
Abstract
There are no current treatments for autism, despite its high prevalence. Deletions of chromosome 16p11.2 dramatically increase risk for autism, suggesting that mice with an equivalent genetic rearrangement may offer a valuable model for the testing of novel classes of therapeutic drug. 16p11.2 deletion (16p11.2 DEL) mice and wild-type controls were assessed using an ethological approach, with 24 h monitoring of activity and social interaction of groups of mice in a home-cage environment. The ability of the excitation/inhibition modulator N-acetyl cysteine (NAC) and the 5-HT1B/1D/1F receptor agonist eletriptan to normalise the behavioural deficits observed was tested. 16p11.2 DEL mice exhibited largely normal behaviours, but, following the stress of an injection, showed hyperlocomotion, reduced sociability, and a strong anxiolytic phenotype. The hyperactivity and reduced sociability, but not the suppressed anxiety, were effectively attenuated by both NAC and eletriptan. The data suggest that 16p11.2 DEL mice show an autism-relevant phenotype that becomes overt after an acute stressor, emphasising the importance of gene-environmental interactions in phenotypic analysis. Further, they add to an emerging view that NAC, or 5-HT1B/1D/1F receptor agonist treatment, may be a promising strategy for further investigation as a future treatment.
Similar content being viewed by others

Introduction
Autism is extremely common, affecting males more than females (estimated to affect roughly 4/1,000 boys and 1/1,000 girls)1,2, and characterised by communication difficulties, social dysfunction, and repetitive or restricted behaviour patterns, with a high rate of comorbid conditions such as anxiety. There are no available drug treatments for autism. Development of improved treatments will only be enabled by increased understanding of the causes of the disease and how they impact on neurobiology, informed by better preclinical models of facets of the disease.
The genetic architecture of autism is complex3. While a large number of common sequence variations increase disease risk, each has only a very small effect individually, and it is the cumulative burden of a range of risk, and protective, gene variants that underlies the aetiological mechanisms ultimately resulting in the manifestation of the common, sporadic disease. However, it is now clear that very rare copy number variants (CNVs), where small numbers of genes are present in one or three, rather than two, copies, substantially increase disease risk. For example, carriers of the deletions of the 16p11.2 locus have dramatically increased risk of autism-spectrum disorders (ASD) and also intellectual disability (8–40x)4,5,6. Interestingly, a high proportion of carriers of the corresponding 16p11.2 duplication develop schizophrenia, suggesting that studying the neurobiological impact of CNVs at this locus may be particularly informative. The 16p11.2 deletion is one of the most powerful genetic risk factors for autism3.
Various drug classes have been tested in mouse models relevant to ASD, for efficacy in reversing behavioural deficits in social paradigms. These social paradigms typically involve placing unfamiliar pairs of rodents in a novel environment (e.g. open field or 3 chambered apparatus) and recording their interactions over a short timeframe7. These relatively simple, high-throughput tests involve separating an animal from its cage mates and placing it in an unfamiliar apparatus. This results in stress-associated social disruption, which in the case of genetically-modified animals may interact with the environmental stressor to reveal a phenotype. Hence the outcomes determined in this testing paradigm may reflect a gene-environmental interaction phenotype, rather than a basal phenotype caused by the model8. While this is not necessarily a confound to the disease-relevance of the model, it is important to dissociate the phenotypes that arise directly from the genetic manipulation and those that arise in response to gene-environment interactions, as this offers new insight into the basis of disease symptoms and aetiology, and is important in the context of drug validation and therapeutic relevance.
Abnormal phenotypes in mouse models relevant to ASD have reportedly been improved by statins9, Akt/mTOR inhibitors10, mGlu5 receptor negative allosteric modulators11, mGlu5 receptor positive allosteric modulators12,13, and GABA-B receptor agonists14,15,16. However, these findings have not translated successfully to clinical trials17,18,19; see also20. The reasons for this are unclear, but arguably are more likely to relate to the questionable translatability of the behavioural assays employed, rather than the construct validity of the genetic models.
In the specific case of mice engineered to reproduce the 16p11.2 deletion (16p11.2 DEL mice), behavioural deficits have reportedly been normalised by the GABA-B receptor agonist baclofen21, a mGlu5 receptor negative modulator22, a 5-HT2A receptor antagonist23 and a 5-HT1B/1D/1F agonist24. The possibility that serotonergic drugs might have therapeutic potential is strengthened by the report of decreased 5-HT turnover in 16p11.2 DEL mice23, and that syntenic 16p11.2 deletion restricted to serotonergic neurones in mice is reportedly sufficient to decrease sociability24. However, there is also considerable interest currently in the possibility that modifying the balance between glutamatergic excitation and GABAergic inhibition might be a productive strategy. Excitation/inhibition (E/I) balance is thought to be disturbed in autism25,26,27. Accumulating evidence suggests that it is also a feature of mouse models relevant to ASD, including 16p11.2 DEL mice28 and neurexin1 knockout mice (Hughes et al., in press). Recent studies have begun to investigate the behavioural effects of agents such as the E/I modulator N-acetyl cysteine (NAC). NAC is primarily an anti-oxidant, but also stimulates the system Xc- cysteine-glutamate antiporter on glial cells, increasing extrasynaptic glutamate29 and thereby facilitating stimulation of presynaptic mGlu2 receptors to inhibit glutamate release.
In this study, we assess the behaviour of 16p11.2 DEL mice in an ethological context, using an automated home cage monitoring system which permits group housed animals to be assessed simultaneously for social and locomotor behaviours, and where the impact of acute stress can be separated from baseline behavioural responses30,31,32,33. In addition, we assess the ability of NAC, as compared to the 5-HT1B/1D/1F agonist eletriptan, to ameliorate the behavioural impact of the 16p11.2 deletion.
Methods and materials
Animals
Male mice hemizygous for the 0.44-Mb region of mouse chromosome 7, syntenic to the human 16p11.2 deletion, were generated by Mills and colleagues30 (Jackson Laboratory stock No. 013128), and backcrossed onto a C57BL/6 N background to generate experimental mice. It is important to emphasise that, in contrast to some other reports using this genetic modification, the mice in this study have been back-crossed onto the C57BL/6 N background until they are effectively congenic. The 16p11.2 DEL mice, and littermate wild-type (WT) controls, were housed with litter mates in their home cages from weaning. This was to ensure a minimal impact from external environmental factors during the development period from weaning to adulthood which may have influenced their behaviour prior to exposure to the experimental stressor in adulthood. Animals were housed in cages of mixed or same-genotype as previous studies with this line have shown that a range of behaviours including social approach, same-sex social interactions, open field and anxiety-related behaviours were not affected by housing in mixed-genotype versus same-genotype cages34, 35. Animals were housed under standard conditions, with food and water ad libitum. All work was approved by the University of Glasgow Animal Welfare and Ethics Review Board (AWERB) and conducted in accordance with the UK Animals (Scientific Procedures) Act 1986.
Home cage monitoring
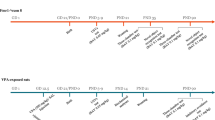
14 male mice (7 WT + 7 16p11.2 DEL) were housed in trios or pairs (2 cages of 2 16p11.2 DEL and 1 WT, 1 cage of 3 WT, 1 cage of 3 16p11.2 DEL, and 1 cage of 2 WT) under a reversed light/dark cycle (lights off at 10.00am). Genotypes and numbers in cages were imposed by the genotype/sex configuration of the original litters, to avoid fighting between unfamiliar males put together after weaning. Note that the presence of 2 rather than 3 mice in a cage does not affect locomotor or anxiety measures (e.g. Supplementary Fig. S1). Equally, there was no evidence that the behaviour of mixed genotypes in a cage differed from that of single genotypes (Supplementary Fig. S2). Under isofluorane anaesthesia, a radiofrequency identification (RFID) transponder was implanted subcutaneously into the lower left abdominal quadrant. Groups of 2–3 mice were then placed in Plexiglas IVC cages with Home Cage Analyser (Actual Analytics Ltd, UK)36,37 monitoring equipment. Mice were placed in the cages for 1.5 h prior to data acquisition to habituate. Recording then commenced at 10am and proceeded for 72 h. On days 3–5 of testing mice received the following treatments (within subjects counterbalanced design, i.p.): vehicle (50% PEG-400, 10% Solutol HS-15, 40% dH2O), NAC (150 mg/kg) and eletriptan (5 mg/kg). All drugs were administered intraperitoneally at an injection volume of 4 ml/kg, 15 min prior to the onset of the dark period (09:45) (Fig. 1). Measures included: total distance travelled (mm), total number of antenna transitions, thigmotaxis, time in centre region of cage, separation (mean Euclidean distance to closest cage-mate (mm)) and social isolation (time spent > 100 mm apart from other cage-mates (s)).

Schedule of home-cage monitoring and drug administration. Days are numbered from introduction to experimental home cage. Dark and light bars represent 12 h dark and light phases. Drugs (vehicle, NAC or eletriptan) were administered in a counterbalanced design on days 3–5, at onset of dark period.
Data analysis
For the measures of social behaviour (separation and isolation), data from pair-housed mice were excluded, as mice housed in pairs showed greater scores on these measures simply due to there being fewer mice in the cage. Statistical significance was assessed by ANOVA (Minitab), with prior Box-Cox normalisation where data deviated substantially from normality. Mann–Whitney tests were used for specific planned comparisons between groups of particular interest. For the main hypothesised effects, figures also show effect size estimation (estimationstats.com), with mean differences shown as Gardner-Altman estimation plots38,39, along with Bayes factors, estimated using JASP40, with default (Cauchy) priors. JASP was used to estimate Bayes factors for t-tests (WT mice: vehicle treatment versus 16p11.2 DEL mice: vehicle treatment) or one way repeated-measures ANOVA (16p11.2 DEL mice: vehicle treatment versus NAC treatment versus eletriptan treatment) with post-hoc testing. In all cases, Bayes factors proved to be robust against choice of priors (JASP). Bayes factor magnitudes were interpreted according to standard practice in life sciences research41.
Results
Impaired social functioning is a key feature of autism, but there are limitations in capturing the complexity and multidimensional nature of this domain in rodent models8, in part because standard paradigms involve elements of stress responsivity, as behaviour is monitored in a novel environment often between unfamiliar animals, over a limited time frame. We therefore monitored behaviours in group-housed mice in a home cage environment over 5 days33. Measures of locomotor activity and anxiety were monitored in parallel.
Basal locomotor and social activity (habituation, days 1–2)
Both WT and 16p11.2 DEL animals exhibited normal circadian activity whereby their active phase was during the dark period (Fig. 2A). Mice were significantly more active during the dark phase versus light phase (F(1, 27) = 34.50; p < 0.0001) but there was no genotype effect (p = 0.63) (Fig. 2B). This finding is in slight disagreement with previous authors: Horev et al.,30 reported mild hyperactivity in DEL animals when placed in novel environment and Arbogast et al.,31 reported increased ambulatory activity in DEL mice during the dark phase.

Locomotor activity and isolation from cage-mates during first 2 days. (A) Circadian pattern of LMA over days 1–2. Results are shown as mean + /− s.e.m. for 15 min time bins. (B) Total LMA shown over days 1 and 2 in dark and light phases. Difference between phases: (F(1, 27) = 34.50; p < 0.0001). (C) Circadian pattern of isolation over days 1–2. Results are shown as mean + /− s.e.m. for 15 min time bins. (D) Total isolation shown over days 1 and 2 in dark and light phases. Difference between phases: (F(1, 27) = 5.30; p = 0.03). Box plots show interquartile range with “Tukey” whiskers.
16p11.2 DEL mice showed few signs of social impairment under baseline conditions. Time spent isolated (> 100 mm from all cage-mates) was significantly reduced during the light versus dark phase (F(1, 27) = 5.31; p = 0.03) and this was expected, as mice tend to sleep together when the lights are on (Fig. 2C). There was no overall effect of genotype (p = 0.78) (Fig. 2D).
There was no evidence that the effects of being moved to the monitoring cage had any interaction with genotype. Both WT and 16p11.2 DEL mice were more active in the first hour after being moved to the monitoring cage, as compared to the equivalent first hour of dark phase on the second habituation day (Supplementary Fig. S3). However, there was no evidence that this, or any of the other behavioural measures, was affected by genotype (Supplementary Fig. S3).
Drug treatment (days 3–5)
The lack of previously reported hyperlocomotor and reduced sociability phenotypes, under home-cage monitoring conditions, prompted us to examine the response to an acute stressor (handling and injection).
Locomotor activity
Following injection of vehicle, the distance moved by 16p11.2 DEL mice was substantially greater than that of WT controls for 30–45 min, supporting a stress-induced hyperlocomotor phenotype in these animals (Fig. 3A). By the end of an hour, the increased activity had subsided back to control levels (Fig. 3A). There was no effect of genotype on activity 60–120 min after injection (Supplementary Fig. S4). The elevated activity immediately following injection in 16p11.2 DEL mice was almost completely suppressed when the injection contained either NAC or eletriptan (Fig. 3A–D). Neither NAC nor eletriptan injection modified locomotor activity significantly in WT mice.

Locomotor activity (distance moved) during first 60 min after injection at start of dark phase on days 3–5. (A) Data shown for 15 min time bins. Effect of genotype: (F(1, 167) = 0.02; p = 0.88); effect of drug treatment: (F(1, 167) = 6.49, p = 0.002; genotype x drug interaction: (F(1, 167) = 12.77, p < 0.001; (B)–(D) Locomotor activity over 60 min after injection on days 3–5. (B) Total distance moved: Effect of genotype: (F(1, 167) = 0.02; p = 0.88); effect of drug treatment: (F(1, 167) = 6.49, p = 0.002; genotype x drug interaction: (F(1, 167) = 12.77, p < 0.001. (C) The mean difference for total distance moved in 60 min following vehicle injection between WT and 16p11.2 DEL mice, plotted as a bootstrap sampling distribution. The Bayes factor for the alternative hypothesis (BF10) of a difference between the vehicle-treated groups, is also shown. (D) The mean difference for NAC compared to vehicle, and eletriptan compared to vehicle, groups, in 16p11.2 DEL mice only, are shown in the above Cumming estimation plot. The mean difference is plotted as a bootstrap sampling distribution. The Bayes factors (BF10) are also shown for one-way ANOVA of data from 16p11.2 DEL mice (left) (blue shading emphasises strong evidence for an overall effect of drug treatment), and for post-hoc tests of NAC vs vehicle (middle) and eletriptan vs vehicle (right). In all cases, Box plots show interquartile range with “Tukey” whiskers. For effect size plots, the mean difference is depicted as a dot; the 95% confidence interval is indicated by the ends of the vertical error bar. ~p < 0.05, ~~p < 0.01 vs corresponding vehicle group, same genotype, same time bin (post-hoc Tukey test).
When we analysed a different measure of locomotor activity—the number of transitions between different sectors of the home cage, the increased activity in the 16p11.2 DEL mice was less clear (Supplementary Fig. S5), but the effect of the drugs to restore behaviour towards the control condition was still evident (Supplementary Fig. S5).
We also analysed behaviours thought to be related to anxiety—thigmotaxis, and time spent in the centre region of the home cage. Following the injection of vehicle, 16p11.2 DEL mice showed a very clear anxiolytic response, as demonstrated by significantly decreased thigmotaxis, and increased total time in the centre of the cage (Fig. 4A,B,D,E). The inhibitory effect of the drugs was less clear here, with no strong evidence that they were able to normalise the responses (Fig. 4A,C,D,F). In addition, both drugs did not modify anxiety-like behaviour in WT mice.

Anxiety measures [thigmotaxis—(A)–(C)—and time in centre—(D)–(F)] during first 60 min after injection at start of dark phase. (A) Thigmotaxis: Effect of genotype: (F(1, 167) = 3.60; p = 0.06); effect of drug treatment: (F(1, 167) = 0.14, p = 0.87; genotype x drug interaction: (F(1, 167) = 2.29, p = 0.11; ## p = 0.003 vs corresponding WT group (one-sided Mann Whitney test). (B) The mean difference for thigmotaxis in 60 min following vehicle injection between WT and 16p11.2 DEL mice, plotted as a bootstrap sampling distribution. The Bayes factor for the alternative hypothesis (BF10) of a difference between the vehicle-treated groups, is also shown. Green shading emphasises very strong evidence for an effect of genotype. (C) The mean difference in thigmotaxis for NAC compared to vehicle, and eletriptan compared to vehicle, groups, in 16p11.2 DEL mice only, are shown in the above Cumming estimation plot. The mean difference is plotted as a bootstrap sampling distribution. The Bayes factors (BF10) are also shown for one-way ANOVA of data from 16p11.2 DEL mice (left), and for post-hoc tests of NAC vs vehicle (middle) and eletriptan vs vehicle (right). (D) Total time in centre zone: Effect of genotype: (F(1, 167) = 3.65; p = 0.058); effect of drug treatment: (F(1, 167) = 1.26, p = 0.29; genotype x drug interaction: (F(1, 167) = 3.38, p = 0.037; ## p = 0.003 vs corresponding vehicle group, same genotype (one-sided Mann Whitney test). (E) The mean difference for time in centre in 60 min following vehicle injection between WT and 16p11.2 DEL mice, plotted on the right as a bootstrap sampling distribution. The Bayes factor for the alternative hypothesis (BF10), of a difference between the vehicle-treated groups, is also shown. Green shading emphasises very strong evidence for an effect of genotype. (F) The mean difference for time in centre, for NAC compared to vehicle, and eletriptan compared to vehicle, groups, in 16p11.2 DEL mice only, are shown in the above Cumming estimation plot. The mean difference is plotted as a bootstrap sampling distribution. The Bayes factors (BF10) are also shown for one-way ANOVA of data from 16p11.2 DEL mice (left),and for post-hoc tests of NAC vs vehicle (middle) and eletriptan vs vehicle (right). In all cases, Box plots show interquartile range with “Tukey” whiskers. For effect size plots, the mean difference is depicted as a dot; the 95% confidence interval is indicated by the ends of the vertical error bar.
When we analysed measures of sociability—the mean separation between mice in the home cage, and the time spent isolated from other mice in the home cage, there was a trend towards reduced sociability in the 16p11.2 mice. The effect of the drugs to restore behaviour towards the control condition was still evident (Fig. 5). Interestingly, eletriptan showed a trend towards increasing isolation in WT mice, that was significant in the 2nd hour after injection (Supplementary Fig. S4).

Social interaction measures [distance of separation—(A)–(C)—and time spent isolated—(D)–(F)] during first 60 min after injection at start of dark phase. (A) Total separation: Effect of genotype: (F(1, 143) = 0.18; p = 0.67); effect of drug treatment: (F(1, 143) = 0.91, p = 0.41; genotype x drug interaction: (F(1, 143) = 3.14, p = 0.047. (B) The mean difference for separation in 60 min following vehicle injection between WT and 16p11.2 DEL mice, plotted as a bootstrap sampling distribution. The Bayes factor for the alternative hypothesis (BF10) of a difference between the vehicle-treated groups, is also shown. (C) The mean difference for separation for NAC compared to vehicle, and eletriptan compared to vehicle, groups, in 16p11.2 DEL mice only, are shown in the above Cumming estimation plot. The mean difference is plotted as a bootstrap sampling distribution. The Bayes factors (BF10) are also shown for one-way ANOVA of data from 16p11.2 DEL mice (left), and for post-hoc tests of NAC vs vehicle (middle) and eletriptan vs vehicle (right); red shading emphasises moderate evidence for an effect of NAC. (D) Time isolated: Effect of genotype: (F(1, 143) = 0.61; p = 0.44); effect of drug treatment: (F(1, 143) = 0.70, p = 0.50; genotype x drug interaction: (F(1, 143) = 6.96, p = 0.001; ~~p < 0.01 vs corresponding vehicle group, same genotype (post-hoc Tukey test). (E) The mean difference for time spent isolated in 60 min following vehicle injection between WT and 16p11.2 DEL mice, plotted on the right as a bootstrap sampling distribution. The Bayes factor for the alternative hypothesis (BF10), of a difference between the vehicle-treated groups, is also shown. (F) The mean difference for time isolated NAC compared to vehicle, and eletriptan compared to vehicle, groups, in 16p11.2 DEL mice only, are shown in the above Cumming estimation plot. The mean difference is plotted as a bootstrap sampling distribution. The Bayes factors (BF10) are also shown for one-way ANOVA of data from 16p11.2 DEL mice (left), and for post-hoc tests of NAC vs vehicle (middle) and eletriptan vs vehicle (right). In all cases, Box plots show interquartile range with “Tukey” whiskers. For effect size plots, the mean difference is depicted as a dot; the 95% confidence interval is indicated by the ends of the vertical error bar.
Discussion
A number of previous reports have documented hyperactivity, increased or reduced anxiety, and unchanged or reduced sociability, in 16p11.2 DEL mice21,30,31,42,43. In all cases, assessments have been made under conditions of some stress to the mice—with the tests involving experimenter handling, exposure to a novel environment, or injection, or social separation or exposure to unfamiliar conspecifics, depending on the task. Hence the resulting phenotype of the previous data potentially represents a combination of genetic and environmental factors. Here we delineate these factors by reporting the basal phenotypes in a relatively stress-free home-cage environment using a continuous monitoring system, and also the effect of an acute injection stressor, to parse these effects in the behavioural phenotypes seen in 16p11.2 DEL mice. In addition, we have characterised the ability of two drugs of interest with respect to future therapeutic options for autism—NAC and eletriptan—to normalise the stress-induced phenotypes observed in 16p11.2 DEL mice, showing dissociable effects on hyperactivity, anxiety-like behaviour and sociability. Both NAC and eletriptan are rapidly absorbed in rodents and other species44,45,46, allowing the assessment of their effects on the post-injection phenotypes observed. Both drugs are also completely cleared by 24 h after administration44,46,47, so no carry-over effects are expected in our counter-balanced repeated measures design.
The within subjects, counterbalanced, repeated measures design is a strength of our study, allowing reductions in sample size while maintaining statistical power. For example, a retrospective power analysis from the social isolation data obtained, using Glimmpse48, estimated a power of 0.83 to detect a genotype x drug interaction at p < 0.05 by ANOVA. Further, in accordance with recent and evolving recommendations38,39,49, we include information on the confidence intervals for the main effects, and also some preliminary Bayesian analysis. The use of Cauchy priors for the Bayesian analysis might be seen as over-conservative, considering the previous reports of hyperactivity and hyposociability phenotypes in 16p11.2 DEL mice21,30,31,50. While our results clearly align with these previous reports, due to the novel use of ethological behaviours in this study, we preferred to regard the results as entirely distinct from previous work, although in fact the Bayes factors obtained turned out to be relatively robust against varying the prior odds. For example, for the social measures in vehicle-injected mice (separation distance and isolation time), an informed prior based on existing literature with this genetic mutation30,31,43 (mean 1.0, std 0.307) yields BF10 values of 3.6 and 3.8 respectively (moderate evidence for a genotype effect).
We have examined the effect of an acute stressor in this study. It will be of great interest in future work to compare the effects of some of the widely-used chronic stress models on the phenotype of this and other mouse genetic models of aspects of ASD.
Comparison with previous research in 16p11.2 DEL mice
The original report of this strain demonstrated a transient increase in locomotor activity relative to controls, lasting for less than 2 h after transfer to a novel environment30. This seems broadly equivalent to our current observations. From testing in a novel environment and/or after an injection, others have observed rather subtle hyperlocomotion21,31,42,43, no obvious locomotor phenotype23,50, or decreased locomotor activity51 in 16p11.2 DEL mice. In another study, Portman et al.35 reported increased home cage activity and decreased activity in a novel environment. The reasons for this variability are not clear, although many of these studies used mice maintained on a mixed background, which inevitably leads to increased variability in complex behaviours. It may also be an indication that this phenotype is rather subtle. Our data suggest a primary role in the interaction between environmental stress and the 16p11.2 DEL genotype that may also contribute to the variability of these phenotypes under different environments. Interestingly, the corresponding mice with a duplication of the same region show a hypolocomotor phenotype, both in home cage and in a novel environment30,31,52. Hence if there is a gene dosage effect in this phenotype, the 16p11.2 DEL mice would be predicted to show elevated activity. We indeed detect increased locomotor activity after an acute stressor, although the effect is transient (Fig. 3). This emphasises the extent to which particular details of behavioral testing procedures (i.e. degree of environmental stressor) can influence the results obtained. Furthermore, the data highlight the fundamental need for the application of emerging behavioural testing strategies, such as those used in this study, that allow for the parsing of genetic and environmental factors in the context of determining translationally-relevant phenotypes in genetic mouse models relevant to neurodevelopmental disorders.
While we detect genotype effects on locomotion and anxiety measures, the less clear effects of genotype on sociability measures suggest that reduced sociability is a less overt phenotype associated with 16p11.2 deletion. Indeed, most of the previous studies failed to detect altered social behaviours in 16p11.2 DEL mice (using a novel environment, 3 chamber test)30,31,35,43. Other studies have detected social dysfunction, albeit using mice on a mixed background21,50. Our data are indicative of subtle social dysfunction that is revealed under conditions of acute stress. Importantly, assessing social interaction between littermates in the home cage environment allows us to characterise sociability in the ethological context of established social hierarchies that exist between mice rather than the sociable behaviour displayed in the context of a novel social interaction with an unfamiliar conspecific (as employed in the 3 chamber test). As mice usually display higher levels aggression in the context of novel social interactions, with lower levels in the context of established hierarchies in the home cage, the potentially confound of this stressor is removed by home cage monitoring of littermate sociability. Future studies will allow the application of both the home cage and 3 chamber social testing paradigms to assess the role of stress and social familiarity in the sociability deficits seen in 16p11.2 DEL, and in other mouse models relevant to autism. It will be of interest to investigate whether exposure to alternative /more sustained stressors produces a larger impact on the social deficits observed.
In terms of the anxiolytic phenotype, other studies of 16p11.2 DEL mice have reported decreased anxiety21,31,43, no change42,50 or increased anxiety51. Again this lack of consistency may at least partially reflect the use of mice on a mixed background in many cases, or the interaction between different stressors in the induction of this phenotype, as supported by our own findings.
Contribution of individual genes within the 16p11.2 region
There is a paucity of information concerning the influence of specific genes within the 16p11.2 CNV on behaviour. Kctd13 gene knockout in mice reportedly does not affect locomotion, anxiety or social behaviour53. However, mice with a genetic deletion specifically of the Taok2 gene exhibit hyperactivity and reduced anxiety in a novel environment54. Hence it seems likely that Taok2 gene haploinsufficiency contributes to these phenotypes observed in the current study, especially since reduced JNK signalling (the downstream target of TAOK2) also decreases anxiety and increases locomotion55,56. Increased locomotor activity is seen in Mapk3 knockout mice57,58, so decreased ERK1 activity due to hemi-deletion of the Mapk3 gene in 16p11.2 DEL mice may also contribute to the hyperlocomotor phenotype.
Restorative effects of NAC
Perturbed E/I balance is thought to be a core neurobiological feature of autism25,26,27, detectable also in various genetic mouse models of autism28. Indeed, optogenetic correction of E/I balance restores normal locomotor and social behaviour in CNTNAP2 knockout mice, another mouse genetic model of aspects of autism59. In vitro studies suggest increased E/I ratios in hippocampus from 16p11.2 DEL mice60, as do in vivo studies in cerebral cortex28. Our data illustrate an ability of acute NAC administration, at a dose previously shown to reduce CNS glutamate levels61, to attenuate the increased locomotor response to injection seen in the 16p11.2 DEL mice. This is quite a striking result, and may reflect a strong link between E/I balance and locomotor activity, since system Xc–deficient mice (system Xc- being the target of NAC) show abnormal locomotion62. While a trend for NAC to attenuate the reduced sociability of 16p11.2 mice was observed, no effect of NAC was detected on the reduced anxiety-like behaviour manifest by these mice. A previous study reported that the same dose of NAC in mice decreases locomotion, and increases anxiety-like behaviour (decreased centre time in open field)61, while Xc–deficient mice show reduced anxiety, as reflected in increased centre time in the open field arena62. The lack of effect of NAC on anxiety measures in 16p11.2 DEL mice is slightly surprising in that context. It may be that the mechanisms underlying the reduced anxiety, which is not considered a core symptom of ASD, are distinct from those underlying the reduced sociability and the increased locomotor activity. Alternatively, the beneficial effects of NAC may not be due to lowering of glutamatergic activity. Abnormal ERK and CREB activity is a feature of many autism-related mouse models, and also of some patients63,64,65, and NAC can modulate this pathway effectively66, in addition to its well-known anti-oxidant properties.
Whatever the mechanisms involved, our data support further investigation of NAC in preclinical models of autism. Small scale clinical studies have noted some improvements in individuals with ASD with NAC, including potential effects on stereotypies and social cognition67,68.
Restorative effects of eletriptan
Panzini et al. reported evidence for decreased 5-HT turnover in the CNS of 16p11.2 DEL mice23, suggesting that serotonergic hypofunction may contribute to the phenotypes seen in these animals. Therefore we tested the ability of the 5-HT1B/D/F agonist to restore these deficits. We found that eletriptan treatment effectively reversed the stress-induced hyperactivity seen in 16p11.2 DEL mice with no effect on the stress-induced reduced anxiety-like and sociability deficits seen in these animals.
5-HT1B agonists increase mouse locomotor activity (although locomotion is essentially normal in 5-HT1B receptor knockout mice)69. We saw a trend for eletriptan to increase locomotion in WT mice (Fig. 3A). Our evidence that eletriptan could suppress the hyperlocomotion observed after injection in the 16p11.2 DEL mice was clear. In view of the tendency for 5-HT1B agonists to enhance locomotion in WT mice, this is more likely to reflect an action of the drug on 5-HT1D or 5-HT1F receptors, neither of which are well-studied in terms of their behavioural effects in rodents.
5-HT1B knockout mice do however show reduced thigmotaxis in an open field69. Consistent with this, 5-HT1B/1D agonists are generally considered to be anxiogenic, so the greatest efficacy of eletriptan was expected to be seen in the anxiolytic phenotype. However, we saw little evidence for an anxiogenic effect of eletriptan in WT mice (Fig. 4A,B), or of an ability to normalise the anxiolytic phenotype observed after an injection in the 16p11.2 DEL mice, indicating a more complex role for eletriptan in anxiogenesis.
It was interesting to note that eletriptan increased social isolation in WT mice in the 2nd hour after injection (Supplementary Fig. S4)—a time when eletriptan will still be present in the CNS44. This is to some extent expected, as 5-HT1B/1D agonists increase social fear in rodents and humans70,71, but the effect is not observed in 16p11.2 DEL mice. Syntenic 16p11.2 deletion restricted to serotonergic neurones in mice reportedly decreases sociability—an effect that can be rescued by stimulation of 5-HT1B receptors24. The present finding of an ability of electriptan to alleviate a social behavioural deficit in 16p11.2 DEL mice suggests further investigation of 5-HT subtype specific agonists in ASD is warranted.
Conclusions
Clinically, the appearance and exacerbation of autistic symptoms are associated with acute or chronic stress72,73,74. An acute stress revealed locomotor, anxiolytic and impaired social behaviours in 16p11.2 DEL mice, that were not present under low stress conditions, suggesting that environmental factors such as stress interact with the genetic variant to reveal a disorder-relevant phenotype. Autistic subjects however tend to be hyperanxious rather than the opposite75, so the translational significance of the anxiolytic response seen in 16p11.2 DEL mice is unclear. NAC and eletriptan showed intriguing efficacy to attenuate the more disease-relevant responses. Our data support evolving concepts that the correction of an unbalanced relationship between excitation and inhibition in the CNS, or, most strikingly, stimulation of 5-HT1B/1D/1F receptors to counteract serotonergic hypofunction, could be important future therapeutic strategies. In particular these data are important in a translational context given that drug-effectiveness was revealed in conditions where a stressor impacted on the behavioural outcome.
References
Taylor, B., Jick, H. & MacLaughlin, D. Prevalence and incidence rates of autism in the UK: time trend from 2004–2010 in children aged 8 years. BMJ Open 3, e003219. https://doi.org/10.1136/bmjopen-2013-003219 (2013).
Sun, X. et al. Autism prevalence in China is comparable to Western prevalence. Mol. Autism 10, 7. https://doi.org/10.1186/s13229-018-0246-0 (2019).
Coe, B. P., Girirajan, S. & Eichler, E. E. The genetic variability and commonality of neurodevelopmental disease. Am. J. Med. Gen. C Semin. Med. Genet. 160, 118–129. https://doi.org/10.1002/ajmg.c.31327 (2012).
Weiss, L. A. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. https://doi.org/10.1056/NEJMoa075974 (2008).
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 43, 838–846. https://doi.org/10.1038/ng.909 (2011).
Stefansson, H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366. https://doi.org/10.1038/nature12818 (2014).
Moy, S. S. et al. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res. 176, 4–20. https://doi.org/10.1016/j.bbr.2006.07.030 (2007).
Millan, M. J. & Bales, K. L. Towards improved animal models for evaluating social cognition and its disruption in schizophrenia: the CNTRICS initiative. Neurosci. Biobehav. Rev. 37, 2166–2180. https://doi.org/10.1016/j.neubiorev.2013.09.012 (2013).
Osterweil, E. K. et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. https://doi.org/10.1016/j.neuron.2012.01.034 (2013).
Xing, X. et al. Suppression of Akt-mTOR pathway rescued the social behavior in Cntnap2-deficient mice. Sci. Rep. 9, 3041. https://doi.org/10.1038/s41598-019-39434-5 (2019).
Silverman, J. L. et al. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 4, 131–151. https://doi.org/10.1126/scitranslmed.3003501 (2012).
Vicidomini, C. et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22, 689–702. https://doi.org/10.1038/mp.2016.30 (2017).
Gogliotti, R. G. et al. mGlu5 positive allosteric modulation normalizes synaptic plasticity defects and motor phenotypes in a mouse model of Rett syndrome. Hum. Mol. Genet. 25, 1990–2004. https://doi.org/10.1093/hmg/ddw074 (2016).
Silverman, J. L. et al. GABAB receptor agonist r-baclofen reverses social deficits and reduces repetitive behavior in two mouse models of autism. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 40, 2228–2239. https://doi.org/10.1038/npp.2015.66 (2015).
Sinclair, D. et al. GABA-B agonist baclofen normalizes auditory-evoked neural oscillations and behavioral deficits in the Fmr1 knockout mouse model of fragile X syndrome. eNeuro. https://doi.org/10.1523/eneuro.0380-16.2017 (2017).
Henderson, C. et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128. https://doi.org/10.1126/scitranslmed.3004218 (2012).
Berry-Kravis, E. M. et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 17, 280–299. https://doi.org/10.1038/nrd.2017.221 (2018).
Overwater, I. E. et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 93, e200–e209. https://doi.org/10.1212/wnl.0000000000007749 (2019).
Youssef, E. A. et al. Effect of the mGluR5-NAM basimglurant on behavior in adolescents and adults with fragile X syndrome in a randomized, double-blind, placebo-controlled trial: FragXis phase 2 results. Neuropsychopharmacology 43, 503–512. https://doi.org/10.1038/npp.2017.177 (2018).
Veenstra-VanderWeele, J. et al. Arbaclofen in children and adolescents with autism spectrum disorder: a randomized, controlled, phase 2 trial. Neuropsychopharmacology 42, 1390–1398. https://doi.org/10.1038/npp.2016.237 (2017).
Stoppel, L. J. et al. R-baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology https://doi.org/10.1038/npp.2017.236 (2017).
Tian, D. et al. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat. Neurosci. 18, 182–184. https://doi.org/10.1038/nn.3911 (2015).
Panzini, C. M., Ehlinger, D. G., Alchahin, A. M., Guo, Y. & Commons, K. G. 16p11.2 deletion syndrome mice perseverate with active coping response to acute stress—rescue by blocking 5-HT2A receptors. J. Neurochem. 143, 708–721. https://doi.org/10.1111/jnc.14227 (2017).
Walsh, J. J. et al. 5-HT release in nucleus accumbens rescues social deficits in mouse autism model. Nature 560, 589–594. https://doi.org/10.1038/s41586-018-0416-4 (2018).
Lee, E., Lee, J. & Kim, E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847. https://doi.org/10.1016/j.biopsych.2016.05.011 (2017).
Dickinson, A., Jones, M. & Milne, E. Measuring neural excitation and inhibition in autism: different approaches, different findings and different interpretations. Brain Res. 1648, 277–289. https://doi.org/10.1016/j.brainres.2016.07.011 (2016).
Horder, J. et al. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [(1)H]MRS study. Transl. Psychiatry 3, e279. https://doi.org/10.1038/tp.2013.53 (2013).
Antoine, M. W., Langberg, T., Schnepel, P. & Feldman, D. E. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101, 648–661. https://doi.org/10.1016/j.neuron.2018.12.026 (2019).
Baker, D. A. et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 6, 743–749. https://doi.org/10.1038/nn1069 (2003).
Horev, G. et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. 108, 17076–17081. https://doi.org/10.1073/pnas.1114042108 (2011).
Arbogast, T. et al. Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet. 12, e1005709. https://doi.org/10.1371/journal.pgen.1005709 (2016).
Pratt, J. A., Winchester, C., Dawson, N. & Morris, B. J. Advancing schizophrenia drug discovery: optimizing rodent models to bridge the translational gap. Nat. Rev. Drug Discov. 11, 560–579 (2012).
Mitchell, E. J., Brett, R. R., Armstrong, J. D., Sillito, R. R. & Pratt, J. A. Temporal dissociation of phencyclidine-induced locomotor and social alterations in rats using an automated homecage monitoring system: implications for the 3Rs and preclinical drug discovery. J. Psychopharm. In press (2020).
Yang, M., Lewis, F., Foley, G. & Crawley, J. N. In tribute to Bob Blanchard: divergent behavioral phenotypes of 16p11.2 deletion mice reared in same-genotype versus mixed-genotype cages. Physiol. Behav. 146, 16–27. https://doi.org/10.1016/j.physbeh.2015.04.023 (2015).
Portmann, T. et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. 7, 1077–1092 (2014).
Bains, R. S. et al. Analysis of individual mouse activity in group housed animals of different inbred strains using a novel automated home cage analysis system. Front. Behav. Neurosci. https://doi.org/10.3389/fnbeh.2016.00106 (2016).
Bains, R. S. et al. Assessing mouse behaviour throughout the light/dark cycle using automated in-cage analysis tools. J. Neurosci. Methods 300, 37–47. https://doi.org/10.1016/j.jneumeth.2017.04.014 (2018).
Calin-Jageman, R. J. & Cumming, G. Estimation for better inference in neuroscience. eNeuro https://doi.org/10.1523/eneuro.0205-19.2019 (2019).
Ho, J., Tumkaya, T., Aryal, S., Choi, H. & Claridge-Chang, A. Moving beyond P values: data analysis with estimation graphics. Nat. Methods 16, 565–566. https://doi.org/10.1038/s41592-019-0470-3 (2019).
Quintana, D. S. & Williams, D. R. Bayesian alternatives for common null-hypothesis significance tests in psychiatry: a non-technical guide using JASP. BMC Psychiatry 18, 178. https://doi.org/10.1186/s12888-018-1761-4 (2018).
Lee, M. D. & Wagenmakers, E. J. Bayesian Cognitive Modeling: A Practical Course (Cambridge University Press, Cambridge, 2013).
Angelakos, C. C. et al. Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res. Off. J. Int. Soc. Autism Res. 10, 572–584. https://doi.org/10.1002/aur.1707 (2017).
Brunner, D. et al. Comprehensive analysis of the 16p11.2 deletion and null Cntnap2 mouse models of autism spectrum disorder. PLoS ONE 10, e0134572. https://doi.org/10.1371/journal.pone.0134572 (2015).
Patel, H. et al. Differential pharmacokinetic drug–drug interaction potential of eletriptan between oral and subcutaneous routes. Xenobiotica 49, 1202–1208. https://doi.org/10.1080/00498254.2018.1540805 (2019).
Zhou, J. et al. Intravenous administration of stable-labeled N-acetylcysteine demonstrates an indirect mechanism for boosting glutathione and improving redox status. J. Pharm. Sci. 104, 2619–2626. https://doi.org/10.1002/jps.24482 (2015).
Buur, J. L., Diniz, P. P., Roderick, K. V., KuKanich, B. & Tegzes, J. H. Pharmacokinetics of N-acetylcysteine after oral and intravenous administration to healthy cats. Am. J. Vet. Res. 74, 290–293. https://doi.org/10.2460/ajvr.74.2.290 (2013).
Hematheerthani, N., Siva Reddy, C., Rani, A. P. & Kumar, P. S. Effect of quercetin on the pharmacokinetic profile of eletriptan, a cyp3a substrate, in rat model. Int. J. Pharm. Sci. Res. 11, 907–915 (2020).
Kreidler, S. M. et al. GLIMMPSE: online power computation for linear models with and without a baseline covariate. J. Stat. Softw. 54, i10. https://doi.org/10.18637/jss.v054.i10 (2013).
Halsey, L. G., Curran-Everett, D., Vowler, S. L. & Drummond, G. B. The fickle P value generates irreproducible results. Nat. Methods 12, 179. https://doi.org/10.1038/nmeth.3288 (2015).
Yang, M. et al. 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res. https://doi.org/10.1002/aur.1465 (2015).
Pucilowska, J. et al. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 35, 3190–3200. https://doi.org/10.1523/jneurosci.4864-13.2015 (2015).
Bristow, G. C. et al. 16p11 duplication disrupts hippocampal-orbitofrontal-amygdala connectivity, revealing a neural circuit endophenotype for schizophrenia. Cell Rep. 31, 107536. https://doi.org/10.1016/j.celrep.2020.107536 (2020).
Arbogast, T. et al. Kctd13-deficient mice display short-term memory impairment and sex-dependent genetic interactions. Hum. Mol. Genet. 28, 1474–1486. https://doi.org/10.1093/hmg/ddy436 (2018).
Richter, M. et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry https://doi.org/10.1038/s41380-018-0025-5 (2018).
Mohammad, H. et al. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol. Psychiatry 23, 362–374. https://doi.org/10.1038/mp.2016.203 (2018).
Stefanoska, K. et al. Neuronal MAP kinase p38α inhibits c-Jun N-terminal kinase to modulate anxiety-related behaviour. Sci. Rep. 8, 14296. https://doi.org/10.1038/s41598-018-32592-y (2018).
Selcher, J. C., Nekrasova, T., Paylor, R., Landreth, G. E. & Sweatt, J. D. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn. Mem. 8, 11–19 (2001).
Mazzucchelli, C. & Brambilla, R. Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell Mol. Life Sci. 57, 604–611 (2000).
Selimbeyoglu, A. et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aah6733 (2017).
Lu, H.-C., Mills, A. A. & Tian, D. Altered synaptic transmission and maturation of hippocampal CA1 neurons in a mouse model of human chr16p11.2 microdeletion. J. Neurophysiol. 119, 1005–1018. https://doi.org/10.1152/jn.00306.2017 (2018).
Durieux, A. M. S. et al. Targeting glia with N-acetylcysteine modulates brain glutamate and behaviors relevant to neurodevelopmental disorders in C57BL/6J Mice. Front. Behav. Neurosci. https://doi.org/10.3389/fnbeh.2015.00343 (2015).
Bentea, E. et al. Absence of system xc- in mice decreases anxiety and depressive-like behavior without affecting sensorimotor function or spatial vision. Prog. Neuropsychopharmacol. Biol. Psychiatry 59, 49–58. https://doi.org/10.1016/j.pnpbp.2015.01.010 (2015).
Yoo, H. J. et al. Whole exome sequencing for a patient with Rubinstein–Taybi syndrome reveals de novo variants besides an overt CREBBP mutation. Int. J. Mol. Sci. 16, 5697–5713. https://doi.org/10.3390/ijms16035697 (2015).
Guilding, C., Stone, T. W. & Morris, B. J. Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur. J. Neurosci. 25, 99–105 (2007).
Amal, H. et al. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry https://doi.org/10.1038/s41380-018-0113-6 (2018).
Sun, L. et al. N-acetylcysteine protects against apoptosis through modulation of group I metabotropic glutamate receptor activity. PLoS ONE 7, e32503. https://doi.org/10.1371/journal.pone.0032503 (2012).
Hardan, A. Y. et al. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol. Psychiatry 71, 956–961. https://doi.org/10.1016/j.biopsych.2012.01.014 (2012).
Ghanizadeh, A. & Moghimi-Sarani, E. A randomized double blind placebo controlled clinical trial of N-Acetylcysteine added to risperidone for treating autistic disorders. BMC Psychiatry 13, 196. https://doi.org/10.1186/1471-244x-13-196 (2013).
Malleret, G., Hen, R., Guillou, J. L., Segu, L. & Buhot, M. C. 5-HT1B receptor knock-out mice exhibit increased exploratory activity and enhanced spatial memory performance in the Morris water maze. J. Neurosci. 19, 6157–6168 (1999).
Bell, R., Donaldson, C. & Gracey, D. Differential effects of CGS 12066B and CP-94,253 on murine social and agonistic behaviour. Pharmacol. Biochem. Behav. 52, 7–16. https://doi.org/10.1016/0091-3057(95)00077-a (1995).
de Rezende, M. G., Garcia-Leal, C., Graeff, F. G. & Del-Ben, C. M. The 5-HT1D/1B receptor agonist sumatriptan enhances fear of simulated speaking and reduces plasma levels of prolactin. J. Psychopharmacol. 27, 1124–1133. https://doi.org/10.1177/0269881112472560 (2013).
Fuld, S. Autism spectrum disorder: the impact of stressful and traumatic life events and implications for clinical practice. Clin. Soc. Work J. 46, 210–219. https://doi.org/10.1007/s10615-018-0649-6 (2018).
Stack, A. & Lucyshyn, J. Autism spectrum disorder and the experience of traumatic events: review of the current literature to inform modifications to a treatment model for children with autism. J. Autism Dev. Disord. 49, 1613–1625. https://doi.org/10.1007/s10803-018-3854-9 (2019).
Hoover, D. W. & Kaufman, J. Adverse childhood experiences in children with autism spectrum disorder. Curr. Opin. Psychiatry 31, 128–132. https://doi.org/10.1097/yco.0000000000000390 (2018).
White, S. W., Oswald, D., Ollendick, T. & Scahill, L. Anxiety in children and adolescents with autism spectrum disorders. Clin. Psychol. Rev. 29, 216–229. https://doi.org/10.1016/j.cpr.2009.01.003 (2009).
Acknowledgements
This research was supported by the Medical Research Council (UK), Grant MR/N012704/1. We are very grateful to John Craig for performing the genotyping for these studies.
Author information
Authors and Affiliations
Contributions
E.J.M. performed the experiments, conducted most of the data analysis, and contributed to writing the manuscript, with input to methodological optimisation, study design and interpretation of results from D.M.T., G.B., and R.L.O. B.J.M. performed some additional data analysis. J.A.P., N.D. and B.J.M. conceived the study, provided guidance on experimental design, and wrote most of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mitchell, E.J., Thomson, D.M., Openshaw, R.L. et al. Drug-responsive autism phenotypes in the 16p11.2 deletion mouse model: a central role for gene-environment interactions. Sci Rep 10, 12303 (2020). https://doi.org/10.1038/s41598-020-69130-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69130-8
This article is cited by
-
Severity of Autism Spectrum Disorder Symptoms Associated with de novo Variants and Pregnancy-Induced Hypertension
Journal of Autism and Developmental Disorders (2024)
-
16p11.2 deletion mice exhibit compromised fronto-temporal connectivity, GABAergic dysfunction, and enhanced attentional ability
Communications Biology (2023)
-
Genetic and environmental mouse models of autism reproduce the spectrum of the disease
Journal of Neural Transmission (2023)
-
The Potential of N-Acetyl-L-Cysteine (NAC) in the Treatment of Psychiatric Disorders
CNS Drugs (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
