
Abstract
Here we report a pilot-sized study to compare the taxonomic composition of sputum microbiome in 17 newly-diagnosed lung cancer (LC) patients and 17 controls. Another object was to compare the representation of individual bacterial genera and species in sputum with the frequency of chromosomal aberrations in the blood lymphocytes of LC patients and in controls. Both groups were male; average age 56.1 ± 11.5 in patients and 55.7 ± 4.1 in controls. Differences in the species composition of bacterial communities in LC patients and controls were significant (pseudo-F = 1.94; p = 0.005). Increased prevalence in LC patients was detected for the genera Haemophilus and Bergeyella; whereas a decrease was observed for the genera Atopobium, Stomatobaculum, Treponema and Porphyromonas. Donors with high frequencies of chromosomal aberrations had a significant reduction in the microbiome of representatives of the genus Atopobium in the microbiome and a simultaneous increase in representatives of the species Alloprevotella compared to donors with a low level of chromosomal aberrations in lymphocytes. Thus, a comparison of the bacterial composition in the sputum of donors with cytogenetic damages in theirs lymphocytes, warrants further investigations on the potential role of microorganisms in the process of mutagenesis in somatic cells of the host body.
Similar content being viewed by others


Introduction
Lung cancer (LC) is the most common malignant tumor and the leading cause of death in the world. In particular, deaths from LC in men account for about 1/3 of deaths for all malignant neoplasms1. The treatment of LC, despite the enormous efforts reflected in of basic and clinical studies, still shows unsatisfactory results on survival and prevention programs show little effect on its incidence. Complex mechanisms of pathological changes and some key biomarkers of LC are recognized but there is a significant gap in the understanding of their mechanisms of theirs action in the course of LC development and progression. Among causative factors in the etiology of LC, smoking is certainly plays a leading role2. At the same time, 15 to 25% of all cases of lung cancer are observed in never-smoking patients. Among women, this figure reaches 53%3. Therefore, in addition to smoking, there are factors that can significantly affect the risk of developing LC.
In addition to the effects of hereditary components in the causation of LC, a number of contributory factors have been reported: passive smoking4, air pollution5, professional carcinogen exposition6, chronic exposure to high doses of radon7 and, recently, the composition of the lung microbiome8.
In connection with the recent development of metagenomic studies, information has been rapidly accumulated regarding the presence and diversity of the microbiota populating the lungs and the entire respiratory tract.
It was noted that alpha diversity (richness and uniform distribution of taxa in samples) is significantly higher in non-cancerous lung tissues than in tumor lung tissues9,10, while community similarity (beta diversity) varies widely. Researchers of the LC microbiota agree that Firmicutes accounts for the main contribution to the development of LC11,12,13. At the level of taxa below type, the results today are mixed. In most cases, scientists have observed correlations for some individual genera. In particular, two genera: Veillonella and Megasphaera were noted as possible LC biomarkers13. Erb-Downward and colleagues observed a link between Acidovorax and small cell carcinoma14, other researchers attribute an increased risk of developing LC to the detection of Granulicatella10,15, Abiotrophia10, Streptococcus10,12, Haemophilus influenzae, Enterobacter spp., Escherichia coli16, Capnocytophaga, Selenomonas, Veillonella, Neisseria17. Given the inconsistency of the previously obtained results, and taking also into account the fact that the taxonomic composition of the respiratory microbiota depends on environmental factors, it is important to continue research, including the analysis of microbiota in patients with LC from different regions of the world. This is especially true in regions with high levels of environmental pollution, for example, due to coal burning, as was previously shown for one of the provinces of China18.
It was shown that changes that offset the balance in the community of microorganisms inhabiting the lungs lead to the predominance of Haemophilus influenza, Acidovorax, Klebsiella, Moraxella catarrhalis, Mycobacterium tuberculosis, and Granulicatella adiacens19.
It is also known that inflammatory reactions can be triggered by opportunistic species such as
Enterobacter spp., E. coli, Pneumococcus16, Legionella20 and Moraxella21. An equally important contribution to the development of LC can be made by microbiota acting on Th17-positive T-helpers, responsible for a balanced immune response and necessary for the control of autoimmune reactions. It was shown that commensal bacteria have a direct effect on the Th17-dependent pathway and calcineurin expression22.
Another, no less important mechanism of microbiota-induced carcinogenesis is the genotoxic properties of many bacterial metabolites. Bacterial genotoxins, such as colibactin, a cytolethal distending toxin (CDT) and others have been identified as compounds that directly damage DNA in host cells23. In other cases, mutagenesis in the cells of the host organism is associated with the formation of DNA-reactive metabolites due to bacterial activity, the formation of radicals, or the immune modulation of the host cells24. This is a mechanism of action for Helicobacter pylori, Pseudomonas aeruginosa, Enterococcus faecalis, Shigella flexneri, Bacteroides fragilis, Neisseria gonorrhoeae, Listeria monocytogenes, Chlamydia trachomatis and others. This list is clearly not exhaustive and will be updated over time.
In general, one can suggested that bacteria use different strategies to ensure their survival and replication, which includes inhibiting DNA repair of host cells, contributing to the survival of infected cells, despite the presence of DNA damage25. The genotoxic potential of bacterial microbiota is the basis for the hypothesis that the stability of the genome of somatic human cells, especially under the influence of genotoxic and carcinogenic factors, can directly (or indirectly) depends on the taxonomic composition of the bacterial community.
To test this hypothesis, we have undertaken efforts to analyze the taxonomic composition of the microbiota in the sputum of LC patients and healthy donors living in the environmentally challenged coal-mining region of Western Siberia (Kuzbass), Russia.
Another object was to correlate the representation of individual bacterial genera and species in sputum with the frequency of chromosomal aberrations (CA) in the blood lymphocytes of LC patients and in controls.
Methods
Cohort information
The composition of the bacterial microbiome was studied in 17 patients with newly-diagnosed LC (male only, average age 56.1 ± 11.5 years) who were admitted to the Kemerovo Regional Oncology Center (Kemerovo, Russian Federation) and 17 healthy male donors, residents of Kemerovo (average age 55.7 ± 4.1 years). There were no differences in mean age between patients and control (p = 0.062). Among LC patients 70.6% were active smokers, among the controls, 64.7%. A summary of the information regarding LC and control subjects is shown in Table 1. An individual questionnaire was filled out for each survey participant, containing information about the place and date of birth, profession, exposure to occupational hazards, health status, diet features, taking medications (the use of antibiotics three months before the study), X-ray procedures, and bad habits (smoking and drinking status). For LC patients, the results of clinical and histological analyses were additionally taken into account. The distribution of LC diagnoses of patients analyzed was: squamous cell carcinoma −5 (29.4%); adenocarcinoma −5 (29.4%); large cell lung carcinoma −3 (17.7%); 4 (23.5%) other forms LC: mesenchymal, non-small cell undifferentiated. In addition, for each patient, the stage of the disease was determined in accordance with the TNM classification26. In accordance with this, 6 patients (35.3%) had stage I-II, and 11 patients (64.7%) stage III-IV of the disease. Additionally, metastases in distant organs were present in 23.5% of LC patients.
Ethics statement
All procedures undertaken were in accordance with the ethical standards of the Helsinki Declaration (1964 and amended 2008) of the World Medical Association. All participants (patients and controls) were informed about the aim, methodology and possible risks of the study; informed consent was signed by each donor. The design of this study was approved by the Ethics Committee of the Kemerovo State University.
Sample collection, process and storage
To analyze the composition of the microbiome of the respiratory tract, sputum samples obtained from LC patients and controls were used. The sputum samples from patients were obtained prior to all diagnostic or therapeutic procedures. Sputum samples were collected non-invasively through participant-induced coughing (i.e., without induction) and represent the oropharyngeal secretion. The resulting samples were immediately placed in sterile plastic vials and frozen (−20 °C). Frozen samples were transported to the laboratory and stored at −80 °C.
Analysis of chromosomal aberrations
Baseline frequencies of CA were studied in the same set of lung cancer patients and controls, from whom sputum microbiome was analyzed. The metaphase chromosomes spreads were prepared using the standard semi-micro-method27. The volumes of 1 mL of blood, 0.1 mL of phytohemagglutinin (Pan-Eco, Moscow), 9 mL of RPMI 1640 medium (Pan-Eco, Moscow), and 2 mL of fetal calf serum (HyClone) were added to the culture flask. The duration of the cultivation was 48 h.
Subsequently, colchicine (PanEco, Moscow, Russian Federation) was added to the culture at a final concentration of 0.5 μg/ml, and the flasks were placed in a 37 C incubator for 2 h. At the end of the incubation cycle, the cell cultures were treated with a hypotonic solution of 0.55% KCl for 20 min at 37 °C. The fixation of the material was performed in three changes of cooled fresh Carnoy’s fixative solution (methanol and glacial acetic acid at 3: 1 ratio). The obtained cell suspension was spread onto clean, cooled, and wet slides. The specimens were code labeled and stained with 2% Giemsa solution (Merck).
The registration of metaphases (200 from each donor) included in the analysis and the criteria for cytogenetic damages were consistent with generally accepted recommendations28. The following parameters were taken into account: the proportion of aberrant metaphases (CAs), the frequency of chromatid-type aberrations (CTAs): сhromatid breaks and chromatid-type exchanges, and the frequency of chromosome-type aberrations (CSAs): chromosome breaks, dicentric and ring chromosomes, atypical monocentric chromosomes. Achromatic gaps were not included in the number of aberrations and were not taken into account.
DNA extraction, 16S rRNA gene amplification and 16S rRNA gene sequencing
DNA was extracted from each sample using FastDNA Spin Kit For Soil (MP Biomedicals) based on the manufacturer’s recommendation. Thirty four 16S rRNA gene amplicon libraries were prepared by PCR amplification of an approximate 467 bp region within the hypervariable (V3-V4) region of the 16S rRNA gene in bacteria, from 50 ng of each of the extracted and purified DNA from sputum samples, respectively, according to the Illumina 16S metagenomic sequencing library protocol. PCR was initially performed with broad-spectrum 16S rRNA primers (forward primer: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′, and reverse primer: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′), using BioMaster Hi-Fi LR 2× 2× ReadyMix DNA polymerase (BiolabMix company, Novosibirsk, Russia). Cycle conditions were 94 °C (3 min 30 s), then 25 cycles of 94 °C (30 s), 55 °C (30 s), 68 °C (40 s), then a final extension of 68 °C (5 min). Libraries were purified using Agencourt AMPure XP beads (Beckman Coulter, Bray, USA) according to the Illumina 16 S metagenomic sequencing library protocol. Dual indices and Illumina sequencing adapters from the Illumina Nextera XT index kits v2 B and C (Illumina, San Diego, USA) were added to the target amplicons in a second PCR step using BioMaster Hi-Fi LR 2× 2× ReadyMix DNA polymerase (BiolabMix company, Novosibirsk, Russia). Cycle conditions were 94 °C (3 min 30 s), then 8 cycles of 94 °C (30 s), 55 °C (30 s), 68 °C (40 s), then a final extension of 68 °C (5 min). Libraries were again purified using Agencourt AMPure XP beads (Beckman Coulter, Bray, USA) according to the Illumina 16 S metagenomic sequencing library protocol. Sample PCR products were then pooled in equimolar amounts, purified using AMPure XP Beads (Beckman Coulter), and then quantified using a fluorometer (Quantus Fluorometer dsDNA (Promega, Madison, WI, USA). Molarity was then brought to 4 nM, the libraries were denatured, and then diluted to a final concentration of 8 pM with a 10% PhiX spike for sequencing on the Illumina MiSeq29.
Taxonomy quantification using 16S rRNA gene sequences and statistical methods
The processing of the results was conducted with the help of the program QIIME230. A quality check was carried out and a sequence library was generated. The sequences were combined into operational taxonomic units (OTUs) based on a 99% nucleotide similarity threshold using the Greengenes reference sequences library (versions 13–8) and SILVA (version 132), followed by the removal of singletones (OTUs containing only one sequence).
Calculation of indicators of alpha diversity and analysis of community similarity (beta diversity) was carried out according to the method UniFrac31. The total diversity of prokaryotic communities (alpha diversity) of sputum was estimated by the number of allocated OTU (analogue of species richness) and Shannon indices (H = Σpi ln pi, pi – part of i-sh species in community). When calculating sample diversity indices, 386 sequences were normalized (the minimum number of received sequences per sample). The variation in the structure of the bacterial community of different samples (beta diversity) was analyzed using UniFrac31 – a method common in microbial ecology that estimates the difference between communities based on the phylogenetic relationships of the presented taxa. In this study, we used a version of the unweighted UniFrac method that takes into account only the presence of taxa, but not their share in the community. The significance of differences between groups of samples was evaluated by the PERMANOVA method (Adonis).
In addition, to assess the significance of differences in the relative percentage of individual bacterial taxa in sputum, as well as the frequency of chromosomal aberrations in lymphocytes, the Mann-Whitney U test was used. To estimate the difference in the frequencies of occurrence, the Fisher exact test was used. Calculations were performed using the software package STATISTICA.10, Statsoft, USA.
Results
Characterization of sputum bacterial communities
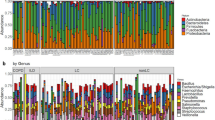
In our sequencing approach (16S rRNA V3–V6) of LC and controls using sputum samples, we were able to identify a total of 11 phyla with relative frequencies above 0.1%. The prevailing phyla in our dataset were Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria (Fig. 1), as it could be expected from previous studies15,18.

Taxonomic structure of microbiomes of upper respiratory tract among healthy and lung cancer patients.
Regarding alpha diversity, neither the number of allocated OTUs nor the Shannon indices, did not showed significant differences between LC and control. Overall, the bacterial communities were both fairly diverse as indicated by Shannon index at genus level (5.632 in LC vs 5.634 in controls). This suggests that any changes to the sputum microbiome as a result of a malignancy are not large-scale community shifts.
Differences in the structure of bacterial communities in sputum samples of lung cancer patients and controls are shown in Fig. 2. The PERMANOVA (Adonis) test using the difference matrix constructed by the unweighted UniFrac method showed a significant difference in the prokaryotic sputum communities of healthy people and LC patients (pseudo-F = 1.94; p = 0.005).

Phylogenetic similarity of prokaryotic sputum communities in LC patients and controls.
Sequencing statistics are summarised in Table 2 (for 67 genera) and in Table 3 (for 32 species), alongside corresponding U-rank Mann-Whitney p values. Among genera, Streptococcus, Prevotella, Anaerosinus and Selenomonas were the most common in the two pools. Seven genera (Actinobacillus, Pediococcus, Abiotrophia, Ruminofilibacter, Elizabethkingia, Psychrobacter and Malus) were found in lung cancer samples, but not in the control. Another eleven genera (Alloscardovia, Lutibacter, Anaerorhabdus, Pyramidobacter, Barnesiella, Gardnerella, Corynebacterium, Scardovia, Olsenella, Eggerthella and Asholeplasma) were found only in control samples, but not in lung cancer. Representatives of 13 genera were not found in the sputum of both groups: Cardiobacterium, Defluviitalea, Finegoldia, Jonquetella, Mobiluncus, Olsenella, Rhodococcus, Rhizobium, Slackia, Sneathia, Sphingobacterium, Spirohaeta, Succinispira. The data in Table 3 refers to the Greengenes reference sequences library (versions 13-8).
In the sputum of LC patients, compared to controls, there was a significant decrease in abundance by percentage of genus Atopobium (0.97 ± 0.9 vs 2.2 ± 1.55; p = 0.002); Stomatobaculum (0.41 ± 0.49 vs 1.13 ± 0.98; p = 0.021); Treponema (0.19 ± 0.49 vs 0.55 ± 0.96; p = 0.017). At the same time, the genus Bergeyella was significantly more represented in the microbiome of LC patients as compared to the controls (0.22 ± 0.27 vs 0.06 ± 0.18; p = 0.038). Representatives of the genus Haemophilus were observed only in 1 of 17 control donors (0.05 ± 0.22) and were present in sputum of 8 LC patients (3.04 ± 9.78; p = 0.006).
In LC, compared to controls, a decrease in the occurence of individual representatives of the same genera was observed at the species level: Atopobium rimae (1.1 ± 0.91 vs 2.27 ± 1.52; p = 0.003), Treponema amulovorum (0.27 ± 0.52 vs 0.05 ± 0.17; p = 0.016), as well as an increase in Bergeyella zoohelcum (0.25 ± 0.25 vs 0.03 ± 0.11; p = 0.004). Additionally, two different species from the genus Prevotella (P.histicola and P. sp. oral clone DO014) were significantly less represented in the sputum of patients than in the controls (Table 3).
Smoking status, as a factor that may have influenced the composition of the bacterial flora in LC patients and controls was studied separately. A significant difference in the occurrence of the species Selenomonas bovis in sputum was revealed in the samples of LC patients differing in smoking status (3.52 ± 2.55% in smokers and 0.91 ± 1.62% in non-smokers; p = 0.044); the genus Bacteroides (4.02 ± 2.51% in smokers and 1.35 ± 1.61% in non-smokers; p = 0.039); the genus Zhouea (0% in smokers and 1.03 ± 1.39% in non-smokers; p = 0.006); the genus Selenomonas (4.07 ± 2.42% in smokers and 1.03 ± 1.55% in non-smokers; p = 0.013); the genus Peptostreptococcus (0% in smokers and 0.73 ± 0.21% in non-smokers; p = 0.031).
Controls differing in smoking status revealed a significant difference in the occurrence of the species Bulleidia moorei in sputum (0.54 ± 0.45% in smokers and 0.12 ± 1.18% in non-smokers; p = 0.035); the genus Granulicatella (1.35 ± 1.43% in smokers and 0% in non-smokers; p = 0.04); genus Neisseria (0.39 ± 0.99% in smokers and 1.82 ± 2.54% in non-smokers; p = 0.026); genus Bulleidea (0.54 ± 0.46% in smokers and 0.13 ± 0.19% in non-smokers; p = 0.035).
Comparison of the species and generic microbiome compositions in the sputum of LC patients of the main pathomorphological forms (squamous, adenocarcinoma and large cell carcinoma) revealed significant differences in the content of the two bacterial genera Veillonella and Leptotrichia. In the sputum of patients diagnosed with large cell carcinoma, Veillonella representatives were recorded with a higher frequency than in patients with adenocarcinoma (15.35 ± 3.39% vs 7.68 ± 3.01%, respectively; p = 0.036). Representatives of the genus Leptotrichia were also recorded with a greater frequency in the sputum of patients with large-cell carcinoma as compared to adenocarcinoma (4.78 ± 2.81% vs 1.11 ± 1.13%, respectively; p = 0.036).
Comparison of the species and generic microbiome compositions in patients with different stages LC (I-II and III-IV) revealed a significant difference in only one species, Porphyromonas endodontalis, the content of which was significantly higher in the sputum of patients with stage I-II (0.53 ± 0.6%) compared with patients at the III-IV stage of RL (0.06 ± 0.21%; p = 0.021).
The presence of metastases to distant organs was associated with an increase in the content in the sputum of representatives of the genus Capnocytophaga (3.05 ± 1.58%) vs 0.71 ± 1.05% in LC patients without metastases (p = 0.016). In addition, Atopobium rimae showed a decrease in sputum in patients with metastases compared with patients without metastases (0.21 ± 0.43% vs 1.37 ± 0.84%, respectively; p = 0.017).
Chromosomal aberrations in lymphocytes and sputum microbiome composition
Lung cancer patients had a significantly increased total frequency of CAs in comparison with controls (4.31 ± 1.86% vs. 2.27 ± 1.03%, p = 0.002). In LC patients, as compared to the control group, significant increases in the frequency of chromosome-type aberrations, such as сhromosome breaks (1.16 ± 0.94% vs. 0.38 ± 0.45%, p = 0.012) in comparison with the control group were detected (Table 4). No significant differences in chromatid-type aberrations were found between LC patients and controls. The Spearman correlation coefficient for the CAs, CTAs and CSAs frequencies with age (for the patients and the controls), were not significant (p > 0.05). No significant difference in the frequencies of CAs was detected between squamous cell lung cancer, adenocarcinoma, large cell lung carcinoma and TNM stage. Additionally, smoking status was not a factor significantly affecting the CAs, CTAs and CSAs frequencies for the two groups studied.
To analyze the possible effect of the taxonomic composition of the microbiome on the level of genetic instability in somatic cells of the host organism, based on the results of cytogenetic analysis, subgroups of patients and controls were formed that differed in the frequency of lymphocytes with CAs.
The subgroup with a low background level of CA (0–3.5%; the average value is 2.26 ± 0.86%) consisted of 7 patients and 16 control groups, and the subgroup with a high level of CA (more than 3.5%; the average value is −5.67 ± 1.15%) consisted of 9 patients and 1 control.
Comparison of the representation of Atopobium rimae type by percentage in the bacterial microbiome revealed a signficant decrease of Atopobium rimae in the sputum from the patients with lung cancer with a high level of CA as compared to the first subgroup (0.59 ± 0.63 versus 1.72 ± 0.9; p = 0.014).
In addition, we compared the content of bacterial genera and species in the combined subgroups of patients and controls with high and low levels of CA in lymphocytes.
In the composition of sputum from donors with high frequencies of CA frequencies, there is a significant decrease in representatives of the genus Atopobium and particularly of species of Atopobium rimae, as well as a decrease in representatives of Prevotella sp. oral clone DO014 (Fig. 3).

Representation of bacterial species and genera in the sputum of donors, differentiated by the level of chromosomal aberrations (CA, %) in blood lymphocytes.
At the same time, an increase in the level of genetic instability in donor lymphocytes was associated with a significant increase in the content in the sputum of representatives of the genus Alloprevotella.
Representatives of the genus Actinobacillus were noted only in the sputum of donors with a high frequency of CA (0.29 ± 0.57%) and were not found in donors with a low level of aberrations in lymphocytes.
Discussion
The contribution of the microbiota of the upper respiratory tract to the pathological transformation of the tissues of these organs is a recognized phenomenon in studies of such a widespread disease as LC.
The urgent problem remaining is the formation of adatabase that is as complete as possible on the microbiome of the respiratory organs of people from different geographical regions of the planet, the establishment of strong cause-effect/associative relationships, and the identification of the molecular mechanisms of LC pathogenesis. This will open up prospects for early diagnosis and, ultimately, open up new approaches in the treatment of LC.
This pilot study, for the first time, presents the results of an investigation of the microbiome of the respiratory tract in a small group of patients with lung cancer of various histological types and degree, residing in Kuzbass, an industrial region of Russia.
Through the use of 16S metagenomic sequencing, we determined differences in the composition of the microbiome up to the species level in 17 men with a confirmed diagnosis of lung cancer (LC) compared with 17 age-matched controls.
In our study, we found 12 types, 119 genera, 118 OTU and analyzed 187361 sequences in total.
The taxonomic structure of the upper respiratory tract microbiome at the type level is shown in Fig. 1, from which it follows that the microbiome is represented mainly by representatives of the Bacteroidetes, Firmicutes, Actinobacteria types.
At the species level, differences in the composition of the microbiome in sputum in patients with LC and in control donors (Table 4) are characterized by a statistically significant decrease in the prevalence of four types of bacteria, namely: Prevotella histicola, Prevotella sp. oral clone DO014, Atopobium rimae, Treponema amylovorum and increase in Bergeyella sp. AF14 (Flavibacteriaceae) species in LC patients.
Prevotella histicola and Prevotella sp. oral clone DO014 belong to type Bacteroidetes, whose family members were recently associated with a worse prognosis for patients with LC32. Although overrepresented in LC patients on a type level, an individual species still could be underepresented in patients with LC, such as Prevotella sp. oral clone DO014 in our case, by analogy with Filifactor species. Filifactor bacteria were recently discovered mainly in healthy study participants and was even noted as a good control that allowed to distinguish between healthy and LC patients11, while the type where Filifactor belongs is represented more frequently in patients with lung cancer13. We also detected Filifactor mainly in healthy people.
Our data are consistent with previously published data. Representatives of the genera Treponema and Filifactor were found as ideal biomarkers in healthy participants in LC study11.
Representatives of the genus Atopobium are usually found in the oral cavity. Atopobium rimae species are associated with chronic periodontitis, bacteria of this species can cause bacteremia, are strictly anaerobic and Gram-positive bacteria, short and elliptical in shape, with a low content of G + C nucleotides33. The association of this species with LC has not been noted previously.
In our study, representatives of the genus Stomatobaculum are less represented in the sputum of patients with LC. This species has only recently been described as part of the microbiota of the human oral cavity and is positively associated with chalicytosis34,35,36, and with increased fasting glucose level (which is a sign of an increase in blood glucose after 2 years and the development of insulin insensitivity) It is also associated with a slowdown in bone restoration during implantation37. Stomatobactulum is strictly anaerobic, Gram-positive, non-spore forming, contains inclusions of iron and sulfur. For this species as well, association with LC was not previously noted.
Sequencing of bacterial 16S rRNA genes from the sputum of the lung cancer patients in our study has shown that the genera Atopobium and Stomatobaculum are significantly less abundant in samples from lung cancer patients as compared to the controls. The association of these two genera with the development of LC was not previously noticed. However, it was previously reported that the family Lachnospiraceae, to which Stomatobaculum belongs showed a positive relationship with LC32.
The representation of bacterial species of Atopobium rimae in the sputum of patients with LC decreases with progression of metastases according to our study, but this requires confirmation in a larger sample collection.
Our 16S rRNA sequencing data indicate an increase in the content of bacteria of the species Bergeyella sp. AF14 (Flavibacteriaceae) in the sputum of LC patients with. Previously, this type of bacteria was not associated with LC. Published data link this species of bacteria to animal respiratory disease38,39. Only recently have these bacteria have been identified as the cause of infectious endocarditis in humans40.
Interestingly, Bergeyella is represented to a lesser extent in patients with chalicytosis34. To firmly establish a more reliable association of this bacterium with the diagnosis of LC, a srtudy with more participants will be required.
Regarding the detection of such widespread bacteria in the respiratory tract as Haemophilus inflluenzae in our samples, we were able to detect it by analyzing the bacterial DNA 16S rRNA gene sequences using the SILVA database, which is based on 16S/18S Archaea analysis. We see that Haemophilus inflluenzae is present in 17 patients with LC and only in 1 subject in the control (p = 0.016). This contradicts previously published data that Haemophilus inflluenzae is mainly detected in healthy individuals15. It is known that most strains of H. influenzae are opportunistic pathogens that coexist with the host without causing disease, and only concomitant factors such as viral infections, decreased immunity, or chronically inflamed tissues such in allergies, predispose for pathogenic infections with H. influenzae. Thus, the reproduction of this pathogen and, therefore, higher chances of its detection should be expected precisely in the case of LC, as our data show. In any case, this discrepancy in the data deserves further study, with the involvement of more participants in the study of microbiota of the upper respiratory tract.
The frequencies of bacterial genera detected in our study indicates a decrease in the diversity of the microbiome of LC patients as compared to controls (Table 3). Seven representatives of unique bacterial genera were found in patients with LC compared to 14 in the controls.
Our data are consistent with previously published data showing that the lung microbiota in LC patients has a less complex composition. LC patients are distinguished by a decrease in the alpha diversity of the lung microbiota41.
Comparison of the microbiome composition in the sputum of patients with lung cancer of the main histological types (squamous cell carcinoma, adenocarcinoma and large-cell carcinoma) at the species and genera level did not reveal significant differences, except for the occurrence of bacteria of the genus Bergeyella. In the sputum of patients diagnosed with adenocarcinoma, representatives of Bergeyella were recorded at a higher frequency than in patients with squamous cell carcinoma (0.4 ± 0.24% and 0.04 ± 0.1%, respectively; p = 0.045).
Comparison of the microbiome in the sputum of patients with lung cancer of different stages (I-II and III-IV) at the species and genera level did not reveal significant differences.
Comparison of the microbiome in the sputum of patients with lung cancer with metastases in distant organs and without them in distant organs at the species and genera level did not revealatopobium significant differences, with the exception of the genus Atopobium, whose representatives were much less frequently detected in patients with metastases (0.21 ± 0.43% and 1, 37 ± 0.084%, respectively; p = 0.017), in particular, species of Atopobium rimae (0.21 ± 0.43% and 1.21 ± 0.089%, respectively; p = 0.034).
Interestingly, there was a different and non overlapping representation of bacterial species in the sputum microbiome of smokers as compared to non smokers in both LC patients and healthy participants in the study.
We examined the chromosome aberrations in the blood lymphocytes of all participants in the study and found that there is a significant correlation between the level of bacteria of a certain species in the sputum and chromosomal aberrations (CA).
Representatives of the genus Alloprevotella were most frequently associated with chromosomal aberrations in all participants in our study (Fig. 3). Bacteria of this genus have recently been identified as reliably associated with the metastatic stage of melanoma42.
Interestingly, representatives of the genus Actinobacillus were found only in the group of participants with high levels of CA. Representatives of the genus Actinobacillus (A.ureae and A. hominis) were previously found in the respiratory tract of healthy people and can cause bronchopneumonia and meningitis43.
Previous publications reported an increase in chromosome damage in the lymphocytes of primary LC patients44,45,46. The results obtained in our study confirm this (Table 4).
This increase in the level of cytogenetic lesions cannot be explained by the consequences of radiation or chemical therapy, since biological material from patients was collected before any treatment procedures. Instead, the cytogenetic instability of somatic cells in untreated patients with LC is a sign that reflects the influence of endogenous genotoxic factors on the human genome. In particular, one such factor can be oxidative stress, which is an indispensable attribute of the tumor process. In addition, the possible clastogenic effects of the bacterial environment in the lung tissue cannot be ruled out as causative agent. The influence of lung microbiota on lung carcinogenesis, immunity and immunotherapy is summarized in a recent review, the major points of which agree with our results as follows: the microbiota of the healthy lung is different from neoplastically transformed lung; bacterial products might promote host oncogene activation; the lung immune system is under the influence of the microbiota47.
It would be interesting to conduct a screening of the microbiome of the respiratory tract on a larger scale and for a longer period of time, for example, over three years, in order to identify the types of bacteria detected by us among healthy people from the same age group and region, in order to compare the data with the subsequent diagnosis of lung cancer and other diseases of the respiratory tract.
We plan to confirm the representation of different bacterial species detected by us in the sputum of patients with LC and healthy subjects by other methods, for example, by the method of specific quantitative PCR.
Conclusion
The method of mass parallel sequencing of 16S ribosomal genes was used to determine the taxonomic composition of the microbiome in the sputum of LC patients and healthy donors (for the first time in the Russian population). Bacterial taxonomic groups have been identified where the microbiome composition differs significantly in patients as compared to controls. The discrepancy between our data and the results of previous studies of the LC microbiome probably reflects the specific epidemiological circumstances of the environment in the population we studied, a region with intensive coal mining and processing. The results obtained for the taxonomic composition of the microbiome rely on experimental data that will be further confirmed using a larger number of patients and control groups. The sputum microbiome, although it does not reflect the specific location of the respiratory tract, can potentially serve as an important non-invasive biomarker in LC. Our results show a correlation between chromosomal aberration of host genomes with bacterial representation in pharyngeal microbiome.
Thus, a comparison of the bacterial composition in the sputum of donors with cytogenetic damages in theirs lymphocytes, warrants further investigations on the potential role of microorganisms in the process of mutagenesis in somatic cells of the host body.
Knowledge of the specific composition of the microbiota in the respiratory tract of a patient will make it possible to predict the effectiveness of chemoradiotherapy, gene therapy, immunotherapy and other treatment methods, and may also contribute to the development of innovative strategies for early prevention and personalized treatment of lung cancer.
References
Wong, M. C. S. et al. Incidence and mortality of lung cancer: global trends and association with socioeconomic status. Sci. Rep. 30, 14300, https://doi.org/10.1038/s41598-017-14513-7 (2017).
de Groot, P. & Munden, R. F. Lung cancer epidemiology, risk factors, and prevention. Radiol. Clin. North. Am. 50, 863–876, https://doi.org/10.1016/j.rcl.2012.06.006 (2012).
Sun, S. et al. Lung cancer in never smokers–a different disease. Nat. Rev. Cancer 7, 778–790, https://doi.org/10.1038/nrc2190 (2007).
Brenner, D. R. et al. Lung cancer risk in never-smokers: a population-based case-control study of epidemiologic risk factors. BMC Cancer 14, 285, https://doi.org/10.1186/1471-2407-10-285 (2010).
Eckel, S. P. et al. Air pollution affects lung cancer survival. Thorax 71, 891–898, https://doi.org/10.1136/thoraxjnl-2015-207927 (2016).
Christian, W. J. et al. Exploring geographic variation in lung cancer incidence in Kentucky using a spatial scan statistic: Elevated risk in the Appalachian coalmining region. Public. Health Rep. 126, 789–796, https://doi.org/10.1177/003335491112600604 (2011).
Field, R. W. Radon: A Leading Environmental Cause of Lung Cancer. Am. Fam. Physician 98, 280–282 (2018).
Mao, Q. et al. Interplay between the lung microbiome and lung cancer. Cancer Lett. 28, 40–48, https://doi.org/10.1016/j.canlet.2017.11.036 (2018).
Jin, J. et al. Diminishing microbiome richness and distinction in the lower respiratory tract of lung cancer patients: A multiple comparative study design with independent validation. Lung Cancer 136, 129–135, https://doi.org/10.1016/j.lungcan.2019.08.022 (2019).
Hosgood, H. D. 3rd et al. The respiratory tract microbiome and its relationship to lung cancer and environmental exposures found in rural china. Env. Mol. Mutagen. 60, 617–623, https://doi.org/10.1002/em.22291 (2019).
Wang, K. et al. Preliminary Study of Microbiota Diversity in Saliva and Bronchoalveolar Lavage Fluid from Patients with Primary Bronchogenic Carcinoma. Med. Sci. Monit. 17, 2819–2834, https://doi.org/10.12659/MSM.915332 (2019).
Liu, Y. et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am. J. Cancer Res. 8, 1775–1787 (2018).
Lee, S. H. et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 102, 89–95, https://doi.org/10.1016/j.lungcan.2016.10.016 (2016).
Erb-Downward, J. R. et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 22, e16384, https://doi.org/10.1371/journal.pone.0016384 (2011).
Cameron, S. J. S. et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One 25, e0177062, https://doi.org/10.1371/journal.pone.0177062 (2017).
Laroumagne, S. et al. Incidence and characteristics of bronchial colonisation in patient with lung cancer: a retrospective study of 388 cases. Fr. Rev. Mal. Respir. 28, 328–335, https://doi.org/10.1016/j.rmr.2010.05.020 (2011).
Yan, X. et al. Discovery and validation of potential bacterial biomarkers for lung cancer. Am. J. Cancer Res. 15, 3111–3122 (2015).
Hosgood, H. D. 3rd et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Env. Mol. Mutagen. 55, 643–651, https://doi.org/10.1002/em.21878 (2014).
Chen, J. et al. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin. Immunol. 32, 25–34, https://doi.org/10.1016/j.smim.2017.08.001 (2017).
Yu, G. et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 28, 163, https://doi.org/10.1186/s13059-016-1021-1 (2016).
Jiang, L. X. et al. Simultaneous Detection of 13 Key Bacterial Respiratory Pathogens by Combination of Multiplex PCR and Capillary Electrophoresis. Biomed. Env. Sci. 30, 549–561, https://doi.org/10.3967/bes2017.074 (2017).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022, https://doi.org/10.1038/nm.2015 (2009).
Grasso, F. & Frisan, T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 11, 1762–1782, https://doi.org/10.3390/biom5031762 (2015).
Chumduri, C. et al. Subversion of host genome integrity by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 17, 659–673, https://doi.org/10.1038/nrm.2016.100 (2016).
Druzhinin, V. G. et al. Induction and modulation of genotoxicity by the bacteriome in mammals. Mutat. Res. 776, 70–77, https://doi.org/10.1016/j.mrrev.2018.04.002 (2018).
Goldstraw, P. New staging system: how does it affect our practice? J. Clin. Oncol. 31, 984–991, https://doi.org/10.1200/JCO.2012.42.7922 (2013).
Hungerford, P. A. Leukocytes cultured from small inocula of whole blood and the preparation of metaphase chromosomes by treatment with hypotonic KCl. Stain. Techn 40, 333–338 (1965).
Savage, J. R. Classification and relationships of induced chromosomal structural changes. J. Med. Genet. 13, 103–122 (1976).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857, https://doi.org/10.1038/s41587-019-0209-9 (2019).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. microbiology 71, 8228–8235, https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Peters, B. A. et al. The Microbiome in Lung Cancer Tissue and Recurrence-Free Survival. Cancer Epidemiol. Biomarkers Prev. 28, 731–740, https://doi.org/10.1158/1055-9965.EPI-18-0966 (2019).
Angelakis, E. et al. Human case of Atopobium rimae bacteremia. Emerg. Infect. Dis. 15, 354–355, https://doi.org/10.3201/eid1502.071399 (2009).
Ye, W. et al. Relationship of tongue coating microbiome on volatile sulfur compounds in healthy and halitosis adults. J. Breath. Res. 19, 016005, https://doi.org/10.1088/1752-7163/ab47b4 (2019).
Duan, X. et al. Smoking May Lead to Marginal Bone Loss Around Non-Submerged Implants During Bone Healing by Altering Salivary Microbiome: A Prospective Study. J. Periodontol. 88, 1297–1308, https://doi.org/10.1902/jop.2017.160808 (2017).
Sizova, M. V. et al. Stomatobaculum longum gen. nov., sp. nov., an obligately anaerobic bacterium from the human oral cavity. Int. J. Syst. Evol. Microbiol. 63, 1450–1456, https://doi.org/10.1099/ijs.0.042812-0 (2013).
Demmer, R. T. et al. Subgingival Microbiota and Longitudinal Glucose Change: The Oral Infections, Glucose Intolerance and Insulin Resistance Study (ORIGINS). J. Dent. Res. 98, 1488–1496, https://doi.org/10.1177/0022034519881978 (2019).
Lorenzo de Arriba, M. et al. Characterisation of Bergeyella spp. isolated from the nasal cavities of piglets. Vet. J. 234, 1–6, https://doi.org/10.1016/j.tvjl.2018.01.004 (2018).
Decostere, A. et al. Bergeyella (Weeksella) zoohelcum associated with respiratory disease in a cat. Veterinary Record 151, 392, 0.1136/vr.151.13.392 (2002).
Chen, Y. et al. Bacteremia caused by Bergeyella zoohelcum in an infective endocarditis patient: case report and review of literature. BMC Infect. Dis. l2, 271, https://doi.org/10.1186/s12879-017-2391-z (2017).
Kovaleva, O. V. et al. Human Lung Microbiome on the Way to Cancer. J. Immunol. Res. 29, 1394191, https://doi.org/10.1155/2019/1394191 (2019).
Vétizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 27, 1079–1084, https://doi.org/10.1126/science.aad1329 (2015).
Jain, V. & Bhardwaj, A. Pneumonia Pathology. Treasure Island (FL): StatPearls Publishing, http://www.ncbi.nlm.nih.gov/books/NBK526116/ (2019).
Lou, J. et al. Investigating the genetic instability in the peripheral lymphocytes of 36 untreated lung cancer patients with comet assay and micronucleus assay. Mutat. Res. 617, 104–110, https://doi.org/10.1016/j.mrfmmm.2007.01.004 (2007).
Minina, V. I. et al. Chromosome aberrations in peripheral blood lymphocytes of lung cancer patients exposed to radon and air pollution. Eur. J. Cancer Prev. 27, 6–12, https://doi.org/10.1097/CEJ.0000000000000270 (2018).
El-Zein, R. A. et al. Identification of Small and Non-Small Cell Lung Cancer Markers in Peripheral Blood Using Cytokinesis-Blocked Micronucleus and Spectral Karyotyping Assays. Cytogenet. Genome Res. 152, 122–131, https://doi.org/10.1159/000479809 (2017).
Ramirez-Labrada, A. G. et al. The Influence of Lung Microbiota on Lung Carcinogenesis, Immunity, and Immunotherapy. Trends Cancer 2, 86–97, https://doi.org/10.1016/j.trecan.2019.12.007 (2020).
Acknowledgements
We would like to thank all patients and controls for donating their samples and for collaborating in this study. This work was supported by Russian Science Foundation Grant No. 18-14-00022. Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
D.V.G. and M.L.V. conceived the study; D.V.G. and M.L.V. wrote the manuscript; B.E.D., V.V.P., M.V.I., A.S.V., C.M.A. and R.S.A. performed laboratory work; D.P.S. and I.V.I. carried out bioinformatics and statistical analyses; D.V.G. organized the supply of clinical samples, S.S.G. and L.A.V. critically revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Druzhinin, V.G., Matskova, L.V., Demenkov, P.S. et al. Taxonomic diversity of sputum microbiome in lung cancer patients and its relationship with chromosomal aberrations in blood lymphocytes. Sci Rep 10, 9681 (2020). https://doi.org/10.1038/s41598-020-66654-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66654-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
