
Abstract
Sphingopyxis granuli strain TFA is an α-proteobacterium that belongs to the sphingomonads, a group of bacteria well-known for its degradative capabilities and oligotrophic metabolism. Strain TFA is the only bacterium in which the mineralisation of the aromatic pollutant tetralin has been completely characterized at biochemical, genetic, and regulatory levels and the first Sphingopyxis characterised as facultative anaerobe. Here we report additional metabolic features of this α-proteobacterium using metabolic modelling and the functional integration of genomic and transcriptomic data. The genome-scale metabolic model (GEM) of strain TFA, which has been manually curated, includes information on 743 genes, 1114 metabolites and 1397 reactions. This represents the largest metabolic model for a member of the Sphingomonadales order thus far. The predictive potential of this model was validated against experimentally calculated growth rates on different carbon sources and under different growth conditions, including both aerobic and anaerobic metabolisms. Moreover, new carbon and nitrogen sources were predicted and experimentally validated. The constructed metabolic model was used as a platform for the incorporation of transcriptomic data, generating a more robust and accurate model. In silico flux analysis under different metabolic scenarios highlighted the key role of the glyoxylate cycle in the central metabolism of strain TFA.
Similar content being viewed by others
Introduction
Members of the Sphingomonadales order, which includes the Sphingomonadaceae and Erythrobacteraceae families, are known as degraders of a large diversity of recalcitrant compounds1. Within the Sphingomonadaceae family, the sphingomonads group, composed of the ubiquitous genera Sphingomonas, Sphingobium, Novosphingobium and Sphingopyxis2, contains metabolically versatile bacteria capable of using a large number of recalcitrant compounds, mainly aromatic compounds and their derivatives, as sole carbon and energy source3. In addition, the genus Sphingopyxis includes organisms adapted to oligotrophic environments, which show specific genomic characteristics such as a low number of rRNA operons, prophages or CRISPRs sequences4. Moreover, oligotrophs show overrepresentation of Clusters of Orthologous Groups (COGs) for lipid transport and metabolism (I) and secondary metabolite biosynthesis, transport, and catabolism (Q) among others4.Thus there is a need to better understand the metabolism of these degradative bacteria and to potentially reveal new insights into their degradative capabilities and lifestyle. To this end, we set out to establish a GEnome-scale metabolic Network REconstruction (GENRE) for a member of this genus that was likely to prove a powerful tool for future studies5,6,7,8. A GENRE is a database with specific information about the species of interest generated from genomic data, and that contains detailed information about metabolism, the network of reactions, including stoichiometry and reversibility, the Gene-Protein-Reaction (GPR) associations and other available biochemical and physiological data6,9. The development of COnstraint-Based Reconstruction and Analysis (COBRA) methods10 has allowed the generation of quantitative GEnome-scale metabolic Models (GEMs), that can be used to understand phenotypes in terms of metabolic fluxes. GEMs allow the integration of additional information, such as gene expression data, compartmentalisation of the pathways and probabilistic transcriptional regulation data, resulting in more robust metabolic models with enhanced predictive capabilities. Nowadays there are tools that automatically generate GEMs, like Path2Models11, CarveMe12 or AGORA13. However, manual curation, although tedious and time-consuming, always provides a high-quality and more reliable metabolic model. A recent review of GEMs shows that only 1.9% of the bacterial models have been manually reconstructed8. Some applications of well-curated GEMs include the design of microorganisms for the production of chemicals, the prediction of enzyme functions or the discovery of drug targets in pathogens8.
According to the NCBI Genome database (Genome List)14, 198 and 813 genome sequences of Erythrobacteraceae and Sphingomonadaceae, respectively, are available. However, despite the environmental significance of these bacterial groups, only one metabolic reconstruction using the genomic information of Zymomonas mobilis, iEM439, has been generated thus far15. This species is a well-known member of the Sphingomonadaceae family because of its ability to produce ethanol in a broad pH range, its ethanol tolerance and its use in other biotechnological applications16. In this report, we present a genome-scale metabolic model of another Sphingomonadaceae, Sphingopyxis granuli strain TFA, a small, rod-shaped, facultative anaerobic, streptomycin-resistant bacterium that possesses the capability to grow using tetralin, a volatile toxic compound that consists of an aromatic and an alicyclic ring, as sole carbon and energy source17. The metabolic pathway for tetralin degradation of strain TFA is the only one completely characterized at both biochemical and genetic levels18. This pathway, which seems to have been horizontally acquired by TFA, represents a paradigm of how a bicyclic compound can be metabolised with one set of genes mainly involved in the degradation of aromatic compounds. Moreover, the mechanisms involved in the regulation of the expression of the pathway structural and regulatory genes at both transcriptional and translational levels have been elucidated18 showing that genes are induced in the presence of tetralin but subjected to carbon catabolite repression in the presence of preferential carbon sources. The genome of strain TFA has been sequenced, assembled into a single circular DNA molecule and functionally annotated19. No free plasmids have been detected in TFA either experimentally or during genome sequencing19. In this report, biochemical data and gene annotations have been put together to generate a GEM of strain TFA, that has been manually curated and whose characteristics and predictive potential are presented. New potential carbon and nitrogen sources and metabolic pathways that could support growth of strain TFA have been discovered. Moreover, the metabolic model has been improved by incorporating transcriptomic data obtained under two different growth conditions. Flux Balance Analysis (FBA) has revealed the importance of the glyoxylate cycle in the central metabolism of this bacterium. Overall, the model presented here constitutes the first GEM generated for a member of the Sphingopyxis genus and the second within the Sphingomonadales order.
Results
Characterization of the genome-scale metabolic network in S. granuli strain TFA
The genome-scale metabolic model of strain TFA, called iIG743, was constructed following a procedure that includes four steps (Fig. 1). Firstly, an initial draft model based on the Escherichia coli K12 model iJO136620 and the Pseudomonas putida KT2440 model iJN141121 was constructed (see Methods). It contained 810 reactions, classified into 84 functional subsystems, 952 metabolites and 386 genes. In this initial draft, 260 reactions and 407 metabolites were added indistinctly from iJN1411 or iJO1366 based on orthologous genes for those reactions present in both models and in the TFA genome. Interestingly, 410 reactions and 421 metabolites were assigned exclusively from iJN1411 whereas only 140 reactions and 124 metabolites came from iJO1366. In total, 670 reactions and 828 metabolites were common between iJN1411 and the initial TFA draft, whilst 400 reactions and 531 metabolites were shared with iJO1366. The higher number of reactions, genes and metabolites taken from iJN1411 suggests that TFA is metabolically more similar to P. putida KT2440 than to E. coli K12 (Supplementary Fig. S1).

Steps for the construction of the genome-scale metabolic model of TFA iIG743. Using the genomic annotation obtained with Sma3s and the E. coli and P. putida models, the initial reconstruction of the TFA metabolic model was obtained using the server GEMSiRV-MrBac. An exhaustive manual curation was carried out using bibliographic, biochemical and metabolic databases. The curated model was converted into a mathematical model using the COBRA package. Biomass production was the reaction used to evaluate the model and to identify and fill the possible gaps.
Secondly, an exhaustive revision and manual curation of reactions was carried out based on the careful analysis of each GPR included in the draft (see Methods). Therefore, each individual GPR was evaluated based not only on sequence identity, but also considering physiological evidences. For instance, the transport reaction 3_4DHBZ1t_pp_ (3,4-dihidroxybenzoate reversible transporter via proton symport) included in the draft from iJN1411 was eliminated since no enzymes for 3,4-dihydroxybenzoate catabolism were found in TFA. Similarly, the transport reactions for 3-hydroxybutyrate (3-HB) and sebacic acid were included as it is known that TFA grows using these fatty acids as sole carbon and energy sources. Then, the initial model was converted into a mathematical representation and was tested for biomass production (a detailed description as to how biomass composition was determined can be found in the methods section). Finally, the network gaps across the metabolic pathways were filled in by adding new reactions based on the information stored in biological databases, scientific literature and in the functional annotation of strain TFA19. During the manual curation, known metabolic features of strain TFA, either experimentally tested or inferred from the genome annotation, were introduced into the metabolic network, including the utilization of tetralin as sole carbon and energy source, the synthesis and degradation of poly-hydroxybutyrate (PHB), the production of the compatible solute ectoine, and the biosynthesis of sphingolipids as components of the cell envelope, which is a characteristic of the Sphingomonadales order. To model tetralin degradation, all available biochemical and genetic data were used18 (Supplementary Fig. S2A). It is worth noting that iIG743 is the first bacterial model in which the aerobic degradation pathway of tetralin has been included providing a platform for further studies on its optimization and integration into bacterial metabolism. For the modelling of PHB synthesis and degradation, the corresponding genes of strain TFA were incorporated into pathways similar to those described for Ralstonia eutropha and other bacteria in which the monomer (R)-3-hydroxybutyryl-CoA originates from two molecules of acetyl-CoA via acetoacetyl-CoA22,23. Additionally, since strain TFA is able to grow on 3-HB as carbon and energy source, an additional pathway that directly activates 3-HB into its CoA derivative in a reaction catalysed by putative acyl-CoA ligases (SGRAN_2644 or SGRAN_3728) was included (Supplementary Fig. S2B). Genes likely to be involved in ectoine and 5-hydroxyectoine production from L-aspartate were located in an operon (etcABCDask, SGRAN_2345 to SGRAN_2349) that however lacked a gene for the expected L-aspartate-β-semialdehyde dehydrogenase (Asd, SGRAN_0849) (Supplementary Fig. S2C). A second aspartokinase (Ask) encoded by SGRAN_1547 was also included. One of the main characteristics of members of the Sphingomonadaceae family is the lack of lipopolysaccharides in their outer membrane, which are replaced by glycosphingolipids24. Since sphinganine is part of the central structure of sphingolipids, its synthesis was also included in the model of strain TFA (Supplementary Fig. S2D). Additionally, genes involved in polyvinyl alcohol (PVA) degradation, which show a high similarity with those encoded by the pva operon located on a Sphingopyxis sp. 113P3 megaplasmid25, were annotated in strain TFA’s genome. Thus, the metabolic pathway was reconstructed based on the Sphingopyxis sp. 113P3 information (Supplementary Fig. S2E). The identification of a PQQ repeat (Interpro IPR002372, IPR018391) within the amino acid sequence of the putative PVA dehydrogenase of strain TFA (SGRAN_2681) suggests that this enzyme is pyrrolo-quinoline quinone (PQQ)-dependent. However, genes encoding a pathway for the biosynthesis of this cofactor have not been found in the genome of strain TFA. Thus, PVA degradation by strain TFA would need an external supply of PQQ, either directly added to the medium or by being secreted by a syntrophic bacterium as has been described for Pseudomonas26 and Sphingomonas27. Although no growth of strain TFA in co-culture with P. putida KT2440 and PVA as carbon and energy source has been detected thus far (data not shown), a PVA degradation pathway has been included in iIG743.
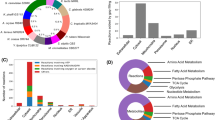
The final model consists of 1397 reactions, 1114 metabolites and 743 genes (Table 1). Compared to other models constructed for α-proteobacteria until date, iIG743 contains a large number of reactions and metabolites. Moreover, the manual curation resulted in high or very high (3 or 4) confidence values for 27.8% of the reactions included in iIG743. Reactions were classified into fourteen major categories (Fig. 2), of which lipid metabolism is the largest by number of reactions (321 reactions). This model has a large number of reactions for which a gene can be associated (74.9%) although for transport reactions this percentage is lower (58.2%). On the other hand, the exchange reactions are by definition non-gene associated.

Functional classification of the reactions in iIG743. Reactions in the final TFA metabolic model are classified into 115 functional subsystems, which were grouped into the fourteen main categories shown in the figure.
Only 2.6% of the reactions were assigned to the catabolism of aromatic compounds, such as those involved in the tetralin degradation pathway, the catabolism of the aromatic amino acids phenylalanine, tyrosine and tryptophan, and the degradation pathway of 3-phenylpropanoate and 3-(3-hydroxyphenyl) propanoate. Analysis of essential genes to predict growth on tetralin showed that all the genes identified previously experimentally to be involved in tetralin degradation18 (thn genes) were essential except thnN, thnO and thnP, which code for a glutaryl-CoA dehydrogenase and an α- and β-flavoprotein subunit, respectively. However, other non-essential genes such as those involved in β-oxidation of pimelic acid (thnH, thnI, thnJ and thnK) that are dispensable for growth with tetralin or pimelic acid were not predicted. The large number of β-oxidation pathways and aredundant pimelic semialdehyde dehydrogenase activity (ThnG) found in strain TFA28 might explain this discrepancy.
Genes encoding enzymes for the utilization of 3-phenylpropanoate and 3-(3-hydroxyphenyl) propanoate as carbon and energy sources, converting these compounds into pyruvate and acetyl-CoA, were also found19 and the corresponding pathways included into iIG743. However, no growth of strain TFA was supported by any of these compounds (Table 2).
Qualitative and quantitative of iIG743 validation
To simulate growth of strain TFA in oxic conditions through iIG743, a minimal medium was established (see Methods) and the flux through the exchange reaction of each metabolite, including O2, was not limited. To test whether iIG743 was able to predict cellular growth, the biomass reaction was selected as the objective function and was maximised through the Flux Balance Analysis algorithm (FBA) implemented in the COBRA package. The model was able to predict growth on compounds previously shown to be carbon and energy sources for strain TFA, such as tetralin, 3-HB and sebacic acid, supporting the absence of gaps in the predicted catabolic pathways. Furthermore, the model was able to predict growth in anoxic conditions using nitrate as electron acceptor and 3-HB or sebacic acid as carbon and energy source as had been shown experimentally19. To evaluate quantitatively the predictive capability of iIG743, the uptake rate of three carbon sources (3-HB, sebacic acid, and L-lactate) for the growth of strain TFA in oxic conditions was experimentally calculated (Table 3; see Methods). Each experimental value of uptake was established as entrance flux in the corresponding exchange reaction and the growth rates were obtained. As shown in Table 3, the average values of the experimentally calculated growth rates of strain TFA with sebacic acid and L-lactate and those predicted by iIG743 were in good agreement. However, iIG743 predicted significantly faster growth of strain TFA on 3-HB than observed, indicating that adjustments of the model will be required for accurate prediction of growth on this carbon source.
Model-based exploration of the metabolic versatility of S. granuli strain TFA
Like other members of the genus Sphingopyxis, strain TFA is expected to be adapted to oligotrophic environments. Under laboratory conditions, strain TFA grows on a limited number of carbon sources, such as tetralin and its degradation intermediate pimelic acid, 3-HB or sebacic acid. Moreover, ammonia is the preferred nitrogen source and the strain does not grow with nitrate or urea29. To explore the metabolic versatility of strain TFA, the complete set of external metabolites present in iIG743 was used to identify additional carbon and/or nitrogen sources. As shown in Table 2, model-based analyses identified a wide range of new compounds that strain TFA could use as carbon sources, including mainly fatty acids and amino acids, some of which were experimentally confirmed (Table 2). Additionally, iIG743 predicted several amino acids, subsequently confirmed (Table 2), that could be used as sole nitrogen and carbon sources. On the other hand, the model predicted biomass formation with some carbon sources that strain TFA cannot grow on in vivo, including ethanol, formaldehyde, 3-phenylpropanoate, 3-(3-hydroxyphenyl) propanoate and fructose. In the case of glucose, the model predicts growth only if a transporter, which has not been annotated in the genome of strain TFA, is artificially included. For growth on PVA, the model predicts that PQQ has to be included as a component of the minimal medium (see above) and shows that for a fixed PVA uptake, PQQ uptake rate determines TFA growth rate on PVA.
When predicted growth rates were normalized according the numbers of carbon atoms present in each carbon source, the model strongly suggested fatty acids as the preferred and more efficient carbon and energy sources over amino acids and other compounds such as lactate or tetralin (Supplementary Fig. S3). Overall these results are in good agreement with the proposed trophic strategy of marine oligotrophic bacteria4, where the overrepresentation of genes involved in lipid transport and metabolism in oligotrophic bacteria was related to the higher ATP yield obtained from fats compared to sugars4.
Model-driven analysis of aerobic metabolism in strain TFA
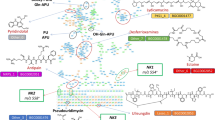
To analyse the metabolic features of strain TFA when using different carbon sources, we performed a careful flux analysis using iIG743 with tetralin, sebacic acid and 3-HB as sole carbon and energy source under oxic conditions (Fig. 3). Since no reliable value for 3-HB and tetralin uptake could be obtained, these uptakes rates (required to constrain the model) were estimated from the experimental growth rates, which were 0.19 h−1 for 3-HB (Table 3) and 0.055 h−1 for tetralin. The predicted uptake rates were −3 and −0.375 mmol·gDW−1·h−1 for 3-HB and tetralin, respectively. In the case of sebacic acid, we applied the experimentally calculated uptake rate, −1.61 mmol·gDW−1·h−1 (Table 3). The analysis of the fluxes showed that the flux through glyoxylate was higher when strain TFA grew on sebacic acid and 3-HB compared to tetralin. In all cases, glyoxylate was directed to glycerol-3-phosphate, although in 3-HB and tetralin its conversion to malate was also predicted. Moreover, the Krebs cycle seemed to operate to provide succinyl-CoA for the activation of acetoacetyl-CoA during 3-HB catabolism (Fig. 3A), while sebacic acid and tetralin could be completely oxidised through this cycle (Fig. 3B,C).

Flux ratio (carbon uptake/reaction flux) through the reactions involved in the catabolism of 3-hydroxybutyrate (A) sebacic acid (B) and tetralin (C). Gene names involved in each reaction are written in grey and the thickness of the green arrows is proportional to the flux ratio. The dashed grey arrows show reactions without flux, dashed green arrows indicate multiple reactions in which intermediates are not represented in the figure.
Model-driven analysis of anaerobic metabolism in strain TFA
Strain TFA is a facultative anaerobe that can grow using either oxygen or nitrate as the terminal electron acceptors19. Anaerobic growth of strain TFA was predicted in silico using 3-HB or sebacic acid as carbon sources. The value for the experimentally calculated nitrate uptake rate (−2.82 mmol·gDW−1·h−1, see Methods) was used as constraint in the exchange reactions. The uptake of the rest of the metabolites in the minimal medium was not limited, except O2, whose uptake was set to 0. Additionally, since a lower value for the non-growth associated ATP maintenance reaction (NGAM) under anoxic conditions has been described in E. coli30, NGAM was reduced from 0.92 (aerobic conditions, value obtained from the iJN1411 model) to 0.15 mmol·gDW−1·h−1 to give predicted growth rates of 0.038 h−1 on 3-HB and of 0.037 h−1 on sebacic acid, while the experimentally calculated values were 0.055 h−1 and 0.020 h−1, respectively. Concomitant nitrite secretion during anaerobic growth of strain TFA using nitrate as electron acceptor has been experimentally demonstrated19. To establish the predictive capacity of iIG743, the uptake of nitrate and 3-HB and the excretion of nitrite were simulated by dynamic FBA (dFBA) throughout the cultivation time. For the simulation, the initial concentrations of nitrate and 3-HB were set to 20 mM and 40 mM, respectively, as previously used under experimental conditions19, and metabolite uptake rates were set as described above. Initial biomass was experimentally calculated to be 0.038 g·L−1.
As visualized in Fig. 4, nitrate was the limiting compound and an equimolar relation between nitrate uptake and nitrite secretion was predicted and experimentally observed19. Similar results were obtained when anaerobic growth with sebacic acid was simulated (data not shown). The results show that iIG743 model contains all the information to simulate TFA growth under anoxic conditions.

Dynamic validation of TFA under anaerobic growth. Initial concentrations for both in vivo and in silico growth were set to 40 mM of 3-hydroxybutyrate (3-HB) and 20 mM of nitrate (NO3). The left panel shows the combination of the graphs of the simulated growth and metabolite concentrations through the growth curve predicted by iIG743 and the right panel the experimental data adapted from García-Romero et al.19.
Using iIG743 as a computational platform for -omics data integration
The predictive capability of a metabolic model for a given condition can lack precision since regulation at the levels of both gene expression and enzymatic activity are not considered, and the fluxes for certain reactions might therefore not reflect physiological behaviour. Consequently, GEMs predictions often benefit from the inclusion of regulatory-derived data such as those obtained by transcriptomics. In addition, beyond providing more accurate predictions, GEMs provide useful computational workflows for the incorporation of -omics data within biological networks31. We thus included available transcriptomic data for strain TFA grown aerobically with tetralin and 3-HB32 in an attempt to optimize iIG743 predictions for the strain grown on each of the two carbon sources. To do this, we reduced the solution space of iIG743 by using the GIMME method33. The expression of each gene was used as a new constraint in the metabolic model since only genes with a FPKM value higher than the cut-off (see Methods) were considered as expressed under each growth condition. Thus, GIMME constructed condition-specific models by removing reactions encoded by unexpressed genes in each condition and finding a flux distribution consistent with the biological objective. To explore the feasible metabolic states, random sampling was applied to each new model. In this process, the possible fluxes and the probability of each one were determined for all of the reactions in the network, (as visualized in the Supplementary Fig. S4), the range of flux distribution for some reactions involved in the central catabolism was reduced and the probability of their optimal flux was increased after the application of the new constraints (green line).This means that the introduction of transcription data narrows the feasible metabolic states in the conditions analysed suggesting that the flux predictions would be more accurate.
In fact, when introducing the transcription data as the constraint, the predicted fluxes in central metabolic pathways for growth of strain TFA changed significantly (Fig. 5). Thus, with 3-HB, the glycolytic enzymes were directed to gluconeogenesis and supplies of pyruvate and phosphoenolpyruvate were obtained from malate and oxaloacetate, respectively. Moreover, no flux to glycerol-3-phosphate synthesis from glyoxylate was predicted. In the case of tetralin, the inclusion of transcription data resulted in an increase in flux through the glyoxylate cycle and the use of pyruvate to produce acetyl-CoA instead of being used as a source of phosphoenolpyruvate (see Fig. 3C), which was obtained from oxaloacetate. In contrast to 3-HB, an enhancement of glycerol-3-phosphate synthesis from glyoxylate was predicted during growth of strain TFA with tetralin. Moreover, the introduction of transcription data modified the prediction of essential genes. For instance, aceA (coding for the isocitrate lyase) was now predicted to be an essential gene for growth on tetralin since glyoxylate became essential for the synthesis of glycerol-3-phosphate as source of glyceraldehyde-3-phosphate (see Fig. 5).

Incorporation of the TFA transcriptomes into iIG743. Flux ratio through reactions (orange box) and gene expression (in FPKM, blue) in the pathways involved in the catabolism of (A) 3-hydroxybutyrate and (B) tetralin. Flux ratio was calculated by dividing the reaction flux by the carbon uptake in the models constrained with expression data. The absence of a FPKM value indicates an orphan reaction without an assigned gene. Arrows thickness is proportional to the flux. The dashed grey arrows show reactions without flux, dashed green arrows indicate multiple reactions in which intermediates are not represented in the figure. Gene names involved in each reaction are written in grey. DHDDA, 2,4-dihydroxydec-2-ene-1,10-dioic acid.
Discussion
In this work, a genome-based metabolic network of S. granuli strain TFA was constructed, successfully validated against some known metabolic states of strain TFA and employed to explore the metabolic versatility of this bacterium. Furthermore, iIG743 was used to generate condition-specific models in two well-known metabolic scenarios, growth on tetralin or 3-HB, by adding gene expression data as new constraints. The resulting models showed a significantly better performance, resulting in two unprecedented tools for a system-level study of the metabolism of tetralin and 3-HB in strain TFA. To our knowledge, iIG743 is the first genome-scale metabolic model constructed within the sphingomonads group and the second one for the overall Sphingomonadaceae family.
Model-based analysis using iIG743 successfully identified new metabolites that strain TFA could use as sole carbon and energy source including some fatty acids and amino acids, most of which have been experimentally validated, thus significantly expanding the range of substrates that this oligotrophic strain can use. Interestingly, the model predicted growth with several compounds that experimentally could not support growth of strain TFA. The toxicity of ethanol34 and formaldehyde35 could explain the incapacity of strain TFA to use these compounds. In the case of glucose, although complete Embden-Meyerhof-Parnas and Entner-Doudoroff pathways were identified, growth was only predicted when a transport reaction was incorporated into the model since no glucose transporter has been annotated in the genome of strain TFA. Moreover, this lack of growth using glucose seems to be characteristic of S. granuli species since S. granuli Kw07 is also unable to use this sugar in contrast to other Sphingopyxis species36. Interestingly, the predictions provided by iIG743 should assist in the engineering of strain TFA to allow growth on commonly used and cheap carbon sources, expanding the use of strain TFA for a variety of biotechnological purposes.
Other compounds identified as carbon sources by the model were PVA, 3-(3-hydroxyphenyl) propanoate (3HPP) and 3-phenylpropanoate (3PP). Although PVA-supported TFA growth was no detected even in the presence of a PQQ producer, the PVA degradation pathway was included in iIG743. The high sequence identity at protein level (>50% with a coverage>85%) and the conservation of the genetic organization of TFA PVA-degradation genes with those present in the well-characterised PVA-degrader Sphingopyxis sp strain 113P3 support the existence of that pathway in TFA. However, more experimental assays, such as the use of PVA with different degree of polymerization, are needed since we cannot rule out that those are silent genes. Genes encoding upper pathways (mhp and hca genes for 3HPP and 3PP, respectively) that converge in 2-oxopent-4-enoate were annotated in the genome of strain TFA. This compound could be further metabolised to pyruvate and acetyl-CoA by the mhpDEF gene products. The lack of growth of strain TFA with 3HPP and 3PP could be explained by (i) the absence of appropriate transport systems, (ii) the lack of a functional regulatory system that allowed gene expression, and/or (iii) inaccuracy in gene annotation on enzyme specificity resulting in an incorrectly predicted substrate for a metabolic pathway. The latter could mean that strain TFA might use the predicted pathways to metabolize other similar compounds. Correct substrate prediction is one of the major challenges of automatic GEM generation and requires extensive experimental data. Interestingly, adaptive laboratory experiments (ALE) have shown that some in silico predicted phenotypes can be achieved in vivo after short-term evolution37 by activating putative silent pathways. Alternatively, the presence of these silent metabolic pathways might represent the first step of an evolutionary process to lose the ability to degrade particular aromatic compounds. Consistent with this, it has been proposed that Sphingopyxis are specialised to degrade particular compounds rather than exhibiting the ability to utilise a wide range of aromatic substrates38.
Although the in silico prediction proposed growth with compounds that have been verified in vivo, in some cases, such as the use of 3-HB in oxic conditions, in silico growth rates were higher than the experimentally calculated. A possible explanation for this discrepancy could be that these compounds were not immediately respired in vivo but captured in an intracellular pool of compounds, such as PHB granules, as it has been described in S. alaskensis RB2256 during growth with glucose39. In fact, 3-HB catabolism implies its conversion into acetoacetyl-CoA and acetyl-CoA (Fig. 3A), which are intermediates in PHB synthesis40. Moreover, direct activation of 3-HB with CoA yielding the monomer for PHB synthesis in a reaction catalysed by an acyl-CoA ligase might also take place in strain TFA (Supplementary Fig. S2B). Although PHB synthesis and degradation were incorporated into iIG743, all predictions were performed without synthesis of this carbon storage granule. However, it has been reported previously that TFA cells accumulate PHB up to approximately 7% of the total biomass dry weight during growth in minimal medium plus 40 mM 3-HB41. As observed in Supplementary Fig. S5A, the demand of PHB in the model leads to a decrease of the growth rate in 3-HB. Therefore, the accumulation of PHB could explain, at least partially, the differences between predicted and measured growth rate in 3-HB.
The demand of PHB synthesis could also explain the discrepancy between the growth rate in vivo and the growth rate predicted by the model with sebacic acid under anoxic conditions. Thus, when PHB synthesis was included, the growth rate of strain TFA using sebacic acid as carbon and energy source was more negatively affected than when using 3-HB in anoxic conditions (Supplementary Fig. S5A). Conversely, the same level of reduction was predicted for the growth rate of strain TFA with both compounds under oxic conditions (Supplementary Fig. S5B). Thus, iIG743 offers a plausible explanation for these observed discrepancies that will require further experimental analysis of PHB synthesis for confirmation.
iIG743 provides an accurate prediction for the growth of strain TFA on tetralin. However, while most of the tetralin degradation genes are predicted to be essential for growth on this compound some of them are dispensable in vivo18. This incorrect prediction might reflect the inaccurate genome annotation or a broad specificity for some enzymes that cannot be inferred from the annotation. In any case, false positive essential genes are often found in GEM-based analysis. For instance, false positive essential genes were found with the model of Pseudomonas aeruginosa strain PA14 (iPau1129)42, where the dispensability of gnyA for L-isoleucine utilization and of scoA and scoB for growth on 4-hydroxybenzoic acid has been recently demonstrated by Dunhpy et al.43. In E. coli, aspC, argD, and gltA were incorrectly predicted as essential since isoenzyme functions have been described44. Thus, as a dynamic process, a genome-scale metabolic reconstruction is expected to undergo continual refinement as inconsistences are found between predictions and experimental data.
As most aromatic compound pathways, tetralin degradation is under carbon catabolite repression in TFA, being thn genes repressed in the presence of preferential carbon sources such as 3-HB or sebacic acid18. Interestingly, iIG743 is able to simulate this phenomenon when oxygen uptake is limited to values predicted by the model when using different carbon sources. For instance, when oxygen uptake was limited to -5 mmol·gDW-1·h-1, the model discriminates between a good carbon source for TFA, such as 3-HB, and tetralin and establishes a hierarchy of use (Supplementary Fig. S6). Although the involvement of oxygen sensing in carbon catabolite repression in TFA has to be experimentally proven, this is another example of the predictive capability of iIG743.
Anaerobic growth of strain TFA using nitrate as electron acceptor could also be modelled using iIG743. According to the prediction, nitrate was the limiting substrate for biomass formation and the concomitant reduction of nitrate to nitrite was observed. When the nitrate concentration was increased from 20 to 40 mM, growth of strain TFA for a longer period and with more biomass formation was predicted (data not shown). However, it is known that in vivo strain TFA shows similar growth rates and final biomass accumulations in the presence of 20 or 40 mM nitrate as electron acceptor19. This may be due to the inhibitory effect of nitrite concentrations higher than 20 mM in the growth of strain TFA19. In fact, the difficulty of predicting the toxicity of metabolic intermediates or final products of pathways is one of the major drawbacks when heterologous pathways are incorporated into the metabolism of a target host45.
The incorporation of transcriptomic data into metabolic models was expected to result in a more accurate metabolic scenario by defining which genes will be transcribed under a certain environmental condition, which could push fluxes into some specific pathways or reactions. When expression data were incorporated into the model, pathways of central metabolism were modified in different ways depending on the carbon source. However, in all cases, the central role of the glyoxylate shunt was maintained or even enhanced. This shunt bypasses the Krebs cycle conserving carbon atoms for gluconeogenesis and diminishing the number of electrons funnelled into respiration. It is known that this cycle is upregulated when acetyl-CoA is the direct product of a metabolic pathway46, which is the case for tetralin, 3-HB and sebacic acid. Moreover, induction of the glyoxylate shunt by oxidative stress has been shown in P. aeruginosa47. This could be the case in strain TFA when grown on tetralin since this compound is toxic because of the formation of hydroperoxides in the cell48. Moreover, the high demand for oxygen during its degradation, which requires high oxygenase activity, might also lead to the formation of hydrogen peroxide49 resulting in the induction of glyoxylate synthesis to deal with the generated oxidative stress47.
In conclusion, we have developed and enhanced a powerful tool for further metabolic studies of strain TFA that will allow us to better explore and exploit the unstudied metabolic characteristics of this oligotrophic bacterium.
Methods
Microorganism and culture conditions
Sphingopyxis granuli strain TFA was used as a model organism for this work17. Strain TFA was grown aerobically at 30 °C in MML rich medium and in MM minimal medium17 containing an appropriate amount of carbon source (Supplementary Table S1). When testing the compound as a nitrogen or carbon and nitrogen source, ammonium was omitted from the MM. For anaerobic growth, strain TFA was pre-cultured aerobically in MM supplied with the same carbon used for the anaerobic culture. The pre-culture at an OD600 of 1 was used to inoculate at an initial OD600 of 0.1 in standing stoppered bottles filled to the top with MM containing an adequate concentration of the carbon source. Sodium nitrate was added to a final concentration of 20 mM as the terminal electron acceptor for the anaerobic cultures and both the nitrate and nitrite present in the medium were monitored as described19. Streptomycin 50 µg/mL was used as antibiotic selection, since strain TFA is naturally resistant to this compound. Growth was determined by measuring optical density at 600 nm.
Model reconstruction
The metabolic reconstruction of S. granuli strain TFA was done using a 4-step protocol as described by Thiele and Palsson6 using the genomic annotation obtained with Sma3s19.
Firstly, two initial models of strain TFA were constructed based on E. coli K12 and P. putida KT2440 models (iJO1366 and iJN1411, respectively)20,21, using the GEMSiRV-MrBac server as described in50. Two lists of orthologous genes, one between strain TFA and K12 and another between strain TFA and KT2440 were obtained by Reciprocal Best Hits using BlastP (≥40% identity, ≥80% coverage and e-value less than 10-20). These lists and the corresponding model in SBML format were used as input files for the GEMSiRV-MrBac server to obtain two draft models of strain TFA in SBML format which included the reactions present in iJO1366 and iJN1411 assigned to the orthologous genes identified in TFA. Both drafts were then merged into one model eliminating redundancies.
Secondly, as part of the manual curation, all the gene-protein-reaction (GPR) associations were manually reviewed. The assignment of each gene-protein to a reaction was validated by BLASTP against the Swiss-Prot database51 and/or Pseudomonas database (http://www.pseudomonas.com/). To increase the number of GPRs, proteins of strain TFA were considered orthologous to the best hit in the BLASTP alignment when the e-value was less than 1e-5, the identity higher than 30% and the coverage of both proteins in the alignment higher than 70%52. Additional criteria such as the absence or presence of a proper catabolic pathway were used to eliminate or to include transport reactions for some metabolites. Furthermore, a confidence value (between 1 and 4) was assigned to each reaction, which indicated the level of biological knowledge. This value was established according to Thiele and Palsson6 with 4 being the highest level of knowledge about the metabolic reaction. Metabolic/biochemical databases such as KEGG53, BRENDA54, MetaCyc55 and BiGG56 were used to find the information for the confidence value assignation. In addition, notes and bibliographic references were associated to some GPRs, which were used, together with the BLASTP result and the confidence value, to evaluate the inclusion of a reaction into the network.
Additionally, pathways for well-known metabolic features of strain TFA were manually added, as well as the biomass reaction, based on the biomass of iJN141157 and iJO1366. Since experimental data for macromolecular composition were not available for TFA, standard values were considered for the biomass (Supplementary Table S5). Moreover, the stoichiometric coefficients for amino acids, DNA and RNA were determined computationally using the genomic information (Supplementary Tables S6, S7, and S8, respectively). The composition of murein, lipids, inorganic ions and soluble pool were taken from P. putida and E. coli and both NGAM and GAM values from P. putida KT2440 iJN1411 (Supplementary Table S2). Since Sphingomonadaceae have sphingolipids in its outer membrane instead lipopolysaccharide (LPS), the latter was removed from TFA biomass and the synthesis of sphingolipids was added in the form of the precursor sphinganine (Supplementary Table S2).
A second biomass reaction for anoxic conditions was established, which lacks some metabolites that cannot be synthesised anaerobically (Supplementary Table S2).
Thirdly, the model was converted into a mathematical representation using the function xls2model stored in the COBRA package present in the MatLab. Thus, a stoichiometric matrix S was constructed in which each row represented a reaction and each column a metabolite. The components of the matrix were the stoichiometric coefficients of the metabolites used for the reactions. This conversion transformed the model into the SBML format, which was required to perform further analysis using the COBRA package.
Finally, it was confirmed that all reactions were balanced and that all metabolites were annotated with their charge and location ([c], cytosol; [p], periplasm and [e], extracellular space). Furthermore, the synthesis of all biomass components by the metabolic network was evaluated. If any one component was not produced, the corresponding biosynthesis pathway was manually revised to find gaps and to fill them in. In the same way, the reactions within each subsystem were analysed to corroborate that essential metabolites were properly synthetized. When possible, a gene was assigned to the newly added reaction using the criteria described above. Otherwise, it was incorporated as an orphan or spontaneous reaction based on available literature. Other non-gene associated reactions were also added, such as exchange reactions for all the external metabolites, sink and demand reactions and the non-growth ATP maintenance reaction (NGAM), as described by Thiele and Palsson6. Finally, the capacity of the final model to predict growth of strain TFA with different carbon sources was checked.
The reactions and metabolites included in the final iIG743 model can be found in the Supplementary Tables S3 and S4, respectively.
Flux Balance Analysis (FBA) and sampling
The metabolic reconstruction of strain TFA consisted of a network of reactions in which fluxes were constrained in several ways. Firstly, the lower and upper fluxes of each reaction were limited. Secondly, the metabolic matrix S was imposing constraint through the stoichiometric coefficients. And, thirdly, the stationary state was considered in the simulation, which is defined by the equation Sv=0, where v represents the fluxes through the reactions7,20
The Flux Balanced Analysis (FBA)58 was used to evaluate the biomass production (growth prediction) once the biomass reaction was fixed as the objective function (BOF, Biomass Objective Function). The result when executing FBA was the growth rate (h-1) predicted under the specified media conditions. All the exchange reactions were sequentially tested as potential carbon, nitrogen, or both carbon and nitrogen source, establishing an uptake (vi) of -10 mmol·gDW-1·h-1. For each test, the uptake of the rest of nutrients in the defined minimal medium, including oxygen, was set up to -30 ≤ vi ≤ 0 mmol·gDW1·h-1 to avoid uptake limitation other than the tested carbon and/or nitrogen source. For the carbon sources, the growth rate was further normalized considering the number of carbons of each source.
Furthermore, the concentration of metabolites in the extracellular space and the biomass formation along the cultivation time were analysed through the dynamic FBA (dFBA)59 under anoxic conditions. For that, the initial concentration of 3-HB and nitrate were set to 40 and 20 mM and the uptake to -3 and -2.82 mmol·gDW-1·h-1, respectively. As required for anoxic simulation, the entrance of oxygen was set to 0 mmol·gDW-1·h-1. The initial biomass was experimentally calculated to be 0.038 grams, the timestep was establish in 1 and the number of steps in 200.
The distribution of feasible fluxes in the condition-specific models was calculated by Markov chain Monte Carlo sampling60 implemented in COBRA package61.
In all the FBA, dFBA and sampling analyse, a minimal media was defined for both oxic and anoxic conditions in which the uptake of the involved compounds was set to –30 mmol·gDW-1·h-1 to avoid limitation, except for those for which the uptake was experimentally calculated (Table 3).
Construction of condition-specific models (constraints applied)
The in silico defined media provided new constraints in the metabolic system. These media were simulated by giving a value of flux entrance to the exchange reactions of the metabolites present in the minimal medium (MM) and the carbon source in which the predictions were to be done. To apply more specific constraints to the model in MM plus tetralin and MM plus 3-HB, transcriptomic data were incorporated following the protocol described31. A flux of 0 through a reaction was considered when the expression of the associated gene(s), measured in FPKM (Fragments Per Kilobase of transcript per Million mapped reads), was lower than the value of the first quartile of the total FPKM values. However, several cut-offs were analysed around the first quartile value, as recommended37.
Calculation of carbon uptake rates
Concentrations of 3-HB and sebacic acid were measured by GC-MS (gas chromatography–mass spectrometry), establishing an initial concentration of 40 mM and 16 mM, respectively. Briefly, 1 mL of sample collected at different time points during growth was centrifuged at 13000 rpm for 5 minutes. 100 µL of the supernatant were evaporated with nitrogen (50 °C) and resuspended on 100 µL of a 0.5 mg/mL solution of citric acid in pyridine. 200 µl of BSTFA + TMCS (99:1) were added to the sample, and the mixture was stirred for 5 seconds and then incubated at 100 °C for 45 minutes. Finally, 1 µL of sample was injected in a DB-17 column (Agilent) and the areas of the picks corresponding to the carbon source and to citrate, which was used as internal standard, were obtained. For L-lactate uptake, a lactate dehydrogenase assay was carried out to determine the concentration of L-lactate in the supernatant during cultivation. The initial concentration was set to 53 mM. Briefly, 5 µL of supernatant was added to 100 µL of 2x reaction buffer (600 mM glycine, 800 mM hydrazine, 4.8 mM NAD+, pH 9.0) and H2O was added to reach 200 µL. 1.5 U L-lactate dehydrogenase (Roche) was used to start the reaction and the formation of NADH was measured in a Tecan fluorimeter after 1 h of incubation at 30 °C. Lactate concentration was calculated based on a standard curve62. To express the fluxes in mmol·gDW-1·h-1, the biomass of strain TFA was experimentally measured in DW (Dry Weight) units. To do that, 100 mL of culture were taken at different times along the growth curve in MM plus the corresponding carbon source, centrifuged for 15 min at 5000 rpm and the pellet resuspended in 2 mL of phosphate buffer (Na2HPO4·12H2O 0.34 mM, KH2PO4 0.147 mM) and centrifuged for 5 min at 13000 rpm. Finally, the pellet was dried at 80 °C and weighed on a precision balance.
References
Yabuuchi, E. & Kosako, Y. Sphingomonadaceae. Bergey’s Manual of Systematics of Archaea and Bacteria 1–2, https://doi.org/10.1002/9781118960608.fbm00179 (2015).
Takeuchi, M., Hamana, K. & Hiraishi, A. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51, 1405–1417 (2001).
Balkwill, D. L., Fredrickson, J. K. & Romine, M. F. Sphingomonas and Related Genera. in The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass (eds. Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H. & Stackebrandt, E.) 605–629, https://doi.org/10.1007/0-387-30747-8_23 (Springer New York, 2006).
Lauro, F. M. et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 106, 15527–15533 (2009).
Fondi, M. & Liò, P. Genome-Scale Metabolic Network Reconstruction. in Bacterial Pangenomics SE - 15 (eds. Mengoni, A., Galardini, M. & Fondi, M.) 1231, 233–256 (Springer New York, 2015).
Thiele, I. & Palsson, B. Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93 (2010).
Nogales, J. A Practical Protocol for Genome-Scale Metabolic Reconstructions. in Hydrocarbon and Lipid Microbiology Protocols: Genetic, Genomic and System Analyses of Pure Cultures (eds. McGenity, T. J., Timmis, K. N. & Balbina, N.) 197–221, https://doi.org/10.1007/8623_2014_12 (Springer Berlin Heidelberg, 2014).
Gu, C., Kim, G. B., Kim, W. J., Kim, H. U. & Lee, S. Y. Current status and applications of genome-scale metabolic models. Genome Biol. 20, 121 (2019).
Feist, A. M., Herrgård, M. J., Thiele, I., Reed, J. L. & Palsson, B. Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7, 129–143 (2009).
King, Z. A., Lloyd, C. J., Feist, A. M. & Palsson, B. Ø. Next-generation genome-scale models for metabolic engineering. Curr. Opin. Biotechnol. 35, 23–29 (2015).
Büchel, F. et al. Path2Models: large-scale generation of computational models from biochemical pathway maps. BMC Syst. Biol. 7, 116 (2013).
Machado, D., Andrejev, S., Tramontano, M. & Patil, K. R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46, 7542–7553 (2018).
Magnúsdóttir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81 (2016).
Genome [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. (2004). Available at:, https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/. (Accessed: 18th December 2019).
Motamedian, E., Saeidi, M. & Shojaosadati, S. A. Reconstruction of a charge balanced genome-scale metabolic model to study the energy-uncoupled growth of Zymomonas mobilis ZM1. Mol. Biosyst. 12, 1241–1249 (2016).
Wang, X. et al. Advances and prospects in metabolic engineering of Zymomonas mobilis. Metab. Eng. 50, 57–73 (2018).
Hernáez, M. J., Reineke, W. & Santero, E. Genetic analysis of biodegradation of tetralin by a Sphingomonas strain. Appl. Environ. Microbiol. 65, 1806–1810 (1999).
Floriano, B., Santero, E. & Reyes-Ramírez, F. Biodegradation of Tetralin: Genomics, Gene Function and Regulation. Genes (Basel). 10, 339 (2019).
García-Romero, I. et al. Genomic analysis of the nitrate-respiring Sphingopyxis granuli (formerly Sphingomonas macrogoltabida) strain TFA. BMC Genomics, https://doi.org/10.1186/s12864-016-2411-1 (2016)
Orth, J. D. et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism-2011. Mol. Syst. Biol. 7, 1–9 (2011).
Nogales, J., Gudmundsson, S., Duque, E., Ramos, J. L. & Palsson, B. Ø. Expanding The Computable Reactome In Pseudomonas putida Reveals Metabolic Cycles Providing Robustness. bioRxiv 139121+, https://doi.org/10.1101/139121 (2017).
Eggers, J. & Steinbüchel, A. Poly(3-hydroxybutyrate) degradation in Ralstonia eutropha H16 is mediated stereoselectively to (S)-3-hydroxybutyryl coenzyme a (CoA) via crotonyl-CoA. J. Bacteriol. 195, 3213–3223 (2013).
Khetkorn, W., Incharoensakdi, A., Lindblad, P. & Jantaro, S. Enhancement of poly-3-hydroxybutyrate production in Synechocystis sp. PCC 6803 by overexpression of its native biosynthetic genes. Bioresour. Technol. 214, 761–768 (2016).
Kawahara, K. et al. Chemical structure of glycosphingolipids isolated from Sphingomonas paucimobilis. FEBS Lett. 292, 107–110 (1991).
Hu, X. et al. The pva operon is located on the megaplasmid of Sphingopyxis sp. strain 113P3 and is constitutively expressed, although expression is enhanced by PVA. Appl. Microbiol. Biotechnol. 78, 685–693 (2008).
Shimao, M. et al. Pyrroloquinoline Quinone as an Essential Growth Factor for a Poly(vinyl alcohol)-degrading Symbiont, Pseudomonas sp. VM15C. Agric. Biol. Chem. 48, 2873–2876 (1984).
Kim, B. C., Sohn, C. K., Lim, S. K., Lee, J. W. & Park, W. Degradation of polyvinyl alcohol by Sphingomonas sp. SA3 and its symbiote. J. Ind. Microbiol. Biotechnol. 30, 70–74 (2003).
López-Sánchez, A., Floriano, B., Andújar, E., Hernáez, M. J. & Santero, E. Tetralin-induced and ThnR-regulated aldehyde dehydrogenase and β-oxidation genes in Sphingomonas macrogolitabida strain TFA. Appl. Environ. Microbiol. 76, 110–118 (2010).
Martínez-Pérez, O., Moreno-Ruiz, E., Floriano, B. & Santero, E. Regulation of tetralin biodegradation and identification of genes essential for expression of thn operons. J. Bacteriol. 186, 6101–6109 (2004).
Gonzalez, J. E., Long, C. P. & Antoniewicz, M. R. Comprehensive analysis of glucose and xylose metabolism in Escherichia coli under aerobic and anaerobic conditions by 13C metabolic flux analysis. Metab. Eng. 39, 9–18 (2017).
Nogales, J. & Agudo, L. A Practical Protocol for Integration of Transcriptomics Data into Genome-Scale Metabolic Reconstructions. in Hydrocarbon and Lipid Microbiology Protocols: Synthetic and Systems Biology - Tools (eds. McGenity, T. J., Timmis, K. N. & Nogales, B.) 135–152, https://doi.org/10.1007/8623_2015_98 (Springer Berlin Heidelberg, 2016).
García-Romero, I., Förstner, K. U., Santero, E. & Floriano, B. SuhB, a small non-coding RNA involved in catabolite repression of tetralin degradation genes in Sphingopyxis granuli strain TFA. Environ. Microbiol. 20, 3671–3683 (2018).
Becker, S. A. & Palsson, B. Ø. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4, (2008).
Ingram, L. O. Ethanol Tolerance in Bacteria. Crit. Rev. Biotechnol. 9, 305–319 (1989).
Takahashi, K., Morita, T. & Kawazoe, Y. Mutagenic characteristics of formaldehyde on bacterial systems. Mutat. Res. Toxicol. 156, 153–161 (1985).
Kim, M. K., Im, W.-T., Ohta, H., Lee, M. & Lee, S.-T. Sphingopyxis granuli sp. nov., a β-glucosidase-producing bacterium in the family Sphingomonadaceae in α-4 subclass of the Proteobacteria. J Microbiol 43, 152–157 (2005).
Fong, S. S., Nanchen, A., Palsson, B. Ø. & Sauer, U. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J. Biol. Chem. 281, 8024–8033 (2006).
Kaminski, M. A., Sobczak, A., Dziembowski, A. & Lipinski, L. Genomic analysis of γ-hexachlorocyclohexane-degrading Sphingopyxis lindanitolerans WS5A3p strain in the context of the pangenome of Sphingopyxis. Genes (Basel). 10 (2019).
Schut, F. et al. Substrate uptake and utilization by a marine ultramicrobacterium. Microbiology 141, 351–361 (1995).
Uchino, K., Saito, T., Gebauer, B. & Jendrossek, D. Isolated poly(3-hydroxybutyrate) (PHB) granules are complex bacterial organelles catalyzing formation of PHB from acetyl coenzyme A (CoA) and degradation of PHB to acetyl-CoA. J. Bacteriol. 189, 8250–8256 (2007).
Martín-Cabello, G., Moreno-Ruiz, E., Morales, V., Floriano, B. & Santero, E. Involvement of poly(3-hydroxybutyrate) synthesis in catabolite repression of tetralin biodegradation genes in Sphingomonas macrogolitabida strain TFA. Environ. Microbiol. Rep. 3, 627–631 (2011).
Bartell, J. A. et al. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun. 8, (2017).
Dunphy, L. J., Yen, P. & Papin, J. A. Integrated Experimental and Computational Analyses Reveal Differential Metabolic Functionality in Antibiotic-Resistant Pseudomonas aeruginosa. Cell Syst. 8, 3–14.e3 (2019).
Guzmán, G. I. et al. Model-driven discovery of underground metabolic functions in Escherichia coli. Proc. Natl. Acad. Sci. USA 112, 929–934 (2015).
Calero, P. & Nikel, P. I. Chasing bacterial chassis for metabolic engineering: a perspective review from classical to non-traditional microorganisms. Microb. Biotechnol. 12, 98–124 (2019).
Renilla, S. et al. Acetate scavenging activity in Escherichia coli: interplay of acetyl-CoA synthetase and the PEP-glyoxylate cycle in chemostat cultures. Appl. Microbiol. Biotechnol. 93, 2109–2124 (2012).
Ahn, S., Jung, J., Jang, I.-A., Madsen, E. L. & Park, W. Role of Glyoxylate Shunt in Oxidative Stress Response. J. Biol. Chem. 291, 11928–11938 (2016).
Ferrante, A. A., Augliera, J., Lewis, K. & Klibanov, A. M. Cloning of an organic solvent-resistance gene in Escherichia coli: the unexpected role of alkylhydroperoxide reductase. Proc. Natl. Acad. Sci. USA 92, 7617–7621 (1995).
Kim, S.-J., Kweon, O. & Cerniglia, C. E. Proteomic applications to elucidate bacterial aromatic hydrocarbon metabolic pathways. Curr. Opin. Microbiol. 12, 301–309 (2009).
Liao, Y. C., Tsai, M. H., Chen, F. C. & Hsiung, C. A. GEMSiRV: A software platform for GEnome-scale metabolic model simulation, reconstruction and visualization. Bioinformatics 28, 1752–1758 (2012).
Boutet, E. et al. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 1374, 23–54 (2016).
Konstantinidis, K. T. & Tiedje, J. M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264 (2005).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Schomburg, I. et al. BRENDA: a resource for enzyme data and metabolic information. Trends Biochem. Sci. 27, 54–56 (2002).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639 (2018).
King, Z. A. et al. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 44, D515–D522 (2016).
Nogales, J. et al. High-quality genome-scale metabolic modelling of Pseudomonas putida highlights its broad metabolic capabilities. Environ. Microbiol. 22, 255–269 (2020).
Orth, J. D., Thiele, I. & Palsson, B. Ø. What is flux balance analysis? Nat. Biotechnol. 28, 245–248 (2010).
Gomez, J. A., Höffner, K. & Barton, P. I. DFBAlab: a fast and reliable MATLAB code for dynamic flux balance analysis. BMC Bioinformatics 15, 409 (2014).
Schellenberger, J. & Palsson, B. Ø. Use of randomized sampling for analysis of metabolic networks. J. Biol. Chem. 284, 5457–5461 (2009).
Schellenberger, J. et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6, 1290–1307 (2011).
Hamilton, H. L. & Pardue, S. Quantitation of Lactate by a Kinetic Method with an Extended Range of Linearity and Low Dependence on Experimental Variables. 30, 226–229 (1984).
Mellbye, B. L. et al. Genome-Scale, Constraint-Based Modeling of Nitrogen Oxide Fluxes during Coculture of Nitrosomonas europaea and Nitrobacter winogradskyi. mSystems 3, 1–13 (2018).
Zhao, H., Li, M., Fang, K., Chen, W. & Wang, J. In silico insights into the symbiotic nitrogen fixation in Sinorhizobium meliloti via metabolic reconstruction. PLoS One 7 (2012).
Zhang, H. et al. Reconstruction of a Genome-scale Metabolic Network of Komagataeibacter nataicola RZS01 for Cellulose Production. Sci. Rep. 7, 1–9 (2017).
Resendis-Antonio, O., Hernández, M., Mora, Y. & Encarnación, S. Functional Modules, Structural Topology, and Optimal Activity in Metabolic Networks. PLoS Comput. Biol. 8, (2012).
Peyraud, R. et al. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 5, 189 (2011).
Imam, S. et al. iRsp1095: A genome-scale reconstruction of the Rhodobacter sphaeroides metabolic network. BMC Syst. Biol. 5, 116 (2011).
Zou, W. et al. Reconstruction and analysis of a genome-scale metabolic model of the vitamin C producing industrial strain Ketogulonicigenium vulgare WSH-001. J. Biotechnol. 161, 42–48 (2012).
Wu, X., Wang, X. & Lu, W. Genome-scale reconstruction of a metabolic network for Gluconobacter oxydans 621H. BioSystems 117, 10–14 (2014).
Yang, Y., Hu, X.-P. & Ma, B.-G. Construction and simulation of the Bradyrhizobium diazoefficiens USDA110 metabolic network: a comparison between free-living and symbiotic states. Mol. BioSyst. 13, 607–620 (2017).
Bordel, S., Rojas, A. & Muñoz, R. Reconstruction of a Genome Scale Metabolic Model of the polyhydroxybutyrate producing methanotroph Methylocystis parvus OBBP. Microb. Cell Fact. 18, 1–11 (2019).
Acknowledgements
We thank Guadalupe Martín and Nuria Pérez for their technical support and all members of the laboratory for their insights and suggestions. This research was funded by Ministerio de Ciencia, Innovación y Universidades, grant numbers BIO2014-57545-R, BIO2014-59528-JIN, and BIO2016-79736-R.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: I.G.-R., J.N., E.S., B.F.; Performed the experiments: I.G.-R., B.F.; Analysed the results: I.G.-R., J.N., E.S., B.F.; Prepared and edited the manuscript: I.G.-R., J.N., E.D., E.S., B.F. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Romero, I., Nogales, J., Díaz, E. et al. Understanding the metabolism of the tetralin degrader Sphingopyxis granuli strain TFA through genome-scale metabolic modelling. Sci Rep 10, 8651 (2020). https://doi.org/10.1038/s41598-020-65258-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65258-9
This article is cited by
-
Unveiling the potential of systems biology in biotechnology and biomedical research
Systems Microbiology and Biomanufacturing (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
