
Abstract
The safety of microbial cultures utilized for consumption is vital for public health and should be thoroughly assessed. Although general aspects on the safety assessment of microbial cultures have been suggested, no methodological detail nor procedural guideline have been published. Herein, we propose a detailed protocol on microbial strain safety assessment via whole-genome sequence analysis. A starter culture employed in traditional fermented pork production, nham, namely Lactobacillus plantarum BCC9546, was used as an example. The strain’s whole-genome was sequenced through several next-generation sequencing techniques. Incomplete plasmid information from the PacBio sequencing platform and shorter chromosome size from the hybrid Oxford Nanopore-Illumina platform were noted. The methods for 1) unambiguous species identification using 16S rRNA gene and average nucleotide identity, 2) determination of virulence factors and undesirable genes, 3) determination of antimicrobial resistance properties and their possibility of transfer, and 4) determination of antimicrobial drug production capability of the strain were provided in detail. Applicability of the search tools and limitations of databases were discussed. Finally, a procedural guideline for the safety assessment of microbial strains via whole-genome analysis was proposed.
Similar content being viewed by others


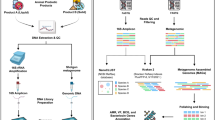
Introduction
In recent years, the importance of safety assessment of microorganisms used for human consumption has been widely recognised. The majority of the studies on this topic usually focused on the safety profile of probiotic cultures. In contrast, the safety of starter cultures used in food fermentation has not been as thoroughly examined. Similar to probiotics, many starter cultures are consumed alive in large quantity. Generally, the safety of starter cultures, notably those belonging to the lactic acid bacteria (LAB) group, is presumed from substantial histories of safe consumption of fermented foods. Regardless, cases of LAB infection have been observed in patients with underlying medical conditions1,2,3. Hence, the safety of all new microbial strains introduced into the food chain should be assessed to improve food safety and public health.
In this study, the safety of Lactobacillus plantarum BCC9546, a starter culture used for fermentation of nham was investigated. Nham is a fermented pork product popularly consumed in Thailand and neighbouring countries. Lactobacillus plantarum is the main LAB responsible for nham fermentation, presenting as the most abundant species in the final fermented product4. The starter culture L. plantarum BCC9546 was isolated from nham in 19995 and commercially available since 2001.
With current advances in genome sequencing technologies, safety evaluation of microbial strains can be done at a much higher resolution through whole-genome sequence analysis. The whole-genome analysis has gained increasing attention and was recommended as a part of strain identification and safety evaluation process by Pariza et al.6. A thirteen-question decision tree proposed by the authors (Fig. 1) was utilized as a guideline for the safety evaluation of starter culture in this study. This study focused on answering the first five questions of the decision tree that can be addressed through the whole-genome analysis. In addition to the safety information of BCC9546, the detailed procedures, resources and precautions in each step were also discussed. Through the application of whole-genome analysis, we propose a procedural guideline for the safety assessment of a microbial strain.

Schematic diagram of a decision tree for safety assessment of microbial cultures to be consumed by humans or animals as proposed by Pariza et al.6.
Results and Discussion
L. plantarum BCC9546 whole-genome sequencing
High molecular weight genomic DNA and plasmid DNA were successfully extracted from the fresh culture of L. plantarum BCC9546. Several bands of plasmid DNA, with the smallest size of approximately 2–3 kb, were visible on the agarose gel (Fig. 2).

Agarose gel electrophoresis of DNA extracts from BCC9546. (a) Genomic DNA running on 1% agarose gel: M, 250 ng DNA size marker (GeneRuler DNA Ladder Mix, Thermo Scientific); 1–2, 250 ng BCC9546 genomic DNA extract. (b) Plasmid DNA running on 0.5% agarose gel: Ms, 250 ng supercoiled DNA Ladder (New England BioLabs); p1, 50 ng BCC9546 plasmid DNA extract; p2, 120 ng BCC9546 plasmid DNA extract. The gel images were cropped for concise visualization. See Supplementary Fig. S1 for the uncropped gel images.
The results obtained from the two whole-genome sequencing platforms were shown in Table 1. The sequences obtained from Pacific Biosciences RS II SMRT (PacBio) sequencing platform yielded three unique contigs, i.e., one chromosome and two mega-plasmids. However, this result was deemed insufficient due to the lack of a 2.2 kb plasmid known to be present in this strain (pLpB9, GenBank accession EU391630.1) in the final assembly. This limitation was most likely due to the highly effective size-selection step in the PacBio protocol. The procedure was highly selective for large DNA fragments while excluding smaller plasmid DNAs from the sequencing reaction. Often, the PacBio sequencing was regarded as the platform providing complete genomic information as both chromosome and large plasmids, usually with the size larger than 10 kb, were detected7,8. Thus, the presence of small plasmids and their associated risk were not determined.
In comparison, the hybrid Oxford Nanopore Technologies (ONT)-Illumina sequencing platform yielded six unique contigs. The largest three contigs were similar to those obtained from the PacBio platform. However, this hybrid platform also identified three additional contigs named plasmid C, D, and E. The smallest plasmid E was nearly identical in size and sequence to the known 2.2 kb pLpB9 plasmid. This finding indicated the completeness of the genome sequenced by hybrid ONT-Illumina platform.
Although the hybrid ONT-Illumina platform was shown to be optimal in providing the complete genome information of the strain harbouring small plasmids such as BCC9546, the chromosome obtained from this technique was 2,400 bp shorter than that obtained from the PacBio platform. This reduced chromosome size was due to minor loss of repetitive sequences with length longer than the Illumina reads (>150 bp) during the polishing step in the hybrid assembly. Some repetitive sequences were identified as redundancy of identical chromosomal section, hence wrongfully omitted from the final assembly. Since this assembly error will affect only the copy number of continuous repetitive sequences, it could be considered as a minor flaw and should not affect the overall safety evaluation outcome of the strain.
Therefore, the limitations of the selected sequencing platform should be noted. Completeness of the genome data is crucial for the safety evaluation of a microbial strain. The selected platform should be able to identify all existing plasmids in the genome since plasmids can be the source or result of horizontal gene transfer that may contain virulence factors and/or antimicrobial resistance (AMR) genes. In the case of the PacBio platform, sequencing should be done in a way that small plasmids, if present in the genome, are included in the sequencing reactions, i.e., omitting the size-selection step. However, it should be noted that this action may compromise the average read length and overall efficiency of the long-read sequencing.
For the safety evaluation of BCC9546, the chromosome and two plasmids (plasmid A and B) obtained from PacBio and three small plasmids (plasmid C, D, and E) obtained from hybrid ONT-Illumina were selected as the complete genome of this strain. The BCC9546 genome consisted of the main chromosome size 3,218,570 bp with a 44.6% GC content, and five plasmids (A-E) ranging from 52,070 bp to 2,271 bp (Fig. 3). Total of 64 tRNA genes and 16 rRNA genes (Five copies of 16S and 23S rRNA genes and six copies of 5 S rRNA genes) were predicted.

Genome map of Lactobacillus plantarum BCC9546. Circles (from outside to inside): circle 1 and 2 are colour-coded according to the COG classification of the genes present on the forward and reverse strands respectively; circle 3 (purple) shows the GC plot; circle 4 (blue and green) represents the GC skew; circle 5 shows incomplete prophage (green), questionable prophage (blue), and intact prophages (red); circle 6 shows antimicrobial resistance genes. The figure is for illustrative purpose only, not drawn to scale.
Species identification
The BCC9546 genome contains five copies of 16S rRNA gene with four sequence variants (≥99.74% identical to one another). The 16 S rRNA gene sequences were highly similar to those of several Lactobacillus species, i.e., 99.80–99.93% to L. plantarum ATCC 14917 T, 99.87–99.94% to L. pentosus DSM 20314 T, 99.68–99.87% to L. paraplantarum DSM 10667 T, 98.73–98.93% to L. xiangfangensis LMG 26013 T, and 98.89–99.09% to L. fabifermentans DSM 21115 T. All of these similarity values were higher than the 98.7% cut-off threshold for species identification as proposed by Chun et al.9. This result indicated that the analysis using only the 16 S rRNA gene alone could not be used to identify species of the bacteria in this group, especially to distinguish between L. plantarum and L. pentosus.
The whole-genome sequence was then used to confirm the species of BCC9546. By considering the average nucleotide identity (ANI) value, this strain had the highest ANI value of 98.74% to L. plantarum ATCC 14917 T. The ANI values to the other four species were much lower than the 95–96% cut-off threshold as proposed by Richter and Rosselló-Móra10, i.e., 85.26%, 79.57%, 75.59%, and 74.68% for the type strain of L. paraplantarum, L. pentosus, L. xiangfangensis, and L. fabifermentans, respectively. Therefore, the whole-genome data supported that the strain BCC9546 indeed belongs to the species L. plantarum.
The accurate taxonomic placement is crucial for the identification of possible risk associated with the taxon. Following the protocol used in this study, the problem of misidentification or inability to distinguish between closely related species as seen in the identification based on biochemical assays and 16 S rRNA gene analysis7,11 can be alleviated.
Determination of virulence factors and toxin genes
The virulence factor database (VFDB)12 was used to identify known virulence factors and toxin genes that may exist in the BCC9546 genome. No virulence genes were found under the stringent criteria of >80% identity and >60% coverage. However, when a set of less stringent criteria (>60% similarity, >60% coverage, and E-value <1e-10) was used, a total of 51 hits were found. Among these hits, hemolysin III (chr_02698) was the only toxin gene identified within the genome. The remaining matches were mostly essential genes for cellular function and adaptation, such as genes involved in cell wall/membrane/envelope biogenesis and attachment (see supplementary file Supplementary_VFDB.xlsx for the complete list of all hits and the manual blast identification of the selected 51 hits). These genes were identified as virulence factors in the virulence factor database as they were also involved in pathogenic bacterial adaptation, survival, or attachment in the hostile/host environment. However, without other pathogenesis mechanisms, these genes could be regarded as beneficial to the bacterium since they increase the bacterial fitness and may be desirable where live cells are needed (i.e., in the case of starter cultures and probiotics).
For the gene identified as hemolysin, the manual investigation through blastp search confirmed the gene identification with 100% identity to “predicted membrane channel-forming protein YqfA, hemolysin III family” of Lactobacillus. Notably, the gene was also observed in several commercial probiotics such as an approved Generally Recognised as Safe (GRAS) probiotic strain L. plantarum 299 V, a widely used commercial probiotic in China L. plantarum JDM1, a kimchi probiotic L. plantarum ST-III, and many other Lactobacillus strains in the GenBank database. Hemolysis test using sheep-blood agar showed a hazy zone of hemolysis around the bacterial growth similar to the area around the probiotic strain 299 V, indicated a similar hemolysis activity of the two strains (see Supplementary Fig. S2). Since the hemolysin III gene is widespread in Lactobacillus spp. and the strains harbouring the gene have been proven for their safety and are commercially available in many countries, the bacterium harbouring this gene should not be of safety concern, provided that no other pathogenesis genes are present in the genome.
In the search for bacterial toxins using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database13, the hemolysin III gene was identified as the only toxin gene in BCC9546 genome. This search strategy facilitated the finding of the relevant toxin genes without a large number of false positives (i.e., the genes involved in adaptation or survival) as seen in the search using VFDB.
In comparison, Zhang et al.14 identified virulence factors in L. plantarum JDM1 using the VFDB database with different stringency criteria, i.e., 70% coverage and 30% identity. The study found as many as 126 hits. However, most of the genes were identified as defensive or non-classical virulence factors, and none were found to be offensive virulence factors. Interestingly, the study failed to identify the hemolysin III gene in the JDM1 genome. This finding raised awareness of the need for consensus criteria or a harmonized protocol for the optimal safety assessment process. In this study, the search using the KEGG database was shown to be accurate and efficient for the purpose.
Biogenic amine production
The ability to produce biogenic amines (BA) should be assessed for microbial strains intended to be used in food, especially in fermented food where high microbial activities may result in undesirable BA accumulation. As recommended by the European Food Safety Authority (EFSA), the BA-nonproducing starter cultures should be used to control the risk due to BA15. A bacterial strain lacking genes involved in BA production can be considered as BA-nonproducing strain and deemed safe for this aspect. In the case that the genes were found, the actual production and accumulated levels of the specific BA at the intended use condition should be determined to quantify the actual risk. To identify the presence of the genes involved in BA production, we found that the KEGG database search was efficient since the enzymes involved in all significant BA-production pathways were included. There were no genes related to the production of BA, including cadaverine, putrescine, spermidine, spermine, ornithine, histamine, tyramine, and tryptamine in the genome of BCC9546. Therefore, this strain can be considered as BA-nonproducer and poses no safety concern in this aspect.
D-lactic acid production
In the search using the KEGG database, two genes responsible for the production of D-lactic acid, i.e., lactate racemase (chr_00083) and D-lactate dehydrogenase (chr_00684 and chr_01677) were identified. The production of D-lactic acid by the strain was confirmed through HPLC method based on chiral analysis of an 18 h-cultured medium. The strain produced approximately 22 g/L lactic acid, in which approximately half were present in the D-configuration (see Supplementary information). Since D-lactic is an essential component in cell wall peptidoglycan of several gram-positive cocci including L. plantarum, production of D-lactic could be seen as an intrinsic property of the bacteria in this group. Therefore, a general precaution for the consumption of D-lactic producing bacteria should be provided to those with a high risk of D-lactic acidosis, such as in patients with short-bowel syndrome or carbohydrate malabsorption16.
Bile salt deconjugation
Through KEGG database search, four copies of the gene encoding choloylglycine hydrolase (chr_00054, chr_02114, chr_02755, chr_02880) were found in the BCC9546 genome. The presence of the choloylglycine hydrolase (also known as bile salt hydrolase) gene is an indication of bile salt deconjugation capability. In the Guidelines for the Evaluation of Probiotics in Food issued by FAO/WHO, bile salt hydrolase activity was included among desirable properties of probiotics such as resistance to gastric acidity, bile acid resistance, and adherence17. However, in the same document, bile salt deconjugation was listed as one of the properties that should be characterised for safety assurance. A comprehensive review by Begley et al.18 reported several studies regarding the beneficial effects of the bile salt deconjugation activity on the survival of bacteria and the cholesterol-lowering impact on the host. However, a high level of deconjugated bile may compromise normal lipid digestion, disrupt normal intestinal conditions, induce gallstone, and may be further modified to carcinogenic secondary bile salts18. After weighing all the benefits and concerns using the scientific evidence shown above, we propose that the bile salt deconjugation property could be seen as desirable when the strain is incapable of modifying the deconjugated biles into the harmful secondary bile products. Aside from the choloylglycine hydrolase, no genes related to the secondary bile salts biosynthesis were found in BCC9546. This concludes the strain’s bile salt deconjugation capability, which may play an essential role in the host digestive system survival. With regards to its inability to produce the harmful secondary bile products, we deem BCC9546 poses no safety concern from this property.
Antimicrobial resistance (AMR) phenotype
The minimum inhibitory concentrations (MICs) and the microbiological cut-off values of the tested antimicrobials were shown in Table 2. BCC9546 exhibited susceptibility to most antimicrobials tested. However, there were two drugs, i.e., chloramphenicol and kanamycin, in which the MICs were above the cut-off values, indicating acquired resistance for these drugs. Thus, these acquired resistances and their possibility of transfer were further investigated.
In this study, we propose the use of a limited number of antimicrobials, i.e., 7–9 depending on the bacterial group, as recommended by the current version of EFSA19 over the more extensive lists documented elsewhere14,20. The EFSA document was chosen as a guideline due to indication of clear microbiological cut-off values that can be used to distinguish between intrinsic and acquired resistance for bacterial groups commonly used in food and feed. It should be noted that the antimicrobial breakpoints widely reported elsewhere usually refer to clinical breakpoints, not microbiological cut-offs. Since the primary purpose of the clinical breakpoints is to identify the choice of drugs for effective treatment, often for infection by specific pathogens, hence they are usually not determined and less relevant in the case of non-pathogenic bacteria. However, in addition to the minimum list as recommended by EFSA, the resistance to other antimicrobials may be investigated if the cut-off values are known or if it fits the purpose of the study.
Antimicrobial resistance gene
The search using two AMR databases, CARD21 and ResFinder22, with the default settings (perfect/strict option for CARD; 90% threshold and 60% minimum length for ResFinder) returned no hits for AMR genes in BCC9546 genome. However, under a less stringent criterion (perfect/strict/loose option in CARD), 273 hits were predicted as AMR genes with ranges from 19–61% identity and 16–307% coverage. Due to the low stringency of the search criterion, the majority of the hits were not actually AMR genes. Nonetheless, we found one gene (Chr_1468) with 100% identity to a gene encoding chloramphenicol acetyltransferase (cat) which is responsible for the chloramphenicol resistance in several Lactobacillus species. This Lactobacillus cat gene possesses 28% best identity match to the cat gene of Enterococcus faecalis that was included in the CARD database. Inability to identify the cat gene in BCC9546 at the default, high stringency setting may be due to the limited repertoire of AMR genes included in the databases. Since both CARD and ResFinder databases mainly focus on AMR determinants of pathogenic bacteria, the AMR genes of non-pathogenic bacteria such as those from Lactobacillus are usually not included. Therefore, the limitation of AMR gene search using current version of CARD and ResFinder databases for non-pathogenic bacteria should be noted.
On the contrary, the KEGG database search yielded ten AMR-related genes in the BCC9546 chromosome (Table 3). The cat gene was promptly identified. While no specific gene for kanamycin inactivation was found, several genes related to the efflux pumps conferring multidrug resistance were identified in the genome. These efflux pumps may contribute to the kanamycin resistance trait of the strain. Additionally, the presence of aadA gene indicated possible streptomycin resistance. Notably, the presence of a macrolide resistance gene msrA did not confer resistance to erythromycin, the macrolide antibiotic used in this study. This may be due to several factors such as the gene expression level and the substrate specificity of the expressed product. Similarly, despite the possession of two beta-lactamase genes, the strain was sensitive to ampicillin. Since beta-lactamase is a large enzyme family with variations in their substrate specificity23, resistance to other beta-lactam drugs cannot be excluded without further investigation.
Mobile genetic elements
The main concern regarding AMR genes in beneficial non-pathogenic bacteria is for their transfer possibility to other possibly pathogenic bacteria which may lead to complications, reducing effectiveness of the antibiotic treatment. To identify this risk, we focused on two types of mobile elements, i.e., plasmids, and bacteriophages, since they are the most likely vehicles involved in inter-cellular genetic exchange through transformation/conjugation and transduction process, respectively. No oriT was found in any of the plasmids, indicating that these plasmids are incapable of self-transmission through conjugative transfer. For the presence of bacteriophage, the PHASTER tool24,25 identified four prophage regions in the main chromosome and two regions in plasmid A (Table 4 and Fig. 3, see Supplementary_prophage.xlsx for details). None of the AMR genes were located within the prophage regions, nor in any of the plasmids. Therefore, we concluded that the AMR genes present in BCC9546 have low risks of being transferred to other bacteria; hence the strain poses no safety concern regarding the functional and transferrable AMR property.
Antimicrobial drug production
To limit the emergence of new AMR sub-populations, the microbial strains used for consumption should not produce antimicrobial drugs, especially those determined as critically important for medical treatment. For this purpose, the World Health Organization’s List of Critically Important Antimicrobials for human medicine (WHO CIA list)26 was used as reference. Based on the pathway search in the current KEGG database, BCC9546 does not possess the ability to produce the antimicrobials of concern; hence poses no safety concern for this aspect.
Safety conclusion of L. plantarum BCC9546
The starter culture L. plantarum BCC9546 was shown to be relatively safe with no transferable AMR genes in the genome. As shown in Table 5, all genes related to the virulence, undesirable metabolites, and AMR identified in BCC9546 were also present in other L. plantarum, including several probiotic strains 299 V, JDM1, ST-III, and the reference strain WCFS1. These genes seem to be ubiquitously present within the species. Therefore, we concluded that the starter culture L. plantarum BCC9546 is safe, at a comparable safety level to the existing probiotic strains.
Procedural guideline for the safety assessment of a microbial strain via whole-genome analysis
To the best of our knowledge, despite suggestions on the issues that should be investigated for microbial safety assessment6,17, no clear procedural guideline have been provided. Despite several studies reported on microbial safety assessements7,11,14,27, no consensual procedure agreement can be observed. Therefore, we would like to propose the guideline for conducting the microbial safety evaluation using the whole-genome analysis as follow:
-
1)
Determination of the smallest extra-chromosomal DNA size. The smallest size of the plasmid, if present, in the genome should be determined. This information is required to confirm the completeness of the whole-genome sequence obtained from the selected sequencing platform. This step can be achieved through common laboratory technique such as agarose gel electrophoresis of the plasmid DNA extracted from the strain of interest.
-
2)
Selection of the whole-genome sequencing platform. The sequencing platform and protocol used should provide complete information of the genome, including all extra-chromosomal DNA. The hybrid sequencing via Oxford Nanopore Technologies and Illumina platform (ONT-Illumina hybrid sequencing) was demonstrated to be suitable, providing complete information of the chromosome and all plasmids. However, the PacBio platform may be used for validation of the chromosome sequence, or when there are no small plasmids in the genome. After sequencing, a complete de-novo genome assembly should be performed to identify the locations of the AMR genes, if present in the genome.
-
3)
Species identification. The 16S rRNA gene may be utilized to identify the most probable species of the unknown strain. The average nucleotide identity (ANI) based on the whole-genome sequence (≥ 95–96% to the type strain of the species)10 should be used to validate the result obtained.
-
4)
Identification of virulence and undesirable genes. Genes responsible for virulence and undesirable properties may be identified using publicly available databases and manually inspected to confirm its identity and function. Care should be taken in the interpretation of the result as genes involved in survival and adaptation should not be considered as virulence genes for non-pathogenic bacteria. The search using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database13 (available at https://www.kegg.jp) for the pathways and genes as outlined in Table 6 was shown to be efficient for this purpose.
Table 6 List of the KEGG pathways that should be inspected for virulence and undesirable genes. -
5)
Identification of functional and transferrable AMR genes. The MICs for specific antimicrobial drugs should be identified as recommended by EFSA, 201219. The AMR genes, especially those responsible for the resistance phenotype, should be determined. The search using the KEGG database under “Brite ko01504: Antimicrobial resistance genes” was shown to be efficient for this purpose. If present, the genes’ location should be determined to assess their transferability. The AMR gene located in conjugative plasmids, plasmids, and intact prophages should be regarded as having a high probability of transfer. A web-based tool such as oriTfinder28 (available at https://bioinfo-mml.sjtu.edu.cn/oriTfinder/) may be used to identify the origin of transfer (oriT), the essential element for self-transmitted conjugative plasmids. Similarly, a web-based tool such as PHASTER24,25 (available at http://phaster.ca/) may be employed for identification of existing prophages in the genome. Strains found to posses functional and transferrable AMR genes should not be used for consumption.
-
6)
Identification of antimicrobial drug production capability. For the antimicrobial drugs of concern, the latest revision of the World Health Organization’s List of Critically Important Antimicrobials for human medicine (WHO CIA list)26 should be used as reference. Since there is no database available for identification of the genes involved in the biosynthesis of all antimicrobials in the list, the KEGG database is currently suggested as the best resource for this purpose. The relevant pathways that should be investigated were provided in Table 7. If production of a specific antimicrobial drug is suspected or was reported for the species, but not yet included in the database, the particular genes involved in the production pathway should be manually searched, and the actual ability of the strain to produce the antimicrobial drugs should be tested.
Table 7 List of the KEGG pathways related to the biosynthesis of antimicrobial drugs with clinical importance.
In conclusion, it should be emphasised that the in silico analysis used in this study represents only the first step in the safety assessment of a microbial strain and cannot fully substitute the in vivo safety assessment and monitoring of the undesirable side effects. However, this analysis can be used to screen for high-risk strains without the need for animal testings. It also provides valuable information for identification of potential risk and specific areas that should be further investigated. The guideline proposed here can be used to facilitate the development of new and safe microbial cultures as well as to ensure public health safety.
Methods
Culture condition
L. plantarum BCC9546 was obtained from BIOTEC culture collection, Thailand. The bacterium was maintained and grown in De Man, Rogosa and Sharpe (MRS) medium (Difco Laboratories Inc., USA). The culture was grown in MRS broth at 30 °C for 12–16 h to obtain cells for DNA extraction.
Genomic DNA extraction
The genomic DNA was extracted using Wizard Genomic DNA Purification kit (Promega Corporation, USA) according to the manufacturer’s protocol with some modifications. The modifications included addition of 25 U/mL mutanolysin (Sigma-Aldrich, USA) in the 1 mg/mL lysozyme resuspending solution, incubation at 37 °C overnight for complete cell lysis, and additional centrifugation at 10,000 × g for 3 min in the protein precipitation step. The DNA pellet was dissolved in DNase/RNase-free water. The concentration and quality of the DNA were measured using NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA). High purity genomic DNA expressing an OD260/OD280 ratio of 1.8–2.0 and an OD260/OD230 ratio of 2.0–2.2 was used for whole-genome sequencing. The integrity of the genomic DNA was visualised using 1% agarose gel electrophoresis in 0.5× TBE buffer. The gel was stained in 5 µg/mL ethidium bromide solution for 5 min and destained in tap-water for 10 min. The gel image was captured using Gel Doc XR + Imaging System (Bio-rad, USA) with Image Lab 5.1 software and setting for optimised exposure time for the intense band.
Plasmid DNA extraction
Plasmid DNA was extracted using ZymoPURE Plasmid Miniprep kit (Zymo Research Corporation, CA, USA). The manufacturer’s protocol was modified to include the addition of 10 mg/mL lysozyme and 25 U/mL mutanolysin to the P1 buffer, and an additional incubation step at 37 °C for one h to ensure complete cell lysis. The possible number of plasmids and their size were visualised using 0.5% agarose gel electrophoresis in 0.5× TBE buffer.
Whole-genome sequencing and genome assembly
The genome of L. plantarum BCC9546 was first sequenced using PacBio sequencing platform (RSII SMRT cell, Pacific Biosciences at McGill University and Génome Québec Innovation Centre, Canada). The sequencing reads were assembled de novo using Celera Assembler in HGAP (Hierarchical Genome-Assembly Process) workflow29. An additional whole-genome sequencing based on hybrid technologies was performed to obtain the full genomic information of the strain. Oxford Nanopore Technologies (ONT) (Rapid sequencing kit, MinIONTM device, Oxford Nanopore Technologies, UK) was used as an alternative long-read sequencing technique, and a short-read high-throughput Illumina platform (NextSeq® 500 high output kit v2 (300 cycles), Illumina, Inc., USA) was used to improve the accuracy of the final sequence. The hybrid ONT-Illumina sequencing was conducted at the University of Arkansas for Medical Sciences, USA. An assembly pipeline for bacterial genomes, Unicycler, was used to assemble and polish the hybrid sequence30.
Gene prediction and functional annotation
Gene prediction and computational annotation of protein-coding genes were performed using MAKER2 annotation pipeline package31. The prokaryotic gene sequences from NCBI database release 232 were used as the training data for the bundled GeneMark HMM within the MAKER2 package.
Species identification
The species designation of the strain was first determined using the 16 S rRNA gene sequences. All copies of the 16S rRNA gene were extracted from the whole-genome data and checked for possible contamination using a web-based tool ContEst16S32 available at https://www.ezbiocloud.net/tools/contest16s. Similarities of the 16S rRNA gene to known bacterial species were searched using the nucleotide basic local alignment search tool (blastn) available at https://blast.ncbi.nlm.nih.gov. The similarity cut-off value at ≥ 98.7% was used for initial species classification using 16S rRNA sequences9. Then, the average nucleotide identity (ANI) to the type strains of selected species were determined using JSpecies Web Server (JSpeciesWS)33 available at http://jspecies.ribohost.com/jspeciesws. The ANI value of ≥95–96% was used as the criterion to confirm the species of the strain10.
Determination of virulence factors and undesirable genes
The presence of virulence factors and toxin genes in the BCC9546 genome were searched using the virulence factor database (VFDB)12 (last updated: Jun 17, 2019) available at http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi. Two search criteria, i.e., a stringent search using the cut-off values at >80% identity, >60% coverage; and a less stringent search with the cut-off values at >60% similarity, >60% coverage, and E-value <1e-10 were used to identify the possible virulence genes for further investigation. In addition, the BlastKOALA search tool in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database13 (Release 90.1) available at https://www.kegg.jp/ was used and inspected for virulence factors and undesirable genes as listed in Table 6. All genes identified from their similarities to those in the databases were manually confirmed using the protein basic local alignment search tool (blastp) suite with non-redundant protein sequences (nr) database available at https://blast.ncbi.nlm.nih.gov/ 34,35.
Determination of antimicrobial resistance (AMR) properties
BCC9546 was investigated for AMR properties by both phenotypic and genotypic methods. The resistance phenotype of the strain was investigated as recommended by the European Food Safety Authority (EFSA)19. The strain’s susceptibility to seven antimicrobial drugs, i.e., ampicillin, gentamicin, kanamycin, erythromycin, clindamycin, tetracycline, and chloramphenicol was determined. The minimum inhibitory concentration (MIC) for each antimicrobial was evaluated through the microdilution method as described in the international standard ISO 10932:201020. In brief, the strain was grown on MRS agar plate for 16–24 h. The colonies were then suspended in 5 mL sterile 0.85% NaCl solution to reach an OD625 of 0.16–0.2. The bacterial suspension was diluted 500 times in double-strength LSM broth (90% IST broth (ISO-sensitest broth, Oxoid Ltd., UK): 10% MRS broth). Fifty microlitres of the diluted bacterial suspension was added into wells containing 50 microliters of double-strength two-fold dilution series of the tested antimicrobials. Ampicillin, erythromycin, and kanamycin were purchased from Bio Basic Inc, USA. Chloramphenicol and clindamycin were purchased from United States Biological, USA. Gentamicin and tetracycline were purchased from AppliChem GmbH, Germany. The antimicrobial solutions were prepared by dissolving each antimicrobial powder in an appropriate solvent and adjusted for the potency as suggested in the ISO standard. Lactobacillus paracasei ATCC 334 and Bifidobacterium longum ATCC 15707 were used as quality control strains to ensure the performance of the prepared antimicrobial solutions. A well containing the test strain and the medium containing the solvent used to dissolve the antimicrobial at the highest concentration was used as the positive control, and a well containing the medium but without the test strain and the antimicrobial was used as the negative control. Since the intended use of BCC9546 is for human consumption, all incubation steps were conducted at 37 °C, under anaerobic condition (10% H2: 10% CO2: 80% N2). The MIC for each antimicrobial was determined, in triplicate, after incubating for 48 h.
The genetic determinants conferring AMR in the genome were searched using three publicly available databases, i.e., Comprehensive Antibiotic Resistance Database (CARD, RGI 5.0.0, CARD 3.0.3) available at https://card.mcmaster.ca/ 21, ResFinder (ResFinder 3.2, database 2019-07-19) available at https://cge.cbs.dtu.dk/services/ResFinder/ 22, and KEGG database (Release 90.1) using BlastKOALA search tool and inspected under “Brite ko01504: Antimicrobial resistance genes”13.
Transferability of the AMR genes found in the genome was investigated by their locations in two mobile elements, i.e., plasmids and bacteriophages. The existence of prophages in the genome was searched using PHASTER tool available at http://phaster.ca/ (Prophage/Virus DB last updated on Aug 3, 2017)24,25. For plasmids, the possibility for self-transmission through conjugation was investigated using oriTfinder, a web-based tool for identification of the origin of transfers in DNA sequences available at https://bioinfo-mml.sjtu.edu.cn/oriTfinder/ (database version: 1.1, May 2017)28.
Determination of antimicrobial drug production capability
To assess the capability of the strain for the production of antimicrobial drugs with clinical importance, the World Health Organization’s complete list of critically important antimicrobials (WHO CIA list)26 was used as a reference for the antimicrobials of interest. The genome was searched and examined for completeness of the pathways involved in antimicrobial drug biosynthesis in the KEGG database as shown in Table 7.
Data availability
The complete genome of L. plantarum BCC9546 consisting of one chromosome and five plasmids were deposited in GeneBank (accession number CP044500-CP044505).
References
Cannon, J. P., Lee, T. A., Bolanos, J. T. & Danziger, L. H. Pathogenic relevance of Lactobacillus: A retrospective review of over 200 cases. Eur. J. Clin. Microbiol. Infect. Dis. 24, 31–40 (2005).
Salminen, M. K. et al. Lactobacillus Bacteremia, Species Identification, and Antimicrobial Susceptibility of 85 Blood Isolates. Clin. Infect. Dis. 42, e35–e44 (2006).
Kayser, F. H. Safety aspects of enterococci from the medical point of view. Int. J. Food Microbiol. 88, 255–262 (2003).
Santiyanont, P. et al. Dynamics of biogenic amines and bacterial communities in a Thai fermented pork product Nham. Food Res. Int. 119, 110–118 (2019).
Valyasevi, R. et al. The microbiology and development of starter culture for nham, the traditional Thai pork sausage. in Seventeenth International Conference of the International Committee on Food Microbiology and Hygiene (ICFMH) 709–711 (1999).
Pariza, M. W., Gillies, K. O., Kraak-Ripple, S. F., Leyer, G. & Smith, A. B. Determining the safety of microbial cultures for consumption by humans and animals. Regul. Toxicol. Pharmacol. 73, 164–171 (2015).
Surachat, K., Sangket, U., Deachamag, P. & Chotigeat, W. In silico analysis of protein toxin and bacteriocins from Lactobacillus paracasei SD1 genome and available online databases. PLoS One 12, e0183548 (2017).
González-Escalona, N., Allard, M. A., Brown, E. W., Sharma, S. & Hoffmann, M. Nanopore sequencing for fast determination of plasmids, phages, virulence markers, and antimicrobial resistance genes in Shiga toxin-producing Escherichia coli. PLoS One 14 (2019).
Chun, J. et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466 (2018).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–19131 (2009).
Senan, S., Prajapati, J. B. & Joshi, C. G. Feasibility of Genome-Wide Screening for Biosafety Assessment of Probiotics: A Case Study of Lactobacillus helveticus MTCC 5463. Probiotics Antimicrob. Proteins 7, 249–258 (2015).
Liu, B., Zheng, D., Jin, Q., Chen, L. & Yang, J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, 687–692 (2018).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 428, 726–731 (2016).
Zhang, Z. Y. et al. Safety assessment of Lactobacillus plantarum JDM1 based on the complete genome. Int. J. Food Microbiol. 153, 166–170 (2012).
EFSA Panel on Biological Hazards (BIOHAZ). Scientific Opinion on risk based control of biogenic amine formation in fermented foods. EFSA J. 9, 2393 (2011).
Bianchetti, D. G. A. M. et al. D-lactic acidosis in humans: systematic literature review. Pediatr. Nephrol. 33, 673–681 (2018).
Joint FAO/WHO Working Group. Report on Drafting Guidelines for the Evaluation of Probiotics in Food. London, Ontario, Canada (2002)
Begley, M., Hill, C. & Gahan, C. G. M. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 72, 1729–1738 (2006).
EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Guidance on the assessment of bacterial susceptibility to antimicrobials of human and veterinary importance. EFSA J. 10, 2740 (2012).
ISO 10932:2010(en). Milk and milk products - Determination of the minimal inhibitory concentration (MIC) of antibiotics applicable to bifidobacteria and non-enterococcal lactic acid bacteria (LAB). International Organization for Standardization (2010).
Jia, B. et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573 (2017).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 (2012).
Philippon, A., Slama, P., Dény, P. & Labia, R. A structure-based classification of class A β-Lactamases, a broadly diverse family of enzymes. Clin. Microbiol. Rev. 29, 29–57 (2016).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 39, W347–352 (2011).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
World Health Organization (WHO). Critically important antimicrobials for human medicine, 6th revision. https://www.who.int/foodsafety/publications/antimicrobials-sixth/en/ (2019).
Hong, H. A. et al. The safety of Bacillus subtilis and Bacillus indicus as food probiotics. J. Appl. Microbiol. 105, 510–520 (2008).
Li, X. et al. OriTfinder: A web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 46, W229–W234 (2018).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Lee, I. et al. ContEst16S: An algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 67, 2053–2057 (2017).
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F. & Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931 (2016).
Altschul, S. F. et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272, 5101–5109 (2005).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Acknowledgements
This work was supported by the National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Thailand (Project Code P-17-52209).
Author information
Authors and Affiliations
Contributions
N.C. and W.V. conceived the experiments; P.S., K.C and K.K. conducted the strain characterisation; N.C., P.S., C.T., S.L., T.S. T.V., T.W., P.J. and I.N. performed the whole-genome sequencing and bioinformatics analysis; N.C. drafted the manuscript. All authors reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chokesajjawatee, N., Santiyanont, P., Chantarasakha, K. et al. Safety Assessment of a Nham Starter Culture Lactobacillus plantarum BCC9546 via Whole-genome Analysis. Sci Rep 10, 10241 (2020). https://doi.org/10.1038/s41598-020-66857-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66857-2
This article is cited by
-
Genome Sequencing Unveils Nomadic Traits of Lactiplantibacillus plantarum in Japanese Post-Fermented Tea
Current Microbiology (2024)
-
Safety assessment of Enterococcus lactis strains complemented with comparative genomics analysis reveals probiotic and safety characteristics of the entire species
BMC Genomics (2023)
-
I am better than I look: genome based safety assessment of the probiotic Lactiplantibacillus plantarum IS-10506
BMC Genomics (2023)
-
Functional genome analysis and anti-Helicobacter pylori activity of a novel bacteriocinogenic Lactococcus sp. NH2-7C from Thai fermented pork (Nham)
Scientific Reports (2023)
-
Integrated meta-omics approaches reveal Saccharopolyspora as the core functional genus in huangjiu fermentations
npj Biofilms and Microbiomes (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
