ferrol (La Coruña, Spain). Enferm. Infecc. Microbiol. Clin. 20, 200–204 (2002).
Rial-Crestelo, D. et al. Effectiveness of direct-acting antiviral therapy in patients with a HCV/HIV coinfection: a multicenter cohort study. Rev. Esp. Enfermedades Dig. 110, 35–42 (2018).
Acero Fernández, D. et al. Changes in the epidemiology and distribution of the hepatitis C virus genotypes in North-Eastern Spain over the last 35 years. Gastroenterol. Hepatol. 41, 2–11 (2018).
Malta, F. et al. Prevalence of naturally occurring NS5A resistance-associated substitutions in patients infected with hepatitis C virus subtype 1a, 1b, and 3a, co-infected or not with HIV in Brazil. BMC Infect. Dis. 17, 1–6 (2017).
Carrasco, I. et al. Baseline NS5A resistance associated substitutions may impair DAA response in real-world hepatitis C patients. J. Med. Virol. 90, 532–536 (2017).
Grandal, M. et al. Prevalence of NS5A resistance associated substitutions in patients with hepatitis C virus genotypes 1a and 3: Impact on current therapeutic strategies. J. Med. Virol. 90, 1094–1098 (2018).
Wang, G. P. et al. Prevalence and impact of baseline resistance-associated substitutions on the efficacy of ledipasvir/sofosbuvir or simeprevir/sofosbuvir against GT1 HCV infection. Sci. Rep. 8, 1–14 (2018).
Organización Médica Colegial de España. Día Mundial de la Hepatitis. (2018).
Parker, C. M., Hirsch, J. S., Hansen, H. B., Branas, C. & Martins, S. S. Facing opioids in the shadow of the HIV epidemic. N. Engl. J. Med. 380, 1–3 (2019).
Konerman, M. A. & Lok, A. S. F. Hepatitis C treatment and barriers to eradication. Clin. Transl. Gastroenterol. 7, e193–e196 (2016).
Acknowledgements
The authors thank patients and the Contributing members of the Spanish Group of Chronic Viral Hepatitis (SI1 Appendix) that have participated in this epidemiological survey and the Multidisciplinary Group of Viral Coinfection HIV/Hepatitis (COVIHEP). The Instituto de Salud Carlos III (ISCIII) provided financial support by the Grant (CP13/00098, PI15CIII/00031 and PI18CIII/00020 to VB). CP is financed by national funds via FCT through Norma Transitória—DL57/2016/CP1376/CT0004.
Author information
Authors and Affiliations
Contributions
V.B. conceived and designed the study; I.M.C., P.M.R., S.A.L., and V.B. acquired the data; S.R. contributed with the samples included in the study; C.P., I.J.E., P.B. and V.B. analyzed and interpreted data; C.P., I.J.E. and V.B. drafted the manuscript; N.T. made a critical revision of the manuscript; C.P, N.T. and V.B. approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palladino, C., Ezeonwumelu, I.J., Mate-Cano, I. et al. Epidemic history and baseline resistance to NS5A-specific direct acting drugs of hepatitis C virus in Spain. Sci Rep 10, 13024 (2020). https://doi.org/10.1038/s41598-020-69692-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69692-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
