
Abstract
β-hemoglobinopathies are caused by abnormal or absent production of hemoglobin in the blood due to mutations in the β-globin gene (HBB). Imbalanced expression of adult hemoglobin (HbA) induces strong anemia in patients suffering from the disease. However, individuals with natural-occurring mutations in the HBB cluster or related genes, compensate this disparity through γ-globin expression and subsequent fetal hemoglobin (HbF) production. Several preclinical and clinical studies have been performed in order to induce HbF by knocking-down genes involved in HbF repression (KLF1 and BCL11A) or disrupting the binding sites of several transcription factors in the γ-globin gene (HBG1/2). In this study, we thoroughly compared the different CRISPR/Cas9 gene-disruption strategies by gene editing analysis and assessed their safety profile by RNA-seq and GUIDE-seq. All approaches reached therapeutic levels of HbF after gene editing and showed similar gene expression to the control sample, while no significant off-targets were detected by GUIDE-seq. Likewise, all three gene editing platforms were established in the GMP-grade CliniMACS Prodigy, achieving similar outcome to preclinical devices. Based on this gene editing comparative analysis, we concluded that BCL11A is the most clinically relevant approach while HBG1/2 could represent a promising alternative for the treatment of β-hemoglobinopathies.
Similar content being viewed by others

Introduction
Sickle cell disease (SCD) and β-thalassemia, commonly known as β-hemoglobinopathies, are inherited blood disorders caused by mutations in the human β-globin gene (HBB)1,2,3,4. In healthy condition, adult human hemoglobin (HbA) consists of 2 α and 2 β chains, whereas fetal hemoglobin (HbF) expressed in early gestation comprises 2 α chains and 2 γ chains. Notably, HbF was observed to bind oxygen with greater affinity than HbA, being functional when reactivated in adults3,5,6.
Recent studies have generated substantial experimental evidence that HbF reactivation by gene disruption of specific transcription factors and regulators could provide a therapeutic benefit for β-hemoglobinopathies7. It has long been appreciated that KLF1 and BCL11A are key regulators involved in the process of γ- to β-globin switching and the repression of these genes leads to HbF resurgence6,7,8,9,10,11. Interestingly, healthy individuals with a benign genetic condition namely hereditary persistence of fetal hemoglobin (HPFH) were observed to exhibit persistent production of functional HbF4,10,12,13. HPFH is caused by large deletions in the δ- and β-globin genes, or point mutations in the γ-globin promoter and γ-globin repressors, such as KLF1 and BCL11A5. Importantly, co-inheritance of HPFH with β-thalassemia was noticed to alleviate the clinical manifestations of the latter14. Therefore, to attain a clinical profit for β-hemoglobinopathies, several attempts were made to re-establish the expression of HbF either by lentiviral transfer of the γ-globin gene or by CRISPR/Cas9-mediated gene disruption of specific regulators14,15,16,17,18,19.
Though lentiviral gene transfer of β-globin exhibited positive effects in treated β-thalassemia patients20, the high volume of semi-random integration sites by lentivirus and the transactivation of the proto-oncogene HMGA2 raised major safety concerns for this approach21,22. Due to the afore-mentioned reasons, CRISPR/Cas9-mediated gene disruption of specific regulators to re-express HbF is a promising alternative7. Thus, several studies have targeted various genetic regulators by CRISPR/Cas9 to reactivate HbF expression, resulting in a profound effect after genetic interference of BCL11A, KLF1, and HBG1/2 promoters14,17,23. Nevertheless, no head-to-head comparison has been performed earlier in CD34+ hematopoietic stem and progenitor cells (HSPCs) for these three targets to assess their therapeutic potential for β-hemoglobinopathies by up-regulating HbF without raising safety issues. Therefore, in the present study, we compared all these targets in parallel for their impact on HbF resurgence and performed safety measurements by molecular analyses in order to select the best candidate for clinical translation.
Results
Gene editing
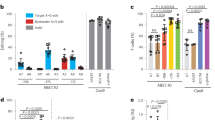
First, we established the optimal electroporation parameters to transfect exogenous mRNA in K-562 cells and CD34+ HSPCs utilizing a DsRed reporter construct. Best electroporation settings were chosen for both K-562 cells (1450 V, 10 ms, 3 pulses) and CD34+ HSPCs (1650 V, 10 ms, 3 pulses) where high transfection efficiency and viability were achieved (>90%; Supplementary Fig. S1a). Further, to validate sgRNAs we electroporated K-562 cells with recombinant pX-330 vector targeting KLF1, BCL11A, and HBG1/2 genomic regions. Each locus was targeted with two different sgRNAs (Fig. 1a) and gene-targeting efficacy was assessed by T7 endonuclease-I (T7E1) assay. Varying levels of mean indel frequencies were observed for KLF1 (T1: 36.2 ± 6.5%; T2: 34.9 ± 5.1%), BCL11A (T1: 22.2 ± 2.2%; T2: 17.0 ± 1.4%), and HBG1/2 (T1: 30.9 ± 14.4%; T2: 21.1 ± 6.0%; Supplementary Fig. S1b).

Gene editing of human CD34+ HSPCs. (a) Schematic representation of the genome-editing strategies and target sequences for each sgRNA. KLF1: targets 1 (T1) and 2 (T2) are localized in the second and third exon, respectively; BCL11A: both targets are situated in the enhancer region of the second intron; HBG1/2: both sgRNAs target HBG1 and HBG2 promoters. (b) Indel percentage in CD34+ HSPCs measured by ICE analysis after electroporation of Cas9 RNP and chemically-modified sgRNAs for T1 and T2 in KLF1, BCL11A, and HBG1/2. (c) Flow cytometry analysis after immunostaining of CD34+ HSPCs to follow differentiation into erythrocytes precursors: percentage of double positive cells for CD34+ (hematopoietic stem cells) and CD45+ (leukocytes), and for CD71 (erythroid precursors) and CD235a (erythrocyte) on day 0 and 21, respectively.
Next, to assess CRISPR/Cas9-mediated HbF up-regulation strategy with bone marrow-derived CD34+ HSPCs, we utilized Cas9 RNP with chemically modified sgRNAs instead of pX-330 vectors as the latter was shown to be less effective. We tested several molar ratios of sgRNA:Cas9 and found that a molar ratio of 2:1 was more effective in generating on-target indels (data not shown). Interestingly, elevated levels of gene editing were noticed in ICE analysis for all the tested sgRNAs (range of 63–91%; Fig. 1b), except for HBG1/2 T1 where lower indels (54.7 ± 10.1%) were spotted. Later, gene-edited CD34+ HSPCs were differentiated towards erythroid lineage for 21 days, confirmed with specific erythroid markers expression (CD71 and CD235a), and molecularly analyzed for HbF expression. None of the treated samples showed proliferation or impaired erythroid differentiation (Fig. 1c).
Transcript analysis of γ-globin, KLF1 and BCL11A
Our qRT-PCR analyses showed that, compared to control samples, HbF up-regulation was noted in KLF1-edited samples (>5 fold) and BCL11A (>4 fold) for both tested targets. Notably, elevated γ-globin transcripts were observed in HBG1/2 gene-targeted samples (>6.5 fold; Fig. 2a). Also, KLF1 and BCL11A transcripts were quantitatively determined by qRT-PCR, showing a marked down-regulation of KLF1 transcripts (KLF1 T1: 4 fold, KLF1 T2: 2 fold; Fig. 2b) with a characterized subsequent BCL11A down-regulation (~2 fold; Fig. 2c) after KLF1 gene disruption. Following the same pattern, a 2-fold down-regulation of BCL11A transcripts was observed only in BCL11A T2 when the enhancer of this gene was genetically disrupted (Fig. 2c).

mRNA and protein analysis of gene-edited CD34+ HSPCs. (a) γ-globin expression analysis by qRT-PCR on day 21. (b) Decline of KLF1 transcripts after treatment with KLF1 T1 and T2. (c) Down-regulation of BCL11A transcripts in KLF1 and BCL11A samples. (d) HPLC histograms of control, KLF1, BCL11A, and HBG1/2 samples. (e) Percentage of HbF for human standard (HS), control, and the different gene editing treatments by HPLC on day 21. (f) HbF Intracellular staining in differentiated CD34+ HSPCs on day 21. (g) Spearman correlation of HPLC and HbF intracellular staining.
HbF quantification by intracellular staining and HPLC
In order to assess HbF expression at protein level in gene-edited CD34+ HSPCs, cells were analyzed by HPLC-mediated hemoglobin electrophoresis and intracellular staining. Notably, hemoglobin electrophoresis revealed that all the edited samples induced higher HbF levels in comparison to the controls (Fig. 2d), while BCL11A T2 and HBG1/2 T2 achieved the most pronounced HbF levels up to 39.5 and 41.9%, respectively (Fig. 2e). Moreover, in vitro differentiation of non-edited CD34+ HSPCs into erythrocyte precursors produced similar amounts of HbF as the standard human controls (Fig. 2e). After flow cytometry analysis, we found elevated numbers of HbF+ CD34+ HSPCs for all the tested target genes (range 50.8–91.7%) where the strongest effect was noted for HBG1/2 T2 (Fig. 2f and Supplementary Fig. S1c). Of note, hemoglobin electrophoresis results strongly correlated with HbF intracellular staining (Spearman’s rho coefficient: ρ = 0.799, p < 0.0001; Fig. 2g).
Expression pattern analysis by RNA-seq
Since KLF1 and BCL11A are transcription factors involved in several signaling pathways, RNA-seq analysis was performed to determine the safety of each gene editing profile. We accounted for relatively similar ICE scores (KLF1 T1: 77 ± 8.9%; BLC11A T2: 86 ± 2.5%; HBG1/2 T2:84.7 ± 9.3%) and HbF levels (KLF1 T1: 23.2 ± 3%; BLC11A T2: 34.3 ± 0.7%; HBG1/2 T2: 39.6 ± 0.2%) to choose the samples for RNA-seq. We noticed that the expression patterns of all three gene-disrupted CD34+ HSPC treatments showed high similarity rates to the control sample (from 92% to 99%). Importantly, a mean value of 1017 genes showed dissimilarity in the BCL11A sample, while KLF1 and HBG1/2 gene disruption led to 2327 and 2129 impaired genes, respectively (Fig. 3a). However, when considering the common differentially expressed genes across all replicates, a clear pattern was observed, where KLF1 resulted in 502 impaired genes, whereas BCL11A and HBG1/2 accounted for 10 and 82 dysregulated genes, respectively (Fig. 3b,c). From those genes, a deeper screening for dysregulated oncogenes or tumor suppressor genes was performed to assess the safety profile of each gene editing approach. These results showed the presence of several disturbed genes involved in cell cycle (E2F2, E2F7), ERK/MAPK and p53 signaling (DUSP2, PPP2R5B, TRIM29), apoptosis (DAPK1), and immune pathways (BCL6) for KLF1- and HBG1/2-treated samples. In contrast, neither oncogenes nor tumor suppressor genes were found for BCL11A samples. Noteworthy, all dysregulated genes for BCL11A-treated samples were found in KLF1 expression panel, except for ALB, IL18R, and MMP25.

RNA-seq analysis. (a) Number of genes down- and up-regulated after targeting KLF1, BCL11A, and HBG1/2. (b) Number of common dysregulated genes for the three individual experiments. (c) Heatmap showing the Log2 fold change of the common differentially expressed genes after gene editing in KLF1, BCL11A, and HBG1/2. R1, R2, and R3 represent the three performed replicates. Dysregulated genes are depicted in a blue-to-red color gradient, where down-regulated genes are shown in blue and up-regulated genes in red. (d) Number of reads indicated as clusters per million (cpm) for hemoglobin genes in edited samples relative to the control sample. (e,f) Cpm values for the most relevant genes regulated by KLF1 (e) and BCL11A (f).
We also assessed the expression values indicated as clusters per million (cpm) for genes of interest involved in hematopoiesis (Fig. 3d). Predictably, adult-to-fetal hemoglobin switching was noticed in our RNA-seq results, where HBG1/HBG2 up-regulation was noted in all treated samples. Furthermore, up-regulation of HBA1/HBA2 was observed in those treatments where high levels of γ-globin transcripts were perceived. Alternatively, no major changes were detected in HBB expression (Fig. 3d). We also screened for important genes regulated by KLF1 and BCL11A with no dissimilarities found when compared to the control, except for ITGA2B (Fig. 3e,f).
Off-target analysis by GUIDE-seq
Most importantly, GUIDE-seq analysis was performed after assessing the optimal dsODN concentration to achieve the highest integration rate with reduced cytotoxicity (Fig. 4a,b). Using 25 pmol of dsODN, we obtained adequate integration index for the most efficient targets (KLF1 T1: 8.55 ± 5.6%, BCL11A T2: 7.45 ± 1.8%; HBG1/2 T2: 5.95 ± 1.5%; Fig. 4c). We also included a sgRNA that targeted the promoter region of HBB (BetaPr) for which numerous in silico off-targets were predicted (Table S3, Fig. 4c). This way, our GUIDE-seq analysis resulted in no detectable off-targets, except for HBG1/2 and BetaPr, where 1 and 39 off-targets were determined (Fig. 4d–g). Interestingly, two on-targets were identified with low number of reads for HBG1/2 T2 after GUIDE-seq (Fig. 4f). We hypothesized that, after gene editing, a 5-kb deletion between HBG1 and HBG2 could restrain GUIDE-seq results. With this aim in mind, we designed ddPCR oligonucleotides within the intergenic region of HBG2 and HBG1 (Table S2) and observed up to 43% of 5-kb excision in gene-edited samples after HBG1/2 T2 transfection utilizing our electroporation devices (Fig. 4h).

GUIDE-seq analysis. (a) dsODN titration in K-562 for GUIDE-seq optimization. The percentage of dsODN integration for each specific dsODN count of particle (5, 15, 25, and 35 pmol) is depicted. (b) Cell viability determined by cell counting on day 5 post-electroporation. (c) Percentage of dsODN integration at the target sites of interest utilizing 25 pmol of dsODN. Mean indel rates determined by ICE analysis are shown for each sgRNA. (d–g) Off-target illustration for KLF1 T1 (d), BCL11A T2 (e), HBG1/2 T2 (f), and BetaPr (g). Target sequences are shown on the top; matches are represented with dots, while mismatches are highlighted underneath. GUIDE-seq reads are shown on the right of each on-target/off-target site. (h) Percentage of CD34+ HSPCs with excised intergenic region of HBG1/2 (5-Kb) after gene editing by Neon System and CliniMACS Prodigy.
Translation of the gene editing platform to CliniMACS Prodigy
Due to the promising outcome of all gene editing strategies, especially HBG1/2 and BCL11A, the next step was to translate this platform to the GMP-grade CliniMACS Prodigy. As previously performed in the Neon System, the best electroporation setting was selected based on DsRed mRNA transfection efficiency in CD34+ HSPCs, where the pulse mode ‘Square’, 600 V/100 µs first pulse, and 300 V/2 ms second pulse, was the setup combination that attained the highest percentage of DsRed+ cells (78.1 ± 7.2%) and cell viability (88.4 ± 13%; Supplementary Fig. S1d). Subsequently, KLF1 T1, BCL11A T2 and HBG1/2 T2 sgRNAs were transfected with the above-explained CliniMACS Prodigy settings, noticing excellent gene editing performance comparable to Neon Transfection System (range of 54–86%; Fig. 5a). Likewise, after erythroid differentiation for 21 days, HSPCs showed similar HbF resurgence by HPLC compared to Neon-transfected cells (KLF1 T1: 19.7 ± 2.8%, BCL11A T2: 40.67 ± 7.8%; HBG1/2 T2: 41.7 ± 3.6%; Fig. 5b), indicating once again the potential of these approaches for clinical translation.

Translation of the CRISPR/Cas9 gene editing platform to the CliniMACS Prodigy GMP-grade device. (a) Percentage of insertions and deletions (InDels) detected by ICE analysis. (b) HbF levels (%) in edited human CD34+ HSPCs measured by HPLC.
Discussion
Earlier studies relied on the fact that β-thalassemia patients with natural mutations in genetic regulators involved in the fetal-to-adult hemoglobin switching, reactivate the expression of HbF, ameliorating the disease symptoms24. Furthermore, mutations or epigenetic modifications in the distal promoter of HBG1/2 can induce a similar outcome, as observed in the rare benign HPFH14,25,26,27,28. Above-mentioned genetic variants trigger a dynamic process, in which the HbF levels increase whereas HbA decreases. When HbF levels are above 20%, it is demonstrated to be therapeutically beneficial, especially in SCD patients due to the reduction of hemoglobin polymerization24,26,29,30. Therefore, owing to its great clinical impact, several investigations were attempted to induce HbF, either by gene disruption or by gene transfer10,14,17,31. Though gene disruption is a promising option, no comparative analyses were performed to date for these genetic loci to choose the best approach for possible clinical applications. Hence, we chose KLF1, BCL11A, and HBG1/2 promoters as the three prominent gene regulators of ‘Globin Switching’ and performed one-to-one comparison for HbF resurgence efficacy and safety using CRISPR/Cas9-mediated gene disruption.
Several mutations have been described for KLF1 in HPFH patients which generates variable levels of HbF (3–30.9%)32. In particular, our study is the first lentiviral-free attempt to use CRISPR/Cas9 to target exon 2 and 3 of KLF1 in CD34+ HSPCs. We successfully induced high indel rates in both exons, down-regulation of KLF1 and BCL11A transcripts, elevated γ-globin mRNA expression, and significant HbF levels comparable to HPFH mutations such as K288X and S270X32,33. Although KLF1 gene disruption resulted in increased HbF (up to 25%) and no off-targets were detected by GUIDE-seq, the negative effect of KLF1 knock-down was observed in our RNA-seq analysis, where several genes with different biological functions were dysregulated compared to the non-edited sample, which could raise major safety concerns. Previous studies have documented that impaired expression of KLF1 might affect the expression of genes involved in cell-cell interaction (CD44 and ITGA2B), microcytosis (AQP1) and cancer (FLI-1)32,34. Nevertheless, in our RNA-seq analysis no differences were perceived for those genes, except for ITGA2B, which its down-regulation is associated to Glanzmann thrombasthenia, a bleeding disorder characterized by a lack of platelets aggregation35.
Another important transcription factor implicated in γ- to β-globin switching is BCL11A24,36,37,38,39, which has become a promising target for HbF resurgence. Likewise, long-term engraftment and normal hematopoiesis could be attained down-regulating BCL11A by targeting its enhancer (GATAA box)16,23,40, whilst impaired erythropoiesis and limited engraftment have been detected in complete knock-down of BCL11A gene23,30,36. Therefore, in our study, we selected two sgRNAs matching the GATAA box and achieved high level of genetic disruption with excellent HbF increment up to ~40% for sgRNA T2. Due to the essential role of BCL11A in globin switch, regulation of proto-oncogenes (MDM2 and TEP1), and genes involved in immune cell development (IL7-R and FLT3)37,41,42, we performed RNA-seq analyses for BCL11A T2 gene-edited samples. BCL11A gene disruption approach resulted in normal expression of the above-mentioned genes and led to the lowest gene expression variation of all different strategies. This low variation was expected since we are targeting the enhancer box located in the second intron of the BCL11A gene, and therefore, its expression declines but there is no total suppression which could affect hematopoiesis. Indeed, dysregulated genes for the BCL11A approach could also be found in KLF1 treated samples, where their expression was highly impaired since KLF1 knock-out induces stronger down-regulation of BCL11A than targeting directly its enhancer. Also, no off-targets were detected for BCL11A T2, hence this strategy showed high level of safety profile. In a complementary manner, a recent study performed by the group of Wu et al. confirmed the safety and efficacy of this sgRNA, which together with our findings could contribute in the current clinical trial for the treatment of β-thalassemia (NCT03655678) and SCD (NCT03745287)43.
The gamma chain of HbF is encoded by the HBG1/2 genes and transcriptionally controlled by several elements in the β-globin gene cluster. Interestingly, the elevation of HbF by natural-occurring deletions in the β-globin cluster, varying from 13 bp, 7.2 kb (Corfu), 12.9 kb (Sicilian) and 13.6 kb, have been identified in HPFH individuals7,14,26,27,28. Furthermore, previous studies using ChIP-seq and CUT&RUN analyses elucidated the consensus binding site of BCL11A (TGACCA) repressor, situated upstream of the transcription start site of the γ-globin gene (−115 bp)28,44. Therefore, similarly to what was previously performed by Traxler et al., we utilized two sgRNAs to target the binding site of BCL11A, mimicking the 13-bp natural-occurring deletion described previously in HPFH individuals. This 13-bp deletion has been described before after gene editing, and might occur via microhomology-mediated end-joining (MMEJ) due to short homology sequences flanking the target site that can be observed in our ICE analysis results (Supplementary Fig. S1e)14,28,44,45 Notably, HBG1/2 T2 induced higher γ-globin and HbF levels compared to KLF1 and BCL11A approaches. Nonetheless, RNA-seq analysis showed that HBG1/2 approach has a better safety profile than KLF1 but lower than BCL11A. After screening for possible oncogenes or tumor suppressor genes, we detected potential genes involved in oncogenesis as also observed in RNA-seq for KLF1-treated samples. However, to definitely confirm that those genes will not promote cancer development, gene dysregulation should also be assessed by other methods such as qRT-PCR, clonal expansion assays, and in vivo models. In fact, a recent publication has reported long-term engraftment of HBG1/2-edited HSPCs in rhesus primates and no toxic effects were found in mature blood lineages after a follow-up of 1.5 years46.
Since HBG1/2 consists of two homologous subunits (HBG1 and HBG2) with just some differences in the upstream region of the distal promoter, this sgRNA cleaves twice in the genome. In the present study, we confirmed that, when the sgRNA cuts simultaneously in both subunits, a 5-kb fragment is excised in high frequencies (up to 43%). Thus, the detection of the on-target reads during GUIDE-seq analysis is hindered and can only be identified when the DSB occurs either in HBG1 or HBG2. These two on-targets were found in our GUIDE-seq results at low number of reads due to the above-mentioned reasons. Also, one off-target at low frequency (5 reads) was detected by GUIDE-seq for HBG1/2 sgRNA. BLAST analysis matched the off-target sequence with an intergenic region (NC_000002.12) located in chromosome 2 downstream of the ATP synthase F(0) complex subunit C3 gene and upstream of the endoplasmic reticulum junction formation factor gene. This region is associated with a long-non coding RNA (lncRNA), which might have several biological roles including epigenetic regulation47. This could elucidate the variation found in the transcript expression profile after gene editing, but the low number of detected reads and the uncertain function of this lncRNA lead to the inability to draw final conclusions.
To evaluate whether these approaches can be transferred to a GMP-grade electroporation device, CliniMACS Prodigy was utilized for the best sgRNAs of our study. The system is noteworthy due to its GMP-compatibility and offers automated electroporation of CRISPR components, cell culture, and direct application into humans with a clinical grade quality. Most importantly, similar results to the Neon Transfection System were attained, demonstrating the clinical potential of these gene therapy approaches. Nonetheless, in vivo experiments must be performed to evaluate the engraftment capacity of gene-edited cells.
Based on this thorough comparative analysis of different HbF-inducing gene editing strategies, we concluded that KLF1 is not a suitable approach for clinical translation due to impaired gene expression after gene editing. On the contrary, BCL11A is a great candidate for the treatment of β-hemoglobinopathies, with high HbF resurgence, no off-targets, and unaltered gene expression. In addition, the above-explained HBG1/2 approach also yielded clinically relevant levels of HbF with mediocre safety profile, and thus, after further investigations, this strategy could be considered a promising alternative gene therapy for β-hemoglobinopathies.
Materials and Methods
Ethics approval
Human mobilized peripheral blood CD34+ HSPCs from individual donors were adquired using protocols approved by the local ethics committee/institutional review board (IRB; ethic number: 829/2016BO2), University Children’s Hospital. Written informed consents were obtained from all the participants in the study. All methods were carried out in accordance with relevant guidelines and regulations.
Cell culture
K-562 cells were acquired from Sigma-Aldrich and cultured at 37 °C with 5% CO2 in RPMI (Biochrom) supplemented with 10% FBS (Gibco), 1% L-glutamine (Biochrom), and 1% Penicillin/Streptomycin (Biochrom).
Immunomagnetic enrichment of HSPCs was performed using magnetic-activated cell sorting system (CliniMACS System, Miltenyi Biotec), according to the manufacturer’s instructions. CD34+ HSPCs were then cultured at 37 °C with 5% CO2 in StemMACS HSC Expansion Media (Miltenyi Biotec) supplemented with human cytokines (Miltenyi Biotec): SCF (100 ng/ml), TPO (20 ng/ml), and Flt3-L (100 ng/ml).
DsRed mRNA in vitro synthesis
Before mRNA transcription, 20 µg pCS2+ DsRed was digested by XbaI (New England Biolabs) for 1 hour at 37 °C. Linearized plasmid was then purified using QIAquick PCR Purification Kit (QIAGEN) following the manufacturer’s instructions. Finally, DsRed mRNA in vitro transcription, poly(A) tailing, and mRNA clean up were performed in accordance with the manufacturer’s protocols of mMESSAGE mMACHINE SP6 Transcription kit (Life Technologies), Poly (A) tailing kit (Ambion), and MEGAclear kit (Ambion), respectively.
Cloning of oligonucleotides in pX330
sgRNAs for each targeted gene were designed (Fig. 1a) and oligonucleotides cloned into the chimeric pX-330 vector (Addgene #42230; Table S1). All constructs were confirmed by Sanger sequencing. The amplification of the vectors was performed in DH5α competent cells (Sigma-Aldrich) and the purification of the plasmids by means of standard plasmid isolation kits (Peqlab Biotechnologie).
In vitro differentiation of CD34+ HSPCs into erythrocyte precursors
CD34+ HSPCs were cultured according to the three-phase differentiation protocol from Dever et al.48. Subsequent erythroid differentiation and maturation were monitored by flow cytometry (BD FACSCalibur) using FITC-conjugated anti-CD34 (Miltenyi Biotec), PE-conjugated anti-CD235a (Miltenyi Biotec), PerCP-conjugated anti-CD45 (Miltenyi Biotec) and APC-conjugated anti-CD71 (Miltenyi Biotec) at two different time points, day 0 and day 21.
K-562 and CD34+ HSPCs cell transfection
To transfect 1 × 106 K-562 cells using the 100 µl Neon transfection kit (Thermo Fisher Scientific), 200 ng of recombinant pX-330 were utilized. Electroporation settings for this cell line were 1,450 V, 10 ms, and 3 pulses. T7 endonuclease-I (T7E1) assay was performed on day 5 after electroporation.
Chemically modified sgRNAs (Synthego; Table S1) and Cas9 ribonucleoprotein (RNP; IDT) were incubated at a molar ratio of 1:2 (45 pmol to 90 pmol) at room temperature for 15 minutes. After complex formation, 1 × 105 CD34+ HSPCs were transfected using the 10 µl Neon transfection kit (Thermo Fisher Scientific) or the Test Cuvette Adaptor (TCA; Miltenyi Biotec) with the following electroporation settings: 1,650 V, 10 ms, 3 pulses (Neon System) or Square mode, 600 V/100 µs, 300 V/2 ms (CliniMACS Prodigy). Subsequently, cells were transferred to stem cell differentiation culture media. On day 5 post-electroporation, cells were harvested for further DNA isolation, T7E assay, and ICE analysis. On day 21, erythrocyte precursors were collected for RNA isolation, qRT-PCR, and HbF quantification. Primer sequences are listed in Table S2.
T7E1 assay and ICE analysis
Genomic DNA was isolated 5 days post-transfection using NucleoSpin Tissue Kit following the manufacturer’s instructions (MACHERY-NAGEL). The target regions were amplified using the GoTaq Colorless Master Mix (Promega). Primers for each target region are listed in Table S2. PCR products were purified by utilizing QIAquick PCR Purification Kit (QIAGEN) and 1 μg of PCR product was used for T7E1 assay in accordance with the manufacturer’s protocol (New England Biolabs). Readouts of the assay were determined on a 2% agarose gel and analyzed by ImageJ (Fiji software). Indel rates for each target were evaluated by the web tool ‘ICE’ (Inference of CRISPR Edits; https://ice.synthego.com/) after Sanger-sequencing of the purified PCR products.
RNA isolation, cDNA synthesis, and qRT-PCR assays
CD34+ HSPCs were harvested on day 21. Total RNA was isolated by using the RNeasy Mini Kit and QiaShredder spin columns (QIAGEN), in accordance with the manufacturer’s protocol. RNA at a concentration of 500 ng was used for cDNA synthesis with the QuantiTect Reverse Transcription Kit (QIAGEN). Amplification and quantification of cDNA were performed with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). PCR was run utilizing the KAPA SYBR FAST 2x MasterMix (KAPA Biosystems). Primer sequences are listed in Table S2. Results were normalized against the expression of the housekeeping gene β2-microglobulin (β2M). The crossing point (CP) values for the unknown samples were evaluated with the formula 2 (CP β2M - CP target gene)49.
HbF quantification
For high-performance liquid chromatography (HPLC), frozen cell pellets were lysed in 200 µl deionized sterile water and ultrasonicated for 5 minutes. Cell debris was removed by centrifugation at 13,000 g. The supernatant was then concentrated to a final volume of 30 µl using a Nanosep molecular filter (PALL Corporation) with a 10 kDa membrane by centrifugation at 13,000 g. Hemoglobin species from cell lysates were separated using a PolyCAT A cation exchanger column (PolyLC Inc). The analysis was performed on an elite-LaChrom HPLC-system (Merck-Hitachi) using a gradient elution mode with a bis-tris buffer system (buffer A: bis-tris 20 mM, NH4-acetate 13 mM, KCN 1 mM and buffer B: bis-tris 20 mM, Na-acetate 38 mM, KCN 1 mM, NaCl 200 mM). Hemoglobin proteins were detected by absorbance measurements at 415 nm. Intracellular HbF was determined 21 days after erythroid differentiation utilizing the kit ‘Monoclonal antibodies directed to HbF’ (Life Technologies).
RNA-seq
Total RNA from edited HSPCs was isolated after 21 days of erythrocyte differentiation by RNeasy Mini Kit (QIAGEN). RNA quality was determined by measuring 260/280 and 230/260 absorbance ratios on a spectrophotometer (Nanodrop ND-1000, Peqlab Biotechnologie) and the RNA concentration using the Qubit Fluorometric Quantitation and RNA Broad-Range Assay (Thermo Fisher Scientific). The RNA Integrity Number (RIN) was determined using the Lab-on-a-Chip-System Bio-analyzer 2100 and the RNA 6000 Nano assay (Agilent).
For library preparation, mRNA fraction was enriched using polyA capture from 100 ng of total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Next, mRNA libraries were prepared using the NEB Next Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) according to the manufacturer’s instructions. Then, the library molarity was determined by measuring the library size (approximately 400 bp) using the Bioanalyzer 2100 with the High Sensitivity DNA assay, and the library concentration (approximately 10 ng/µl) using Qubit Fluorometric Quantitation and dsDNA High sensitivity assay (Thermo Fisher Scientific). For the first experiment, the libraries were denaturized according to the manufacturer’s instructions, diluted to 1.2 pM and sequenced as paired-end 75 bp reads on an Illumina NextSeq500 (Illumina) with a sequencing depth of >22 million clusters per sample. For the second experiment, the libraries were denatured, diluted to 270 pM and sequenced as paired-end 50 bp reads on an Illumina NovaSeq6000 (Illumina) with a sequencing depth of approximately 20 million clusters per sample.
Read quality of RNA-seq data in FASTQ files was assessed using ngs-bits (v.2019_03) to identify sequencing cycles with low average quality, adaptor contamination, or repetitive sequences from PCR amplification. Reads were aligned using STAR v2.7.3a50 to the grch37 and the alignment quality was analyzed using ngs-bits (v.2019_11). Normalized read counts for all genes were obtained using Subread (v2.0.0) and edgeR (v3.26.8). The distribution of logarithmized cpm-normalized expression values showed similar characteristics over all samples. Based on the filtered data set, samples were investigated with respect to their pairwise similarity. Spearman’s rank correlation coefficient was calculated for each pair of samples. A hierarchical clustering was performed on the resulting similarity values. Differential gene expression analysis was conducted based on the filtered gene expression data set and a statistical model incorporating the group property of samples was tested by fitting a negative binomial distribution using a generalized linear model (GLM) approach. For the analysis, genes were classified when their gene expression fold change (log2 fold change) were equal or greater than +1, and equal or minor than −1. Only genes that were impaired in the three independent experiments were considered. Finally, a screening for oncogenes or suppressor genes was performed using Ingenuity Pathway Analysis (IPA; QIAGEN) to determine the safety of each gene therapy strategy. FASTQ files for all replicates were uploaded to Sequence Read Archive (SRA) at NCBI website (http://www.ncbi.nlm.nih.gov/bioproject/606664).
DNA library preparation
DNA library preparation for GUIDE-seq analysis was performed as described earlier in K-562 cells51,52. The optimal dsODN concentration based on integration efficiency by ICE analysis and cell viability by cell counting was determined after electroporation of 100,000 cells with different dsODN concentrations (5, 15, 25, and 35 pmol). 25 pmol of dsODN was used for further transfections together with sgRNA and Cas9 RNP at molar ratio of 2:1. After 5 days in culture, DNA was isolated with DNeasy Blood & Tissue Kit using standard protocols (QIAGEN). DNA fragments of 200–450 bp were generated and subsequently ligated to adaptors by utilizing NEBNext Ultra II kit (New England Biolabs). NEBNext Ultra II Q5 Master Mix (New England Biolabs) was used for the first DNA amplification, whereas KAPA SYBR FAST 2x MasterMix (KAPA Biosystems) was utilized for the second amplification. The libraries were pooled and loaded into 3 lanes of an Illumina GAIIx single-read flow cell and two MiSeq flow cells. Bound molecules were clonally amplified on a cBot instrument. Subsequently, the first 50 nucleotides from each fragment were sequenced followed by a seven nucleotide sequencing run to decipher the barcode sequence in the adapter (Illumina).
GUIDE-seq
Demultiplexing, PCR duplicate consolidation, cleavage site recognition, off-target activity identification, and visualization was performed with the GUIDE-Seq Analysis Package v1.0.153 using the GRCh37.75 human genome as reference. The read alignment step of the pipeline was conducted using BWA-MEM v0.7.1752 and bedtools v2.2854 was used for downstream analysis.
Droplet digital PCR (ddPCR)
PCR mastermix was prepared by adding ddPCR Multiplex Supermix (Bio-Rad Laboratories), primers (950 nM), probes (250 nM), and DNA (350 ng) at a final volume of 20 µl. Next, QX200 ddPCR droplet generator (Bio-Rad Laboratories) was utilized to separate the DNA into 20,000 droplets, which were transferred to a 96-well plate and sealed to avoid evaporation using the PX1 PCR Plate Sealer (Bio-Rad Laboratories). Finally, the PCR was run on the C1000 Touch Thermal Cycler (Bio-Rad Laboratories) with the following thermal parameters: 10 min at 95 °C, 40 cycles comprising 30 s at 95 °C, 1 min at 61 °C, and 2 min at 72 °C, followed by enzyme inactivation at 98 °C during 10 min. Finally, PCR products were examined using the QX2000 droplet reader (Bio-Rad Laboratories) and analyzed with the QuantaSoft 1.6.6 software (Bio-Rad Laboratories).
References
Finotti, A. et al. Recent trends in the gene therapy of beta-thalassemia. J Blood Med 6, 69–85, https://doi.org/10.2147/JBM.S46256 (2015).
de Brevern, A. G. et al. IthaGenes: An Interactive Database for Haemoglobin Variations and Epidemiology. Plos One 9, https://doi.org/10.1371/journal.pone.0103020 (2014).
Thein, S. L. The molecular basis of beta-thalassemia. Cold Spring Harb Perspect Med 3, a011700, https://doi.org/10.1101/cshperspect.a011700 (2013).
Lohani, N., Bhargava, N., Munshi, A. & Ramalingam, S. Pharmacological and molecular approaches for the treatment of beta-hemoglobin disorders. J Cell Physiol 233, 4563–4577, https://doi.org/10.1002/jcp.26292 (2018).
Olivieri, N. F. & Weatherall, D. J. The Therapeutic Reactivation of Fetal Haemoglobin. Human Molecular Genetics 7, 1655–1658, https://doi.org/10.1093/hmg/7.10.1655 (1998).
Akinsheye, I. et al. Fetal hemoglobin in sickle cell anemia. Blood 118, 19–27, https://doi.org/10.1182/blood-2011-03-325258 (2011).
Antony, J. S. et al. CRISPR/Cas9 system: A promising technology for the treatment of inherited and neoplastic hematological diseases. Adv Cell Gene Ther 1, https://doi.org/10.1002/acg2.10 (2018).
Dulmovits, B. M. et al. Pomalidomide reverses gamma-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood 127, 1481–1492, https://doi.org/10.1182/blood-2015-09-667923 (2016).
Wienert, B. et al. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 130, 803–807, https://doi.org/10.1182/blood-2017-02-767400 (2017).
Wilber, A., Nienhuis, A. W. & Persons, D. A. Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood 117, 3945–3953, https://doi.org/10.1182/blood-2010-11-316893 (2011).
Zhou, D., Liu, K., Sun, C. W., Pawlik, K. M. & Townes, T. M. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet 42, 742–744, https://doi.org/10.1038/ng.637 (2010).
Sankaran, V. G., Xu, J. & Orkin, S. H. Advances in the understanding of haemoglobin switching. Br J Haematol 149, 181–194, https://doi.org/10.1111/j.1365-2141.2010.08105.x (2010).
Sankaran, V. G. & Orkin, S. H. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med 3, a011643, https://doi.org/10.1101/cshperspect.a011643 (2013).
Traxler, E. A. et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 22, 987–990, https://doi.org/10.1038/nm.4170 (2016).
Wilber, A. et al. Therapeutic levels of fetal hemoglobin in erythroid progeny of beta-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood 117, 2817–2826, https://doi.org/10.1182/blood-2010-08-300723 (2011).
Canver, M. C. et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197, https://doi.org/10.1038/nature15521 (2015).
Shariati, L. et al. Genetic disruption of the KLF1 gene to overexpress the gamma-globin gene using the CRISPR/Cas9 system. J Gene Med 18, 294–301, https://doi.org/10.1002/jgm.2928 (2016).
Chung, J. E. et al. CRISPR-Cas9 interrogation of a putative fetal globin repressor in human erythroid cells. Plos One 14, e0208237, https://doi.org/10.1371/journal.pone.0208237 (2019).
Zhao, H. F. et al. Lentiviral Transfer of gamma-Globin with Fusion Gene NUP98-HOXA10HD Expands Hematopoietic Stem Cells and Ameliorates Murine beta-Thalassemia. Mol Ther 25, 593–605, https://doi.org/10.1016/j.ymthe.2017.01.019 (2017).
Negre, O. et al. Gene Therapy of the beta-Hemoglobinopathies by Lentiviral Transfer of the beta(A(T87Q))-Globin Gene. Hum Gene Ther 27, 148–165, https://doi.org/10.1089/hum.2016.007 (2016).
Ronen, K. et al. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat beta-thalassemia. Mol Ther 19, 1273–1286, https://doi.org/10.1038/mt.2011.20 (2011).
Cavazzana-Calvo, M. et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 467, 318–322, https://doi.org/10.1038/nature09328 (2010).
Chang, K. H. et al. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34(+) Hematopoietic Stem and Progenitor Cells. Mol Ther Methods Clin Dev 4, 137–148, https://doi.org/10.1016/j.omtm.2016.12.009 (2017).
Bauer, D. E., Kamran, S. C. & Orkin, S. H. Reawakening fetal hemoglobin: prospects for new therapies for the beta-globin disorders. Blood 120, 2945–2953, https://doi.org/10.1182/blood-2012-06-292078 (2012).
Fluhr, S. et al. Epigenetic dysregulation of the erythropoietic transcription factor KLF1 and the beta-like globin locus in juvenile myelomonocytic leukemia. Epigenetics 12, 715–723, https://doi.org/10.1080/15592294.2017.1356959 (2017).
Ye, L. et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta-thalassemia. Proc Natl Acad Sci USA 113, 10661–10665, https://doi.org/10.1073/pnas.1612075113 (2016).
Antoniani, C. et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 131, 1960–1973, https://doi.org/10.1182/blood-2017-10-811505 (2018).
Martyn, G. E. et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet 50, 498–503, https://doi.org/10.1038/s41588-018-0085-0 (2018).
Powars, D., Weiss, J., Chan, L. & Schroeder, W. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? 63, 921–926 (1984).
Brendel, C. et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest 126, 3868–3878, https://doi.org/10.1172/JCI87885 (2016).
Canver, M. C. & Orkin, S. H. Customizing the genome as therapy for the beta-hemoglobinopathies. Blood 127, 2536–2545, https://doi.org/10.1182/blood-2016-01-678128 (2016).
Siatecka, M. & Bieker, J. J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118, 2044–2054, https://doi.org/10.1182/blood-2011-03-331371 (2011).
Borg, J., Patrinos, G. P., Felice, A. E. & Philipsen, S. Erythroid phenotypes associated with KLF1 mutations. Haematologica 96, 635–638, https://doi.org/10.3324/haematol.2011.043265 (2011).
Arnaud, L. et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet 87, 721–727, https://doi.org/10.1016/j.ajhg.2010.10.010 (2010).
Nurden, A. T., Fiore, M., Nurden, P. & Pillois, X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood 118, 5996–6005, https://doi.org/10.1182/blood-2011-07-365635 (2011).
Funnell, A. P. et al. 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood 126, 89–93, https://doi.org/10.1182/blood-2015-04-638528 (2015).
Wu, X. et al. Bcl11a controls Flt3 expression in early hematopoietic progenitors and is required for pDC development in vivo. Plos One 8, e64800, https://doi.org/10.1371/journal.pone.0064800 (2013).
Masuda, T. et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 351, 285–289, https://doi.org/10.1126/science.aad3312 (2016).
Gasparello, J. et al. BCL11A mRNA Targeting by miR-210: A Possible Network Regulating gamma-Globin Gene Expression. Int J Mol Sci 18, https://doi.org/10.3390/ijms18122530 (2017).
Smith, E. C. et al. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 128, 2338–2342, https://doi.org/10.1182/blood-2016-08-736249 (2016).
Basak, A. et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest 125, 2363–2368, https://doi.org/10.1172/JCI81163 (2015).
Xu, L., Wu, H., Wu, X., Li, Y. & He, D. The expression pattern of Bcl11a, Mdm2 and Pten genes in B-cell acute lymphoblastic leukemia. Asia Pac J Clin Oncol 14, e124–e128, https://doi.org/10.1111/ajco.12690 (2018).
Wu, Y. et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776–783, https://doi.org/10.1038/s41591-019-0401-y (2019).
Liu, N. et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173(430–442), e417, https://doi.org/10.1016/j.cell.2018.03.016 (2018).
Magrin, E., Miccio, A. & Cavazzana, M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood, https://doi.org/10.1182/blood.2019000949 (2019).
Humbert, O. et al. Therapeutically relevant engraftment of a CRISPR-Cas9–edited HSC-enriched population with HbF reactivation in nonhuman primates. Science Translational Medicine 11, eaaw3768, https://doi.org/10.1126/scitranslmed.aaw3768 (2019).
Marchese, F. P., Raimondi, I. & Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biology 18, 206, https://doi.org/10.1186/s13059-017-1348-2 (2017).
Dever, D. P. et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389, https://doi.org/10.1038/nature20134 (2016).
Johnson, R. L., Milenkovic, L. & Scott, M. P. In vivo functions of the patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Molecular cell 6, 467–478 (2000).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2012).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197, https://doi.org/10.1038/nbt.3117 (2015).
Tsai, S. Q., Topkar, V. V., Joung, J. K. & Aryee, M. J. Open-source guideseq software for analysis of GUIDE-seq data. Nature Biotechnology 34, 483–483, https://doi.org/10.1038/nbt.3534 (2016).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Vol. 1303 (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Acknowledgements
Authors would like to thank the Stem Cell Laboratory in the University Children’s Hospital of Tuebingen, for providing cells for our experiments. This study was financially supported by the research funding programs Jürgen Manchot Stiftung, Fortüne Tuebingen (N°. 2412-0-0; N°. 2485-0-0), Clinician Scientist Program (N°. 440-0-0), Förderverein für krebskranke Kinder Tübingen e.V., and the University Children’s Hospital of Tübingen. We also acknowledge support by Open Access Publishing Fund of University of Tübingen.
Author information
Authors and Affiliations
Contributions
Performed experiments and analyzed data: A.L.C. and A.D.M. Performed hemoglobin electrophoresis: T.E. Analyzed GUIDE-seq data: L.H. and P.B. RNA-seq data: J.A. and N.C. Contributed reagents/materials/analysis tools: J.S.A., N.L., D.M.S., M.S.D.K., R.H. and M.M. Wrote the paper: A.L.C., A.D.M., J.S.A. and M.M. Conceived, designed and supervised the experiments: M.M., A.L.C., A.D.M. and J.S.A. All authors read and approved the final manuscript. A.L.C. and A.D.M. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lamsfus-Calle, A., Daniel-Moreno, A., Antony, J.S. et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci Rep 10, 10133 (2020). https://doi.org/10.1038/s41598-020-66309-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66309-x
This article is cited by
-
Detection of CRISPR/Cas9-Mediated Fetal Hemoglobin Reactivation in Erythroblasts Derived from Cord Blood-Hematopoietic Stem Cells
Molecular Biotechnology (2024)
-
Identification of a genomic DNA sequence that quantitatively modulates KLF1 transcription factor expression in differentiating human hematopoietic cells
Scientific Reports (2023)
-
Heterochromatin rewiring and domain disruption-mediated chromatin compaction during erythropoiesis
Nature Structural & Molecular Biology (2023)
-
CRISPR/Cas-based gene editing in therapeutic strategies for beta-thalassemia
Human Genetics (2023)
-
Genome-based therapeutic interventions for β-type hemoglobinopathies
Human Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
