
Abstract
We report the molecular basis of the largest Tunisian cohort with inherited retinal dystrophies (IRD) reported to date, identify disease-causing pathogenic variants and describe genotype–phenotype correlations. A subset of 26 families from a cohort of 73 families with clinical diagnosis of autosomal recessive IRD (AR-IRD) excluding Usher syndrome was analyzed by whole exome sequencing and autozygosity mapping. Causative pathogenic variants were identified in 50 families (68.4%), 42% of which were novel. The most prevalent pathogenic variants were observed in ABCA4 (14%) and RPE65, CRB1 and CERKL (8% each). 26 variants (8 novel and 18 known) in 19 genes were identified in 26 families (14 missense substitutions, 5 deletions, 4 nonsense pathogenic variants and 3 splice site variants), with further allelic heterogeneity arising from different pathogenic variants in the same gene. The most common phenotype in our cohort is retinitis pigmentosa (23%) and cone rod dystrophy (23%) followed by Leber congenital amaurosis (19.2%). We report the association of new disease phenotypes. This research was carried out in Tunisian patients with IRD in order to delineate the genetic population architecture.
Similar content being viewed by others


Introduction
Inherited retinal dystrophies (IRD) are a large group of inherited eye disorders which affect photoreceptors and lead to visual impairment. The prevalence of IRD has been estimated in one case for each 2,500–7,000 persons among the general population1. IRDs are further classified into as retinitis pigmentosa (RP), cone rod dystrophy (CRD), and cone dystrophy (CD). Initial symptoms include night blindness, photophobia and/or progressive loss of the peripheral vision2. Clinical symptoms vary across different IRD subtypes and different disease genes.
Genetically, different IRD can be caused by pathogenic variants in more than 300 genes, over 100 of these have been linked to syndromic IRD (https://sph.uth.edu/retnet/), displaying three form of inheritance: autosomal dominant (AD), autosomal recessive (AR) and X-linked (XL). Occasionally, mitochondrial variants and digenic inheritance have been identified3.
Molecular genetics is essential for gene-based treatment, clarify diagnoses and to direct appropriate counseling. However, it is currently unknown how many genes are involved in IRDs, and even by using the latest next generation sequencing (NGS) techniques, pathogenic and likely pathogenic variants are identified only in 50% to 75% of patients4. Due to the relatively high frequency of consanguinity in Tunisia, ranging from 20 to 40%, this population could contribute to the identification of new genes responsible for AR-IRD5. To identify causative pathogenic variants in a large cohort of families diagnosed with nonsyndromic (24/26) or syndromic (2/26) AR-IRD, homozygosity mapping of known IRD loci was carried out. Pathogenic variant screening of the identified genes in all 74 families gave an overall idea about the most frequent genes and variants in patients with IRD in Tunisia. We believe it is essential to combine molecular and clinical data to diagnose IRD patients, especially with the emergence of therapeutic options.
Results
Clinical diagnosis and pathogenic and likely pathogenic (P/LP) variants identified
50 affected and 48 unaffected relatives belonging to 26 families with suspected recessive inheritance were included. Pathogenic variants are listed in Table 1. A total of 26 causative P/LP variants in 19 genes were identified in 26 families, including 14 missense substitutions (53.9%), 5 deletions (19.2%), 4 nonsense P/LP variants (15.4%) and 3 splice site pathogenic variants (11.5%). 8 (30.8%) P/LP variants were novel, while the remaining 18 (69.2%) were reported previously. 96.2% of all P/LP variants were homozygous, only one family carried a heterozygous pathogenic variant in PRPH2 in family 17 (F17) (3.8%). Segregation of the mutant allele was confirmed in the majority of the families. For missense variant the substituted amino acid residues are highly conserved across species, and in silico pathogenicity prediction tools PolyPhen2 and SIFT predicted these changes to be deleterious.
After molecular testing, all patients were re-evaluated to monitor whether their retinal phenotype was similar to previously described retinopathies caused by pathogenic variants in the same gene. In case of discrepancy, the respective phenotypes were considered as potential novel genotype–phenotype correlations.
Information for each patient is presented in Table 2. Below we present the families with novel P/LP variants.
LCA (Fig. 1, 2)

Clinical features of LCA patients; A F1 LE fundus of patient IV.7. B F2 RE fundus of patient II.2. C , D F5 fundus of RLE of patient II.1. E F5 fluorescein angiography of LE of patient III.1 showing noevascular membrane. F F4 LE fundus of patient III.1. G F4 OCT of LE of patient III.1.

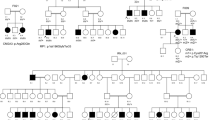
Segregation analysis of disease causing variants in the families with IRD. Affected individuals are indicated with filled symbols (blue), whereas unaffected relatives are indicated by open symbols. +: wild type allele; −: pathogenic variant.
Clinical data of patients from families F1, F2, F3, F4 and F5 revealed an age of onset of disease from birth with nystagmus and photophobia. BCVA was limited to light perception. Patient in F3 was monophthalmic of the right eye (RE) (Fig. 1).
F1-RPGRIP1 (Fig. 1A)
One novel homozygous deletion (NM_020366: c.3113-3114delCT, p.T1038Rfs*8) in RPGRIP1 was identified in F1 with 2 affected members. The deletion of the CT in exon 10 results in a frameshift with a premature stop codon at position amino acid 1046.
F2-GUCY2D (Fig. 1B)
The second novel homozygous likely pathogenic variant (NM_000180: c.2660 T > G, p.V887G) in GUCY2D was found in F2 in one affected individual. This likely pathogenic variant has previously been published in the validation of a targeted array but no phenotype was presented6.
F5-CRB1 (Fig. 1C, 1D, 1E)
The novel homozygous likely pathogenic variant (NM_201253.2: c.3542 + 1G > A, p.?) in CRB1 was found in F5 in one patient (Fig. 2). Fundus appearance in this proband included all clinical characteristics of CRB1 pathogenic variant.
F4-IQCB1
The homozygous likely pathogenic variant (NM_001023570: c.994C > T, p.R332*) in IQCB1 was found in F4 in two affected children (Fig. 1F and G). Fundus appearance in both probands revealed normal structure. Renal function and ultrasound were normal.
CRD patients (Fig. 2,3)

Clinical features of CRD patients; A F6, RE fundus of patient II.1. B F7, RE of patient II.1. C, D, E, F and G Clinical features of patients from F8. C: Fundus imaging of RE of index patient III.2. D FAF showing central macular hypoFAF surrounded by ring of hyper FAF, small areas of hypoFAF in the mid-periphery. E SS-OCT showing vitreo-retinal traction with macular hole. F fundus photo of the right eye of patient III.3. G SS-OCT showing diffuse chorio-retinal atrophy. H, I, J, K, L, M, N, O and P Clinical features of patients from F9. H Fundus imaging of LE of patient IV.4. I FAF showing central macular hypoFAF surrounded by ring of hyper FAF. J SS-OCT showing macular atrophy. K fundus photo of the left eye of the sister IV.6. L FAF showing central macular hypoFAF surrounded by ring of hyper FAF. M SS-OCT showing macular atrophy. N fundus photo of the left eye of IV.2. O FAF large macular atrophy surrounded smaller areas of atrophy. P SS-OCT showing macular atrophy with parafoveolar gliosis. Q, R and S Clinical features of patients from F9 showing cloverleaf maculopathy with peripheral RPE atrophy.
6 families (15 patients) with CRD (F6, F7, F8, F9, F10 and F11) were included. Their mean age was 35 years (14–48 years) with disease onset ranging from 6 to 18 years. All patients had photophobia, visual loss and night blindness. BCVA ranged from light perception to 2/10. Fundus examination showed macular lesions in all patients. We found yellowish macular deposits in 2 patients (F6 and F7), macular atrophy in 3 patients (F8, F11), beaten-bronze macula in 3 patients (F9) and symmetrical cloverleaf maculopathy in three patients (F10). Peripheral retina showed nummular pigment deposits in 2 patients (F6 and F7), few bone spicule shaped deposits in the mid periphery along with atrophy of the periphery in 8 patients (F8, F9 and F10), and hyperplasia of the retinal pigment epithelium (RPE) with yellowish deposits and atrophy in 5 patients (F11) (Fig. 3).
F8-CDHR1 (Fig. 3C, D, E, F and G)
A novel homozygous deletion (NM_033100: c.863-2_863-1delAG, p.?) in CDHR1 was observed in F8 with no other candidate P/LP variants (Fig. 2). This deletion is located in the crucial splice acceptor domain of intron 9 and could impact the normal splicing pattern of CDHR1.
F9-C8ORF37 (Fig. 3H, I, J, K, L, M, N, O and P)
The affected individual in family F9, carried a novel homozygous splice-site pathogenic variant (NM_177965: c.470 + 1G > T, p.?) in C8ORF37 (Fig. 2). This gene has recently been shown to cause RP7, with only 5 cases reported with splice-site variants. This variant was located in the donor splice site of intron 6.
F11-ABCA4
A novel homozygous likely pathogenic variant (NM_000350.2: c.1916A > G, p.Y639C) in exon 13 in ABCA4 was identified in family F11 (Fig.4). Clinical data showed typical hallmarks of CRD.

Clinical features of patients with ABCA4 pathogenic variant (A, B, C, D and E); RP patients (F, G, H, I, J and K); BB patients (L,M); achromatopsia (N, O, P) and CSNB (Q). A F11, LE fundus of patient II.2. B F12, RE fundus of patient II.1. C F13, RE fundus of patient II.2. D F14, RE fundus of patient III.3. E F15, RE fundus of patient II.1. F F16, RE fundus of patient V.4. G F17, RE fundus of patient V.1. H F18, RE fundus of patient IV.1. I F19, RE fundus of patient IV.2. J F20, LE fundus of patient II.5. K F21, RE fundus of patient II.5. L F22, RE fundus of patient II.3. M F23; RE fundus of patient II.1. N F24, LE fundus of patient II.3. O F25, LE fundus of patient II.3. P F25, LE OCT of patient II.3. Q F26, LE fundus of patient II.1.
RP Patients (Fig. 2,4)
The 10 patients (F16, F17, F18, F19, F20 and F21) with RP had a mean age of 35.57 years (21–63 years) with disease onset ranging from 5 to 20 years. All patients had night-blindness. BCVA ranged from hand movement to 10/10. Fundus examination showed typical RP with bone spicule deposits in mid periphery in all patients. In 2 patients, we found large areas of atrophy (F16). Macula was normal in 6 patients and atrophic in four (Fig. 4F).
F16-NMNAT1 (Fig. 4F).
The homozygous pathogenic variant (NM_001297778.1: c.37G > A, p.A13T) in NMNAT1 segregated with the disease in F16, with two affected members (F16-V.1, F16-V.4) (Fig. 2). This pathogenic variant has previously been reported as causing LCA8. In contrary to this report, our affected patients had relatively preserved visual acuity until the third decade (Table 2). On fundus examination, we found normal macular aspect with few bone spicule shaped pigment deposits and white dot deposits, large areas of atrophy in the mid periphery (Fig. 4F).
Bardet-Biedl (Fig. 2,4)
Clinical reassessment of extraocular symptoms was also performed in 4 patients from two families (F22, F23) who were shown to have Bardet-Biedl syndrome with retinal dystrophy, obesity and polydactyly. Fundus examination showed macular atrophy in all patients with bone spicule deposits and atrophy in the peripheral retina (Fig. 4M and N).
Other retinal dystrophies (Fig. 2, 4)
Four patients (F24, F25) had achromatopsia with nystagmus, photophobia and visual impairment since birth. Fundus examination was normal and OCT showed optically empty space with partial retinal pigment epithelium disruption in 2 patients (F25) (Fig. 4O and P). The index patients (F26) had a story of nyctalopia since childhood. Ophthalmic examination showed high myopia, cataract and chorioretinal atrophy (Fig. 4R). A novel likely pathogenic variant NM_002420.5: c.3947 T > G, p.L1316R in TRPM1 (Fig. 2) was identified in the index patient in F26.
Discussion
The data presented here showed that a number of genes can cause IRD in this Tunisian cohort. Taking together with our previous report, the analysis of 73 Tunisian families highlights the mutational load in IRD by identifying likely disease-causing genes in more than 25 genes in 50 families associated with different forms of IRD. A total of 50 likely disease causing alleles were identified, including 8 nonsense pathogenic variants, 10 deletions, 1 duplication, 1 complex rearrangement, 6 splice-site alleles and 24 missense alleles considered potentially pathogenic, 42% of which were novel. In addition, a novel finding from this study was the evidence of high frequency of ABCA4, RPE65, CRB1 and CERKL pathogenic variants in Tunisian families with IRD9,10,11,12 (Fig. 5). Homozygosity mapping combined with systematic screening of known genes resulted in a positive molecular diagnosis in 68.4% families. This is in accordance with several reports13,14 and is similar to the 75% frequency reported in Saudi Arabia15. However, in Spanish cohort studying large sporadic IRD groups (877 patients), the diagnostic yield was 44%16. On one hand, this difference might be explained by the selection of the cohort analyzed, as we chose families with either two affected individuals or sporadic cases with particular phenotype and we excluded patients with Usher syndrome. On the other hand, this may be due to the high frequency of consanguineous marriages in our cohort.

Mutational spectrum in 73 Tunisian cases with confirmed molecular diagnosis.
This work provides an overview of the mutational spectrum of IRD in Tunisian cohort (Fig. 5) which gives an the most frequent genes in our cohort of patients with retinal disorders were: 14% ABCA4 (p.E1087K, p.W782*, p.Y639C, p.P1380L, p.N380K and dup32-40; del45-47), 8% RPE65 (p.R91W, p.H182Y, p.R234* and c.1129-2A > G), 8% CRB1 (p.R764H, p.P836T, p.Y702C and c.3542 + 1G > A) with variable phenotypes of severe IRD, ranging from LCA to RP as previously reported17 and 8% CERKL (c.1133 + 3_1133 + 6delAAGT). As expected, our results look more similar to the Spanish cohort where CRB1 (7%), ABCA4 (7%), CERKL (4%) and EYS (4%) were the most frequent mutated genes16. Compared to other ethnic groups, however, the most prevalent pathogenic genes in Saudi Arabia were KCNV2, RP1, TULP1, RPGRIP1, CRB1 and RPE65 respectively15. In addition, characterized patients in Israeli/ Palestinian populations show high frequency of pathogenic variants in FAM161A, CRB1, USH1C, MAK and DHDDS17. This could be explained by the two countries sharing some ethnic origins.
In this study, we also highlighted the importance of combining molecular and clinical data to correctly diagnose IRDs. We would like to point out that in the subset of families analyzed in this study potential disease-causing variants were detected in 19 genes, out of which 8 have not yet been described in association with the observed IRD phenotype: RPGRIP1, GUCY2D, CRB1, CDHR1, C8ORF37, ABCA4, CNGB1 and TRPM1. Ophthalmic and genetic findings are presented in Table 3.
Ophthalmic investigation identified characteristic signs and symptoms of LCA in five families (F1, F2, F3, F4 and F5). This group of the most severe and the earliest occurring IRD resulting in congenital blindness18 typically becomes evident in the first year of life like in our five families represent 19.2% of our cohort. To date, genetic heterogeneity of LCA is well known, with 24 genes currently implicated in its pathogenesis19. Molecular analysis in our families identified three new pathogenic variants: novel homozygous deletion c.3113_3114delCT identified in RPGRIP1. The second missense pathogenic variant p.V887G is localized in GUCY2D, gene, estimated to account for 20% of LCA cases20 and constitute the most common cause of the disease. The third is a novel splice variant c.3542 + 1G > A in CRB1, the most commonly mutated gene in our cohort (8%) with variable phenotypes of severe IRD, ranging from LCA to RP as previously reported21.
NGS allows for the screening of a large number of genes implicated in the pathophysiology of IRD. In F4, despite having a previously reported homozygous pathogenic variant p.R332* in IQCB1, the phenotypes of our index patient and their affected sister never showed dysplasia in any organ; rather, they only had LCA. Usually, defects in this gene result in Senior-Loken syndrome type 5 (SLSN5), where degenerative phenotypes involving kidneys and eyes are common22, but sometimes the phenotype only shows LCA, as presented in this family23,24.
We report ophthalmic and genetic findings of patients with RP, composed of 10 patients, with AR-RP presented in 8 patients and 2 patients with AD-RP phenotype, which represent 23% of our cohort. patients with AR-RP were carrying four new pathogenic variants: Patients in F8 were carrying a novel homozygous deletion c.863-2_863-1delAG localized in the crucial splice acceptor domain of intron 9 CDHR1. To date, studies have revealed around thirty cases with this particular CDHR1 pathogenic variant; gene known to play a key role in the maintenance of photoreceptor structure and integrity25. We also describe a novel homozygous splicing pathogenic variant c.470 + 1G > T in C8orf37, observed in F9. The localization of genetic abnormalities has previously been described by Rahner et al., where 56% of the pathogenic variants are located in exon 6 in the C-terminal region of C8orf37 and the majority of reported variants are splicing variants26. We identified new homozygous pathogenic variant p.Y639C in family F11. ABCA4 pathogenic variants were responsible for 14% of cases in our cohort for a wide variety of IRD phenotypes from AR Stargardt disease to CRD and, in some advanced cases RP27. Therefore, no clear genotype–phenotype could be established. Three different pathogenic variants were identified in CNGB1, which represent 6% of our cohort. One of these variants is a novel homozygous pathogenic variant p.R765C in F19 where the index patient presented typical symptoms of RP.
Unexpectedly, two probands in family F16 with RP had damaging missense pathogenic variant, p.A13T in nicotinamide adenine dinucleotide (NAD) synthase gene NMNAT1. This pathogenic variant has been previously identified in patients with LCA8. Although the proband’s phenotype is consistent with RP, and the pathogenic variants are predicted to be deleterious, our patients showed well preserved visual acuity. Fundus examination revealed bone spicule-like pigment deposits and white spot deposits at the mid-periphery.
Our data provide an overview of the mutational spectrum of IRD in a Tunisian cohort, which gives an idea about genes spectrum in North Africa. We demonstrate a high degree of genetic complexity in both, the causative disease genes and their associated phenotypes, highlighting uncommon genotype–phenotype correlations and contributing to the current knowledge about disease-causing variants. We realise that this study presents some limitations, such as relatively small number of patients and the lack of complete ophthalmic and other examinations. Ideally, the efficacy of genotype–phenotype correlation could be improved with a complete ophthalmic examination, including ERG in all patients.
Methods
Clinical data and sample collection
150 families were evaluated at the Department B of Hedi Rais Institute of Ophthalmology, Tunis; Tunisia, over the course of 15 years. We selected a subset of families which accepted to be part of the study, with onset of the disease in the first or second decade of life, with clinical diagnosis of AR-IRD excluding patients with unclear or unlabeled diagnosis of retinal dystrophy or for whom multimodal imaging exploration could not be carried out. 73 families fulfilled these criteria.
In this study, we draw up a report on 26 families. Consanguinity involving first-cousin marriages was observed, and in the non-consanguineous families, most marriages were between individuals from the same geographic origin and the highest number of cases was recorded in the region of Nabeul containing 15 families. A questionnaire was used to collect information which included socio-demographic data (age, gender, geographical origin, educational level, occupation, socio-economic level) family medical, surgical and ophthalmological history, age of onset and duration of symptomatology (onset date has been defined by the age of onset of the first visual symptoms), the disease course (defined as either stationary or progressive) and any additional non-ocular findings, such as deafness, mental retardation, polydactyly, obesity, heart disease or other malformations. Each patient had complete ophthalmological examination including best corrected visual acuity (BCVA) using Snellen chart, slit-lamp biomicroscopy, dilated fundus examination, retino-photos, optical coherence tomography (Swept source DRI OCT-A Triton®,Topcon, Tokyo, Japan), fundus autofluorescence (Heidelberg Spectralis; Heidelberg-Engineering, Heidelberg, Germany) and some patients also received full-field electroretinogram (ERG) (Métrovision, France).
This study was approved by the Local Ethics Committee of the Hedi Rais Institute. Peripheral blood samples were obtained from the index patient and from some of the family members, including parents and affected siblings. Informed consent was obtained from all participants and a parent and/or legal guardian for participants under the age of 18 years old. Analyses were done in accordance with local guidelines. DNA was extracted from leukocytes according to the salting-out method28.
Whole exome sequencing (WES)
Exome capture was performed using the Roche Nimble-Gen version 2 (44.1–megabase pair) and paired-end multiplexed sequencing was carried out on an Illumina HiSeq 2000 system (Illumina, San Diego, CA, USA) by Otogenetics Corporation (Norcross, Georgia, USA) using DNA samples from all index cases. Homozygosity was evaluated from SNPs obtained by WES.
Sequence data alignment, variant calling and identification
The Illumina paired-end DNA sequence data were mapped and aligned to the reference human genome NCBI Build 37 (hg19) using the Nextgene software package v.2.3.5. (Soft-genetics, State College, PA). Median coverage of the target region was 95X with 96% of target region covered by at least 10 reads.
Variant assessment
Identified variants were analyzed by PolyPhen-2 (https://genetics.bwh.harvard.edu/pph2/) and SIFT (https://sift.bii.a-star.edu.sg/) to predict the pathogenicity of the respective variants. The variant frequency in the healthy control population was evaluated using ExAC (https://exac.broadinstitute.org/) and gnomAD (https://gnomad.broadinstitute.org/) databases. Variants outside exons and flanking splice regions, synonymous or with a minor allele frequency (MAF) > 1% were filtered out. Amino acid conservation of the altered protein region was analyzed using a multispecies alignment comparing human, monkeys, chicken, fish, frog, fly and worm. Protein sequences were obtained from Uniprot (https://www.uniprot.org) or PolyPhen-2.
To predict the putative impact of the identified splice site variation, in silico analysis was done using Human Splicing Finder (v2.4.1), SKIPPY (https://research.nhgri.nih.gov/skippy) and the Automated Splice Site Analyses (https://www.fruitfly.org/seq_tools/splice.html).
The resulting list of homozygous gene variants was compared to the IRD genes found in RetNet (https://sph.uth.edu/retnet/disease.htm).
Sanger sequencing
Identified variants were validated by Sanger sequencing and segregation analysis. DNA was amplified by PCR using FastStart PCR Master Kit (Roche, Basel, S) and sequenced as previously described11. Primers and condition of each PCR are provided in Table 4. Fragments were sequenced on an ABI 3100XL DNA automated sequencer (Applied Biosystems, Foster City, CA).
References
Parmeggiani, F. Clinics, epidemiology and genetics of retinitis pigmentosa. Curr. Genomics 12, 236–237 (2011).
Ayuso, C. et al. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2, 34 (2010).
Ferrari, S. et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 12, 238–249 (2011).
Chen, Y. et al. Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 consanguineous Pakistani families. Invest. Ophthalmol. Vis. Sci. 58, 2218–2238 (2017).
Kerkeni, E. et al. Association among education level, occupation status, and consanguinity in Tunisia and Croatia. Croat. Med. J. 47, 656–661 (2006).
Schorderet, D. F., Bernasconi, M., Tiab, L., Favez, T. & Escher, P. IROme, a new high-throughput molecular tool for the diagnosis of inherited retinal dystrophies-a price comparison with Sanger sequencing. Adv. Exp. Med. Biol. 801, 171–176 (2014).
Jinda, W. et al. Whole exome sequencing in eight thai patients with leber congenital amaurosis reveals mutations in the CTNNA1 and CYP4V2 Genes. Invest. Ophthalmol. Vis. Sci. 58, 2413–2420 (2017).
Perrault, I. et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 44, 975–977 (2012).
Habibi, I. et al. Identifying mutations in Tunisian families with retinal dystrophy. Sci. Rep. 6, 37455 (2016).
Habibi, I., Chebil, A., Kort, F., El Schorderet, D. F. & Matri, L. Exome sequencing confirms ZNF408 mutations as a cause of familial retinitis pigmentosa. Ophthalmic. Genet. 38, 494–497 (2017).
Chebil, A. et al. Genotype-phenotype correlation in ten Tunisian families with non-syndromic retinitis pigmentosa. J. Fr. Ophtalmol. 39, 277–286 (2016).
Falfoul, Y. et al. Phenotypic progression of stargardt disease in a large consanguineous Tunisian family harboring new ABCA4 mutations. J. Ophthalmol. 2018, 1030184 (2018).
Siemiatkowska, A. M. et al. Molecular genetic analysis of retinitis pigmentosa in Indonesia using genome-wide homozygosity mapping. Mol. Vis. 17, 3013 (2011).
Bocquet, B. et al. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol. Vis. 19, 2487 (2013).
Abu-Safieh, L. et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23, 236–247 (2013).
Martin-Merida, I. et al. Genomic landscape of sporadic retinitis pigmentosa: findings from 877 Spanish cases. Ophthalmology 126, 1181–1188 (2019).
Beryozkin, A. et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci. Rep. 26, 13187 (2015).
Den Hollander, A. I. et al. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 27, 391–419 (2008).
Thompson, J. A. et al. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol. Genet. Genomic Med. 5, 652–667 (2017).
Hunt, D. M. et al. Guanylate cyclases and associated activator proteins in retinal disease. Mol. Cell Biochem. 334, 157–168 (2010).
Bujakowska, K. et al. CRB1 mutations in inherited retinal dystrophies. Hum. Mutat. 33, 306–315 (2012).
Chaki, M. et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 80, 1239–1245 (2011).
Estrada-Cuzcano, A. et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 52, 834–839 (2011).
Stone, E. M. et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol. 129, 81–87 (2011).
Nikopoulos, K. et al. Identification of two novel mutations in CDHR1 in consanguineous Spanish families with autosomal recessive retinal dystrophy. Sci. Rep. 5, 13902 (2015).
Rahner, N. et al. A novel C8orf37 splice mutation and genotype-phenotype correlation for cone-rod dystrophy. Ophthalmic Genet. 37, 294–300 (2016).
Zernanat, J. et al. Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum. Mutat. 35, 1187–1194 (2014).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215 (1988).
Marlhens, F. et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 17, 139–141 (1997).
Otto, E. A. et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior–Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 37, 282–288 (2005).
Den Hollander, A. I. et al. CRB1 has a cytoplasmic domain that is functionally conserved between human and Drosophila. Hum. Mol. Genet. 10, 2767–2773 (2001).
Stone, E. M. et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331 (2017).
Collin, R. W. et al. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 86, 783–788 (2010).
Lewis, R. A. et al. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease.. Am. J. Hum. Genet. 64, 422–434 (1999).
Webster, A. R. et al. An analysis of allelic variation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 42, 1179–1189 (2001).
Ducroq, D. et al. The ABCA4 gene in autosomal recessive cone-rod dystrophies. Am. J. Hum. Genet. 71, 1480–1482 (2002).
Coco, R. M., Tellería, J. J., Sanabria, M. R., Rodríguez-Rúa, E. & García, M. T. PRPH2 (Peripherin/RDS) mutations associated with different macular dystrophies in a Spanish population: a new mutation. Eur. J. Ophthalmol. 20, 724–732 (2010).
Langmann, T. et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am. J. Hum. Genet. 87, 376–381 (2010).
Abd El-Aziz, M. M. et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 40, 1285–1287 (2008).
Hjortshøj, T. D. et al. Novel mutations in BBS5 highlight the importance of this gene in non-Caucasian Bardet-Biedl syndrome patients. Am. J. Med. Genet. A 146A, 517–520 (2008).
Smaoui, N. et al. Screening of the eight BBS genes in Tunisian families: no evidence of triallelism. Invest. Ophthalmol. Vis. Sci. 47, 3487–3495 (2006).
Wissinger, B. et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 69, 722–737 (2001).
Carss, K. J. et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 100, 75–90 (2017).
Simonelli, F. et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest. Ophthalmol. Vis. Sci. 48, 4284–4290 (2007).
Roepman, R. et al. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl. Acad. Sci. 102, 18520–18525 (2005).
Lu, X., Guruju, M., Oswald, J. & Ferreira, P. A. Limited proteolysis differentially modulates the stability and subcellular localization of domains of RPGRIP1 that are distinctly affected by mutations in Leber’s congenital amaurosis. Hum. Mol. Genet. 14, 1327–1340 (2005).
Hanein, S. et al. Evidence of a founder effect for the RETGC1 (GUCY2D) 2943DelG mutation in Leber congenital amaurosis pedigrees of Finnish origin. Hum. Mutat. 20, 322–323 (2002).
Thompson, J. A. et al. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol. Genet. Genomic. Med. 5, 652–667 (2017).
Henderson, R. H. et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br. J. Ophthalmol. 95, 811–817 (2011).
Stingl, K. et al. CDHR1 mutations in retinal dystrophies. Sci. Rep. 7, 6992 (2017).
Ravesh, Z. et al. Novel C8orf37 mutations cause retinitis pigmentosa in consanguineous families of Pakistani origin. Mol. Vis. 21, 236–243 (2015).
Estrada-Cuzcano, A. et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am. J. Hum. Genet. 90, 102–109 (2012).
Van Huet, R. A. et al. Clinical characteristics of rod and cone photoreceptor dystrophies in patients with mutations in the C8orf37 gene. Invest. Ophthalmol. Vis. Sci. 54, 4683–4690 (2013).
Heon, E. et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 25, 2283–2294 (2016).
Zernant, J. et al. Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum. Mutat. 35, 1187–1194 (2014).
Chacón-Camacho, O. F., Granillo-Alvarez, M., Ayala-Ramírez, R. & Zenteno, J. C. ABCA4 mutational spectrum in Mexican patients with Stargardt disease: identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp Eye Res. 109, 77–82 (2013).
Xiang, Q. et al. Identification of a CNGB1 frameshift mutation in a han Chinese family with retinitis pigmentosa. Optom Vis. Sci. 95, 1155–1161 (2018).
Miraldi Utz, V. et al. Presentation of TRPM1-associated congenital stationary night blindness in children. JAMA Ophthalmol. 136, 389–398 (2018).
Audo, I. et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 85, 720–729 (2009).
Acknowledgements
We thank the family members for their invaluable participation and cooperation. We acknowledge the help provided by the ophthalmologist in this study and the colleagues who referred patients to us. We thank Susan E Houghton and Denisa Dzulova for reading the manuscript.
Author information
Authors and Affiliations
Contributions
D.F.S. and I.H. identified the pathogenic variants; Y.F., A.C, A.H, K.E and L.E.M. referred patients and clinical data; D.F.S., I.H. and Y.F. wrote the paper; H.I. and Y.F. prepared the figures; A.T. conducted the DNA extraction experiments; D.F.S. and L.E.M designed the experiments. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habibi, I., Falfoul, Y., Turki, A. et al. Genetic spectrum of retinal dystrophies in Tunisia. Sci Rep 10, 11199 (2020). https://doi.org/10.1038/s41598-020-67792-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67792-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
