
Abstract
Genome–wide association studies (GWAS) have revealed a plethora of putative susceptibility genes for Alzheimer’s disease (AD), with the sole exception of APOE gene unequivocally validated in independent study. Considering that the etiology of complex diseases like AD could depend on functional multiple genes interaction network, here we proposed an alternative GWAS analysis strategy based on (i) multivariate methods and on a (ii) telescope approach, in order to guarantee the identification of correlated variables, and reveal their connections at three biological connected levels. Specifically as multivariate methods, we employed two machine learning algorithms and a genetic association test and we considered SNPs, Genes and Pathways features in the analysis of two public GWAS dataset (ADNI-1 and ADNI-2). For each dataset and for each feature we addressed two binary classifications tasks: cases vs. controls and the low vs. high risk of developing AD considering the allelic status of APOEe4. This complex strategy allowed the identification of SNPs, genes and pathways lists statistically robust and meaningful from the biological viewpoint. Among the results, we confirm the involvement of TOMM40 gene in AD and we propose GRM7 as a novel gene significantly associated with AD.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD) is the predominant form of dementia (50–75%) in the elderly population. Two forms of AD are known: an early-onset (EOAD) that affects the 2–10% of the patients and is inherited in an autosomal dominant way, with three genes APP, PS1 and PS2 mainly involved; a late-onset form (LOAD) that affects the vast majority of the patients in the elderly over 65s, whose causes remain still unknown1. Although LOAD has been defined as a multifactorial disease and its inheritance pattern has not been clarify yet, it is coming out the idea that it could be likely caused by multiple low penetrance genetic variants2, with a genetic predisposition for the patients and their relatives estimated of nearly 60–80%2.
The first well known gene associated to LOAD was APOE3. It encodes three known isoforms proteins (APOE2, APOE3 and APOE4), with APOE4 known to increase risk in familial and sporadic EOAD. This risk is estimated to be threefold and 15-fold for heterozygous and homozygous carriers respectively, with a dose-dependent effect on onset age2.
Large-scale collaborative GWAS and the International Genomics of Alzheimer’s Project have significantly advanced the knowledge regarding the genetics of LOAD1. Anyways, none of the new identified loci reached the magnitude of APOEe4, as predisposing risk factor for AD, with the majority of the hereditable component of AD remaining unexplained4. Several different but not mutually exclusive explanations of such failure could coexist: AD could be caused by the concerted action of independent genetic factors, each having a small effect size that require to adopt multivariate methods and to increase sample size5; or it could be caused by the concerted actions of multiple genes (again characterized by low effect size) that act inter-dependently in still undefined pathways, that would need a pathway-based approach, as done for other complex diseases6. Alternatively, AD could be caused by vary rare but highly penetrant mutations that might be identified through DNA sequencing7.
In order to explore the first two possible scenarios, in this study we proposed an alternative GWAS analysis strategy based on (i) multivariate methods and on (ii) a SNPs-Genes-Pathways ensamble, in order to guarantee the identification of correlated variables, and reveal the possible connections existing among the identified relevant variables at different, but biologically connected levels.
Figure 1 depicts this alternative strategy. We analyzed both datasets at the SNPs, genes and pathways levels: in the SNPs analysis we used a multivariate methods named l1l2FS, in the genes analysis we used an association genetic test named SKAT and in the pathways analysis we considered Group Lasso with overlap. All these methods share the multivariate aspect, because they consider more features simultaneously (i.e., all the SNPs of one chromosome in the first analysis, all the SNPs belonging to one gene in the gene based analysis and all the pathways of one group in the pathway based analysis) differently from the univariate methods, such as the t-test, that evaluate the statistical association of each single feature at the time. The final purpose is to identify lists or signatures of possible causal SNPs, genes and pathways that considered together might provide a convincing picture of heritable factors in the LOAD pathogenesis.

The alternative GWAS’s analysis strategy. ADNI-1 and ADNI-2 datasets were analyzed unimputed at the SNP, Gene and Pathway levels using three machine learning methods [i.e., l1l2FS within PALLADIO framework, SKAT and Group Lasso with overlap (w. o.)]. The global signature represents the summary of the single integrated signatures identified within the proposed GWAS strategy.
In the Results section we show the signatures of SNPs, genes and pathways identified considering both the binary classification tasks, cases@controls and APOEe4, while in the discussion we comment the obtained results considering the possible integration of the signatures across the SNPs, genes and pathways levels and also across ADNI-1 and ADNI-2 dataset that we analyzed separately and considering only the genotype SNPs.
Results
SNP-based results
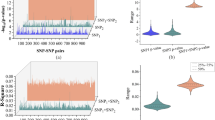
The SNPs analysis performed on unimputed ADNI-1 dataset (Table S1) identified a signature of 14 SNPs relevant for cases@controls task (Fig. 2 and Table S2). These SNPs, mapped on 14 genes or intergenic regions and are located on chromosomes 6 and 20. In particular, chromosome 6 showed higher performance values with respect to chromosome 20, considering both balanced accuracy and MCC (0.61 ± 0.06 and 0.21 ± 0.13) (Fig. 2A). In addition, the higher distance between the regular (light blue) and the permutation (red) distributions of the calculated balanced accuracies, reinforced the robustness of the obtained results (Fig. 2B). Among the genes of this short SNP-signature, only CDKAL1 is known to be associated to AD based on the literature8.

SNP-based results of ADNI-1. (A) The classification performance of SNP based analyses performed in ADNI-1 considering two classification tasks: AD vs. healthy controls (cases@controls) or 1/2 APOEe4 vs. 0 APOEe4 carriers (APOEe4 task). B. ACC, Balanced Accuracy; MCC, Matthews Correlation Coefficient; #genes*, number of genes or intergenic regions. (B) Balanced accuracy distribution plots of the regular (light blue) and the permutation batches (red) related to chromosomes 6 and 20 in the cases@controls task. (C) Balanced Accuracy distribution plots of the regular (light blue) and the permutation (red) batches related to chromosomes 1, 3, 9, 19 and 20 in the APOEe4 task.
It is well recognized that APOE polymorphic alleles are the main genetic determinants of AD risk, being the individuals carrying one or two e4 alleles at higher risk to develop AD3. Considering that APOEe4 polymorphism was harbored in 120 AD of 179 and 58 Control of 214, a further analysis based on the binary classification 1 or 2 APOEe4 vs 0 APOEe4 presence (APOEe4 task) was performed in order to characterize a polygenic profile that could uncover small effect size gene variants associated with the disease in a cumulative manner. 39 SNPs, which map to 47 genes or intergenic regions, have been identified in the APOEe4 task (Fig. 2A and Table S2). Chromosomes 19 and 20 were associated with the highest balanced accuracy and MCC results (Fig. 2A) and the distribution plots underlines this result (Fig. 2C). Based on the literature, 9 genes (i.e., red genes in Table S2) over a total of 47 are known to be involved in AD.
Interestingly, the two classification tasks (cases@controls and APOEe4 task) had in common SHLD1 gene on chromosome 20, involved in the DNA double-strand breaks (DSBs) repairing mechanisms9. This gene is the closest to different SNPs found discriminant in the two tasks: in cases@controls task rs6053572 is located in the intergenic region between GPCPD1 and SHLD1 genes while in the APOEe4 task rs236137 and rs1287032 are located in the intergenic region between SHLD1 and CHGB (Table S2).
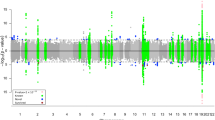
SNP-based analysis on unimputed ADNI-2 dataset (Table S1) identified for cases@controls task a signature of 138 SNPs, which map to 183 genes or intergenic regions harbored on 19 different chromosomes, with a balanced accuracy and MCC values ranging from 0.63 to 0.81 and 0.26 to 0.63 respectively (Fig. 3A and Table S3). In particular, chromosomes 9, 10, 14, 20 and 21 are the most reliable since they showed a higher distance between the two distribution measures (Fig. 3B). Based on the literature 12 genes (i.e., red genes in Table S3) over 138 are already known to be involved in AD.

SNP-based results of ADNI-2. (A) The classification performance of SNP based analyses performed in ADNI-2 considering two classification tasks: AD vs. healthy controls (cases@controls) or 1/2 APOEe4 vs. 0 APOEe4 carriers (APOEe4 task). B. ACC, Balanced Accuracy; MCC, Matthews Correlation Coefficient; #genes*, number of genes or intergenic regions. (B) Balanced accuracy distribution plots of regular (light blue) and permutation (red) batches related to chromosomes 9, 10, 14, 20, 21 in the cases@controls task. (C) Balanced accuracy distribution plots of regular (light blue) and permutation (red) batches related to chromosome 19 in the APOEe4 task.
When we considered the APOEe4 task, only chromosome 19 was found statistically significant, with very high values of both balanced accuracy (0.94) and MCC (0.90) (Fig. 3A and Table S3) and with very high distance between the two distributions (Fig. 3B). The derived SNP signature harbored only four SNPs located in three genes: rs367209 in LOC101928063, rs383133 in ZNF221, rs415499 in ZNF155 and rs365745 that causes a missense mutation in ZNF221 gene. Interestingly, LOC101928063 and its rs367209 SNP, was found statistically relevant also considering ADNI-2-cases@controls task. Although none of these genes are already known to be associated to AD, according to the AlzGene database 10 (https://www.alzgene.org/), these SNPs are located in a linkage region (i.e., 19q13) known to be associated to AD.
Considering SNP signatures of ADNI-1 and ADNI-2, we identified as a common gene GRM7, encoded for metabotropic glutamate receptor 7. ADNI-2 cases@controls task identified the intergenic SNP rs266410 between MRPS35P and GRM7-AS3 (the antisense version of GRM7), while ADNI-1 APOEe4 task the SNP rs9311976, located in an intron inside GRM7.
Gene-based results
In order to identify a gene signature for cases@controls and APOEe4 tasks, both the uninputed ADNI-1 and ADNI-2 datasets were analyzed by using three different tests included in the SKAT software (see Supplementary Information), applying a very conservative threshold for selecting a list of genes and SNPs highly relevant for AD (see Supplementary Information). In ADNI-1 dataset, TOMM40, with rs2075650, was found significantly associated to AD, applying all the tests (Table 1); while TEF gene, and in particular rs738499, was found significant in distinguishing cases@controls only with SKAT test (Table 1).
When the ADNI-1 dataset was analyzed considering the APOEe4,task, the genes or intergenic regions found significantly associated with AD risk were TOMM40, the intergenic region between LOC100129500 and APOC1, and the intergenic region between TOMM40 and APOE (Table 2). In particular, the two SNPs, rs439401 and rs405509 found in the intergenic regions LOC100129500-APOC1 and in TOMM40-APOE respectively, were confirmed by all the SKAT three tests applied.
It is noteworthy that the SNP rs2075650 harbored in the introns of TOMM40 was found in both ANDI-1 classifications tasks and it is confirmed in the literature to be associated to AD11.
Considering ADNI-2 dataset, the gene-based analysis did not give any significant association.
Pathway-based results
For the pathway-based analysis we considered REACTOME database12. In particular, we chose specific pathway groups (Tables S4 S5), whose relevance in neurodegenerative processes were well recognized 13. With the ADNI-1 cases@controls task, no groups reaching statistical significance were found. At variance, different pathway groups, associated with AD risk (APOEe4 task), achieved a good test score (Table 2). In the ADNI-2 dataset the pathway analysis reached a good statistical significance in both the classification tasks addressed (i.e., cases@controls and APOEe4 tasks). Group c1 showed pathways in common across both datasets and classification tasks. In particular, the “detoxification of reactive oxygen species” pathway was found in ADNI-1 APOEe4 task and in both ADNI-2 tasks. The two ADNI-2 tasks share the “cellular senescence” pathway. Although c1 group “PIP3 activates AKT signaling” pathway was found significant only in ADNI-1, it is noteworthy for its relevance in different intracellular processes, including neuronal survival, metabolism, and glucose uptake, (Manning and Cantley 2007), whose down regulation has been associated with neurodegenerative disorders14. In addition, the two pathways, “mitochondrial protein import” and “GPCR ligand binding”, belonging to the group 5a and 9a respectively, were identified in the APOEe4 tasks of both datasets (Table 2). Interestingly, these two latest pathways involved TOMM40 and GRM7 gene respectively, previously identified by SNPs and gene-based analysis.
In addition, we also performed a functional characterization in KEGG database (see Supplementary Information), in order to further biologically characterize the gene lists derived from the SNPs signatures identified before. A successful analysis was obtained only for ADNI-1 APOEe4 task and ADNI-2 cases@controls task, whose SNP signatures reported a long SNPs’ list (Table 3). In ADNI-1 APOEe4 task only the “Neuroactive ligand-receptor interaction” pathway, that includes the genes P2RY13, GRIN3A, LEPR, GRM7, P2RY14, reached a significant adjusted P value (Adj-P value = 5.99e−06).
In ADNI-2 the most important pathways related to AD were “Chemokine signaling”, “Calcium Signaling”, “Axon Guidance” and “Neuroactive ligand-receptor interaction” (Table 3). The latter pathway, in common with ADNI-1, involved GRIN2A, GRM7, GABRG3 and CYSLTR2 genes.
Discussion
Despite the promise of GWAS to reveal the genetic contribution to AD susceptibility, the majority of its heritable component remains unexplained. The major factor contributing to hamper the identification of genetic burden lies in the complexity of GWASs data management, together with the genetic heterogeneity of AD. In fact, although GWAS studies have revealed a plethora of putative susceptibility genes for AD, APOE gene is the sole exception unequivocally validated in independent studies.
The final purpose of the alternative strategy that we present in this study is to contribute in uncovering a robust heritable AD signature in the analysis of GWAS data. The key points of this strategy are the following: a new representation of the genotyped SNPs data; the telescope approach, since the data were analyzed at SNPs, genes and pathways levels; the choice of multivariate machine learning methods ad hoc for the three levels; the analysis of two separate dataset, each addressing two relevant binary classification tasks: cases@controls and APOEe4.
The new data representation have improved the classification performances of the applied machine learning algorithms because (i) it changed the nature of the data (from categorical to continuous), improving their interpretability for the considered machine learning methods and (ii) it made a more evident and biologically sound prioritization of some SNPs with respects to others. The analysis at SNPs, genes and pathway levels allowed the comparison of the results at these three levels and the successive identification of common related features, increasing their robustness in the association with AD. We chose the most appropriate machine learning method based on the characteristics of the analyzed data: l1l2FS was used to analyzed SNPs data because it is a sparse method, meaning that the solution to the classification problem is searched among a precise selection of the most relevant SNPs. This feature is essential to discard all those SNPs that are background noise or that are weakly associated to the addressed classification problems. The precise choice of the algorithm together with the SNP data transformation improved the selection of the most relevant SNPs from both the statistical and biological viewpoints. For the analysis of the gene level we select SKAT method because it provides the user the possibility to weight the SNPs differently based on their frequency occurrence in the subjects of the SNP dataset. Group Lasso with overlap was chosen because it is characterized by a feature that we seek in the analysis of the pathways: while looking for the most discriminant pathways within a group of them, which in this work constitute a single SNPs data matrix, we wanted the algorithm to consider the involvement of a gene in more then one pathway of the group. Finally the motivation behind the choice of addressing two classification tasks and the separate analysis of ADNI-1 and ADNI-2 dataset is to compare the results, between different biological questions and between two independent studies respectively. Besides the cases@controls classification task, where the disease is the discriminant, we considered also the APOEe4 task because studies have shown that individuals with two copies of the e4 allele are at even greater AD risk, and the odds ratios for developing AD based on APOE is 5 times greater in APOEe4 homozygotes compared to heterozygotes15. Therefore, a binary classification based on the presence of almost one APOEe4 allele could (i) uncover a cumulative polymorphic risk variants contributing to AD predisposition, and/or (ii) highlight superimposable genetic fingerprint, allowing a better understanding of APOE genotype contribution in the disease etiology. In addition, this classification might give useful insights for better addressing the therapeutic strategies, since multiple studies over the past two decades have demonstrated that APOE variants may affect the therapeutic response to anti-dementia drugs16,17,18,19. In this context, very recently, Berkowitz et al. 15 claimed that in the prospective of clinical precision strategy, the APOEe4 carrier status could have a very important impact on AD prevention interventions.
Considering the single signatures and datasets, in this study we identified lists of SNPs and genes, some of which are already reported in literature (see red colored SNPs and genes in Tables S2 and S3). But, in the tentative to adopted highly stringent and powerful statistical correction (permutation-regular batch for SNPs analysis and genome wide conservative threshold for genes analysis) to avoid false-positive results and increase the robustness of AD signatures, the numbers of SNPs and genes associated with AD or AD risk is strongly reduced. Even, the gene analysis of ADNI-2 did not give any results.
Furthermore, when we compared the two ADNIs datasets the majority of the signatures identified in ADNI-1 were not confirmed in ADNI-2 for both classifications tasks, although the demographic and clinical characteristics of the subjects enrolled in the two studies were comparable. A possible explanation of the low reproducibility of the results between the two datasets could be due to the following issues: (i) the different Illumina GWAS platforms, (ii), the lack of imputing procedure before the independent analysis of the two dataset (iii) and the difference in the genotype of APOE gene between ADNI-1 and ADNI-2 datasets. ADNI-1 and ADNI-2 datasets measured 620,901 and 730,523 SNPs respectively, of which only 300,000 were in common. An imputation procedure of ADNI-1 and ADNI-2 could have increased the SNPs overlap between the dataset and therefore could have increased the number of relevant SNPs genes and pathways and in turn allowed the validation of all the SNPs signatures identified in ADNI-1 and ADNI-2.
In addition, the fact that in ADNI-1 dataset APOE gene was genotyped separately from the other genes present in the platform, differently from ADNI-2, had surely influenced the obtained results in the SNPs and genes analyses.
The only heritable susceptible gene confirmed across SNPs and genes in ADNI-1 and across pathways in both datasets was TOMM40. On the other hand, the SNP and the pathway analysis of both ADNI-1 and ADNI-2 uncovered GRM7 gene as significantly associated with AD or AD susceptibility genetic profile.
TOMM40 is located in 19q13.32 locus, a known linkage region for AD10. Its encoded protein plays a key role in the mitochondria functionality being essential for import of protein precursors into mitochondria. The SNPs-based analysis identified two SNPs rs2075650 and rs8106922 harbored on TOMM40 gene, for ADNI-1 APOEe4 task. At the same time the SNP rs2075650 located in one intron of TOMM40, has been identified by the genes analysis of both controls@cases and APOEe4 task of ADNI-1 dataset. The literature confirms that TOMM40 gene is deeply involved in AD pathology20,21,22, and in particular that rs2075650 SNP is already known to be a contributing factor for AD (Huang et al. 2016; Potkin et al. 2009).
TOMM40 was also confirmed by pathway analysis in both dataset for the APOEe4 task. In particular, the protein encoded by TOMM40 is involved in the “mitochondrial protein import” pathway. The fact that SNPs, genes and pathways analyses highlighted the strong association between TOMM40 variants and APOEe4 genotype is due to TOMM40 location in the tight gene cluster TOMM40-APOE-APOC1-APOC4-APOC2 that is a strong linkage disequilibrium (LD) block 25. Furthermore, it has been reported that the APOE-TOMM40 genomic region has been associated with cognitive aging26 and with pathological cognitive decline27. APOE-TOMM40 genotypes have been also shown to modify disease risk and age at onset of symptoms28,29.
Interestingly, although ADNI-2 showed a long list of susceptible genes in SNPs analysis, TOMM40 did not emerge, probably due to the lack of datasets imputation. On the other hand, in ADNI-2 APOEe4 task, TOMM40 appeared in the pathway “mitochondrial protein import”.
GRM7 represents a novel possible candidate gene that needs to be experimentally validated, for the association with AD. It is located in 3p26.1 locus, and encodes for the metabotropic glutamate receptors 7, involved in the presynaptic neurotransmitter regulation30. GRM7 was identified in the SNPs and pathways analyses of both ADNI-1 and ADNI-2 dataset. In particular, the SNP rs9311976 on GRM7 gene was found in ADNI-1 APOEe4 task, while rs266410 in the inter region MRPS35P1/GRM7-AS3 (the antisense of GRM7) of ADNI-2 controls@cases task. GRM7 was also identified in “GPCR ligand binding” pathway, belonging to the REACTOME group 9a in the APOEe4 tasks of both datasets (Table 2), and confirmed by the in silico functional characterization that found enriched the same KEGG pathway “neuroactive ligand-receptor interaction” in both ADNI-1 and ADNI-2 SNP signatures. The identification of GRM7 gene in the APOEe4 classification task corroborated the association of glutamate signaling with APOE genotype. In fact, reduced expression of glutamate receptor proteins has been found in APOEe4 carrier AD31 and a defective glutamate synthesis has been shown in presynaptic APOEe4 neurons32. Furthermore, GRM7 has been found involved in schizophrenia33 and other mental disorders34,35. These finding were also confirmed by epidemiologic studies that showed significant associations between GRM7 and depression, anxiety, schizophrenia, bipolar disorder, and epilepsy36,37. Recently it has also been demonstrated that 3xTg-AD mice showed lower GRM7 protein expression in hippocampus, associated with an increased anxiety behavior, compared with the wild-type mice38. The significance of such results were confirmed by a genome-wide gene and pathway-based analyses on depressive symptom burden in the three independent cohort derived from the Alzheimer's Disease Neuroimaging Initiative (ADNI), the Health and Retirement Study (HRS), and the Indiana Memory and Aging Study (IMAS)39. In addition, GRM7 has been confirmed associated with AD in a meta-analysis of GWAS studies where glutamate signaling genes were found overrepresented in KEGG pathway enrichment analysis40.
In conclusion, the alternative GWAS analysis strategy applied in the analysis of two unimputed ADNI datasets, identified TOMM40 and GRM7 polymorphic variants as strongly associated with AD. Their relevance was confirmed by the identification of the mitochondrial import and glutamatergic signaling pathways, identified by pathway analysis, in which TOMM40 and GRM7 are respectively involved. Furthermore, the fact that these genes were found strongly associated with APOEe4 status at the SNPs, genes and pathway levels, corroborated its significance in the context of a cumulative polygenetic susceptibility to AD.
Although a possible limitation of this work could be found in the preprocessing phase and specifically in the absence of the imputation step, that could had improved the overlap of ANDI-1 and ADNI-2 dataset and the validation of the identified SNPs signatures, we strongly believe that this alternative approach of GWAS analysis presented in this study could provide a valuable way to uncover the genetic hereditability of multifactorial diseases like AD. In the future we plan to (i) test our approach applying the imputation step before the reanalysis of ADNI-1 and ADNI-2; (ii) to validate the results obtain in ADNI-1 in ADNI-2 and viceversa, following the strategy adopted in this work; (iii) to validate the identified SNPs, Genes and Pathways signatures in other independent GWAS dataset; (iv) to integrate covariates data, such as clinical characteristics of the patients to the analysis of the most discriminant SNPs, Genes and Pathways identified before.
Material and methods
Datasets
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (https://adni.loni.usc.edu/). The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. In this study GWAS data and APOE genotype obtained in the ADNI-1 and ADNI-2 datasets 41 were used (Table S1), considering the AD and healthy controls (CN) group. The genotyping platforms used by ADNI-1 and ADNI-2 were: Illumina Human 610-Quad BeadChip that measures 620.901 SNPs and CNV markers for ADNI-1 and Illumina Human OminExpress-24v that measured 730.525 SNPs and CNV markers for ADNI-2. Differently from ADNI-2, in ADNI-1 APOE genotyping is provided outside the GWAS platform. In both datasets, we performed two supervised binary classification analyses: AD vs. cognitively healthy subjects (cases@controls task) and subjects at higher risk vs. subjects not at risk of developing AD, according with APOE status (1 or 2 alleles vs. 0 allele of APOEe4) (APOEe4 task) (Table S1). Since the two dataset have been generated by two different Illumina platforms, showing a discrepancy between the number of SNPs and the APOE genotype presence, they were analyzed separately without performing the imputation step before the analysis phase. In addition the validation of ADNI-1 signatures in ADNI-2 dataset and of ADNI-2 signatures in ADNI-1 dataset has also been performed (see Supplemental Results).
Alternative GWAS analysis strategy
An alternative approach was devised to analyze the datasets. Considering the two classification tasks addressed and the SNPs, Genes and Pathways levels, each ADNI dataset was analyzed six times (Fig. 1).
In order to increase the signal over noise ratio, reducing the number of SNPs to analyze, we adopted the following strategy: (1) for the SNP and pathway analyses we employed two sparse methods (i.e., l1l2FS and Group Lasso with overlap), designed to identify the SNPs or pathways which are most discriminative for the classification tasks while restricting the selection of SNPs and pathways, and we considered a different representation of the SNP data (see Supplementary Information); (2) for the SNP analysis we analyzed each chromosome separately while for the gene and pathway analyses we grouped the SNPs considering genes/intergenic regions and pathways relevant for AD respectively.
SNP analysis
For the SNP analysis, we chose l1l2FS, a method that belong to sparse techniques42. This method allows the identification of the most discriminative variables for the problem at hand (classification tasks) while making feature selection (see Supplementary Information). l1l2FS was used within PALLADIO43 (https://slipguru.github.io/palladio/), a machine learning framework that can be customized to consider various combinations of feature selections and classification methods (Figure S1A). In order to ensure the reliability of the results, we used PALLADIO to perform two sets of experiments, which we referred to as regular batch and permutation batch (Figure S1B). The level of distance of the two distributions measured the reliability of the obtained results: the higher the distance, more reliable are the obtained results (see Supplementary Information).
Gene analysis
For the gene analysis, we considered three association tests available in the SKAT package (https://www.hsph.harvard.edu/skat/): Burden, SKAT and SKATO (see Supplementary Information). SKAT44,45 is a supervised regression method that test the association between genetic variants in a region and a dichotomous or a continuous trait while adjusting for covariates. The dichotomous traits considered here were cases@controls and APOEe4 tasks. In the first application of this alternative GWAS analysis strategy we chose to exclude covariates such as age at onset, race, sex. Furthermore we chose to consider genes or intergenic regions leveraging on the mapping files SNPs-to-genes provided by the GWAS platform manufacturer (i.e., “Human610_Gene_Annotation_hg19.txt” for ADNI-1 and “HumanOmniExpress-24v1-1_Annotated.txt” for ADNI-2).
The threshold of genome-wide significance we established, was conservative and in accordance with other studies46,47,48,49 (see Supplementary Information).
Pathway analysis
We selected 9 groups of pathways more relevant for neurodegenerative processes (Tables S4 and S5) inside REACTOME database50(https://reactome.org/).
In this study the pathways we selected and analyzed are the same for both dataset but some groups can differs in the number of pathways between the datasets because there is a low overlap of analyzed SNPs between the used platforms of ADNI-1 and ADNI-2 dataset.
Each group contained two or more pathways and each group represented a SNP matrix that, together with a label that characterizes each subject, was given as input to “Group Lasso with overlap”42. This latter is a machine learning method, able to consider the presence of overlapping groups of SNPs mapped to genes, involved in more than one pathway inside a group. The goal of “Group Lasso with overlap” is to induce a “sparse” selection at the group level, using all the pathways specified in the group. In this way, starting from a possibly long list of pathways inside a group, the algorithm selected a few (but informative) pathways that could be relevant for the problem at hand.
References
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Naj, A. C., Schellenberg, G. D. & Alzheimer’s Disease Genetics Consortium (ADGC). Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 174, 5–26 (2017).
Pericak-Vance, M. A. et al. Complete genomic screen in late-onset familial Alzheimer disease. Evidence for a new locus on chromosome 12. JAMA 278, 1237–1241 (1997).
Gandhi, S. & Wood, N. W. Genome-wide association studies: The key to unlocking neurodegeneration?. Nat. Neurosci. 13, 789–794 (2010).
Moore, J. H., Asselbergs, F. W. & Williams, S. M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 26, 445–455 (2010).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009).
Ng, P. C. et al. Genetic variation in an individual human exome. PLoS Genet. 4, e1000160 (2008).
Herold, C. et al. Family-based association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG and PDCL3. Mol. Psychiatry 21, 1608–1612 (2016).
Gupta, R. et al. DNA repair network analysis reveals Shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173, 972-988.e23 (2018).
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D. & Tanzi, R. E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 39, 17–23 (2007).
Souza, M. B. R., Araújo, G. S., Costa, I. G., Oliveira, J. R. M. & Alzheimer’s Disease Neuroimaging Initiative. Combined genome-wide CSF Aβ-42’s associations and simple network properties highlight new risk factors for Alzheimer’s disease. J. Mol. Neurosci. 58, 120–128 (2016).
Fabregat, A. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655 (2018).
Rosenthal, S. L. & Kamboh, M. I. Late-onset Alzheimer’s disease genes and the potentially implicated pathways. Curr. Genet. Med. Rep. 2, 85–101 (2014).
Manning, B. D. & Toker, A. AKT/PKB signaling: Navigating the network. Cell 169, 381–405 (2017).
Berkowitz, C. L. et al. Clinical application of APOE in Alzheimer’s prevention: A precision medicine approach. J. Prevent. Alzheimer’s Dis. 5, 245–252 (2018).
Cacabelos, R. Pharmacogenomics and therapeutic strategies for dementia. Expert Rev. Mol. Diagn. 9, 567–611 (2009).
Cacabelos, R. et al. Pharmacogenomics & pharmacoproteomics APOE-TOMM40 in the pharmacogenomics of dementia. J Pharmacogenom Pharmacoproteom 5, 3 (2014).
Cacabelos, R. et al. Pharmacogenomic studies with a combination therapy in Alzheimer’s disease. In Molecular Neurobiology of Alzheimer Disease and Related Disorders 94–107 (Karger, 2004). https://doi.org/10.1159/000078531.
Roses, A. D. Pharmacogenetics in drug discovery and development: A translational perspective. Nat. Rev. Drug Discov. 7, 807–817 (2008).
Goh, L. K. et al. TOMM40 Alterations in Alzheimer’s Disease Over a 2-Year Follow-Up Period. J. Alzheimer’s Dis. 44, 57–61 (2015).
Linnertz, C. et al. The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimer’s Dement. 10, 541–551 (2014).
Chiba-Falek, O., Gottschalk, W. K. & Lutz, M. W. The effects of the TOMM40 poly-T alleles on Alzheimer’s disease phenotypes. Alzheimer’s Dement. 14, 692–698 (2018).
Huang, H. et al. The TOMM40 gene rs2075650 polymorphism contributes to Alzheimer’s disease in Caucasian, and Asian populations. Neurosci. Lett. 628, 142–146 (2016).
Potkin, S. G. et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PLoS ONE 4, e6501 (2009).
Gottschalk, W. K. et al. J. Park. Dis. Alzheimer’s Dis. 1 (2014).
Davies, G. et al. A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol. Psychiatry 19, 76–87 (2014).
Hayden, K. M. et al. A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimer’s Dement. 8, 381–388 (2012).
Lutz, M. W., Crenshaw, D. G., Saunders, A. M. & Roses, A. D. Genetic variation at a single locus and age of onset for Alzheimer’s disease. Alzheimer’s Dement. 6, 125–131 (2010).
Bernardi, L. et al. Role of TOMM40 rs10524523 polymorphism in onset of Alzheimer’s disease caused by the PSEN1 M146L mutation. J. Alzheimer’s Dis. 37, 285–289 (2013).
Gee, C. E. et al. Blocking metabotropic glutamate receptor subtype 7 (mGlu7) via the Venus flytrap domain (VFTD) inhibits amygdala plasticity, stress, and anxiety-related behavior. J. Biol. Chem. 289, 10975–10987 (2014).
Sweet, R. A. et al. Apolipoprotein E*4 (APOE*4) genotype is associated with altered levels of glutamate signaling proteins and synaptic coexpression networks in the prefrontal cortex in mild to moderate Alzheimer disease. Mol. Cell. Proteom. 15, 2252–2262 (2016).
Dumanis, S. B., DiBattista, A. M., Miessau, M., Moussa, C. E. H. & Rebeck, G. W. APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J. Neurochem. 124, 4–14 (2013).
Sacchetti, E. et al. The GRM7 gene, early response to risperidone, and schizophrenia: A genome-wide association study and a confirmatory pharmacogenetic analysis. Pharmacogenom. J. 17, 146–154 (2017).
Niu, W. et al. Association study of GRM7 polymorphisms with major depressive disorder in the Chinese Han population. Psychiatr. Genet. 27, 78–79 (2017).
Noroozi, R. et al. Glutamate receptor, metabotropic 7 ( GRM7) gene variations and susceptibility to autism: A case-control study. Autism Res. 9, 1161–1168 (2016).
Chen, X., Long, F., Cai, B., Chen, X. & Chen, G. A novel relationship for schizophrenia, bipolar and major depressive disorder Part 3: Evidence from chromosome 3 high density association screen. J. Comp. Neurol. 526, 59–79 (2018).
Haenisch, S. et al. SOX11 identified by target gene evaluation of miRNAs differentially expressed in focal and non-focal brain tissue of therapy-resistant epilepsy patients. Neurobiol. Dis. 77, 127–140 (2015).
Zhang, Y.-L. et al. Anxiety-like behavior and dysregulation of miR-34a in triple transgenic mice of Alzheimer’s disease. Eur. Rev. Med. Pharmacol. Sci. 20, 2853–2862 (2016).
Nho, K. et al. Comprehensive gene- and pathway-based analysis of depressive symptoms in older adults. J. Alzheimers. Dis. 45, 1197–1206 (2015).
Pérez-Palma, E. et al. Overrepresentation of glutamate signaling in Alzheimer’s disease: Network-based pathway enrichment using meta-analysis of genome-wide association studies. PLoS ONE 9, e95413 (2014).
Saykin, A. J. et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers. Dement. 6, 265–273 (2010).
Hastie, T., Tibshirani, R., Hastie, M. W., Tibshirani, @bullet & Wainwright, @bullet. In Statistical Learning with Sparsity: The Lasso and generalization. (2015).
Barbieri, M., Fiorini, S., Tomasi, F. & Barla, A. PALLADIO: A parallel framework for robust variable selection in high-dimensional data. Work. Pap. https://doi.org/10.1109/PyHPC.2016.13 (2016).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D. & Lin, X. Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 92, 841–853 (2013).
Kraft, P., Zeggini, E. & Ioannidis, J. P. A. Replication in genome-wide association studies. Stat. Sci. 24, 561–573 (2009).
Mukherjee, S. et al. Gene-based GWAS and biological pathway analysis of the resilience of executive functioning. Brain Imaging Behav. 8, 110–118 (2014).
Fadista, J., Manning, A. K., Florez, J. C. & Groop, L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 24, 1202–1205 (2016).
Kanai, M., Tanaka, T. & Okada, Y. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J. Hum. Genet. 61, 861–866 (2016).
Croft, D. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 (2013).
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuro imaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (https://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Author information
Authors and Affiliations
Consortia
Contributions
M.S. contributed to conception and design of the study; analysis and interpretation of the data; preparation of the final manuscript. G.A. contributed to the revision of the manuscript. F.T. and V.T. contributed to the implementation of the method Group Lasso with overlap used in the Pathway-based analysis. A.B. contributed to the revision of the final manuscript. D.U. contributed to the interpretation of the data, to the preparation and revision of the final manuscript. The ADNI consortium provided the two GWA datasets analyzed in this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Squillario, M., Abate, G., Tomasi, F. et al. A telescope GWAS analysis strategy, based on SNPs-genes-pathways ensamble and on multivariate algorithms, to characterize late onset Alzheimer’s disease. Sci Rep 10, 12063 (2020). https://doi.org/10.1038/s41598-020-67699-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67699-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
