
Abstract
Many Alzheimer’s disease (AD) genes including Apolipoprotein E (APOE) are found to be expressed in blood-derived macrophages and thus may alter blood protein levels. We measured 91 neuro-proteins in plasma from 316 participants of the Rotterdam Study (incident AD = 161) using Proximity Extension Ligation assay. We studied the association of plasma proteins with AD in the overall sample and stratified by APOE. Findings from the Rotterdam study were replicated in 186 AD patients of the BioFINDER study. We further evaluated the correlation of these protein biomarkers with total tau (t-tau), phosphorylated tau (p-tau) and amyloid-beta (Aβ) 42 levels in cerebrospinal fluid (CSF) in the Amsterdam Dementia Cohort (N = 441). Finally, we conducted a genome-wide association study (GWAS) to identify the genetic variants determining the blood levels of AD-associated proteins. Plasma levels of the proteins, CDH6 (β = 0.638, P = 3.33 × 10−4) and HAGH (β = 0.481, P = 7.20 × 10−4), were significantly elevated in APOE ε4 carrier AD patients. The findings in the Rotterdam Study were replicated in the BioFINDER study for both CDH6 (β = 1.365, P = 3.97 × 10−3) and HAGH proteins (β = 0.506, P = 9.31 × 10−7) when comparing cases and controls in APOE ε4 carriers. In the CSF, CDH6 levels were positively correlated with t-tau and p-tau in the total sample as well as in APOE ε4 stratum (P < 1 × 10−3). The HAGH protein was not detected in CSF. GWAS of plasma CDH6 protein levels showed significant association with a cis-regulatory locus (rs111283466, P = 1.92 × 10−9). CDH6 protein is implicated in cell adhesion and synaptogenesis while HAGH protein is related to the oxidative stress pathway. Our findings suggest that these pathways may be altered during presymptomatic AD and that CDH6 and HAGH may be new blood-based biomarkers.
Similar content being viewed by others

Introduction
Apolipoprotein E (APOE) is the most common genetic risk factor for Alzheimer’s disease (AD)1,2 and an important driver of the lifetime risk for AD3,4. APOE interacts with other common genetic determinants of AD2,5, suggesting an interaction with specific protein pathways. Despite two decades of research, the role of APOE in determining the risk of AD is far from being understood6. The IMI ADAPTED (The Alzheimer’s Disease Apolipoprotein Pathology for Treatment Elucidation and Development) is an Innovative Medicine Initiative (IMI) that aims to improve the understanding about the role of APOE gene in AD.
AD pathology is characterized by the extracellular deposition of amyloid-beta (Aβ)-42 and intracellular accumulation of phosphorylated tau in the brain. Cerebrospinal fluid (CSF) levels of Aβ-42, phosphorylated (p-tau) and total tau (t-tau) are well-established biomarkers of the central nervous system and brain AD pathology7. However, there is a growing evidence for a relation between other pathologies and AD, such as vascular pathology8. For example, studies integrating epidemiological and vascular research showed that vascular pathology may affect brain function and increase the risk of AD9. APOE and many of the novel genes implicated in AD are expressed in monocytes/macrophages10,11,12 in the blood, and thus these genes may alter the protein signatures in blood. There is also a growing body of evidence indicating that Aβ may disrupt the cerebral microcirculation regulation13,14,15, endothelial function16,17, and brain perivascular macrophages function18. Thus, protein and metabolite homeostasis in blood may also be altered as a consequence of (early) amyloid pathology. Indeed, there is an increasing interest in the relation between protein levels in plasma and AD during presymptomatic stages of AD19. Multiple studies have investigated the association of a range of proteins with AD in plasma, but few have addressed the effect of APOE19,20,21,22,23,24,25. Furthermore, there is lack of investigations connecting molecular signatures of AD in blood to neuropathological AD markers in CSF.
Advances in high-throughput omics technologies have allowed the detection and quantification of several classes of plasma-based biomolecular compounds including circulating metabolites and proteins26. In the present study, we aimed to identify altered levels of proteins in the circulation of presymptomatic AD patients in the overall population and among various genetic risk groups based on the APOE gene, with a view to obtaining insights into molecular signatures in the circulation. To this end, we have examined the association of neurology relevant proteins in a prospective population-based, the Rotterdam Study. Proteins associated with AD were further tested for replication in the BioFINDER study. Next, we conducted a genome-wide association study to find the genetic variants determining the blood levels of AD-associated proteins. Finally, we studied the association of the protein consistently associated with AD to amyloid and tau levels in CSF in the Amsterdam Dementia Cohort (ADC).
Results
Association of plasma proteins with AD
Detailed results of overall and APOE stratified association analysis of proteins with AD are provided in Table 1 and Fig. 1. No significantly associated protein to AD was identified in the overall analysis at an FDR < 0.05. Overall, there is a tendency that protein levels are more likely increased (positive effect size, β) than decreased (negative effect size, β) in AD patients that carry the APOE ε4 allele (Fig. 1a,b) and those homozygous for APOE ε3 allele (Fig. 1c) but not for APOE ε2 patients (Fig. 1d). In APOE stratified discovery analysis, we observed that levels of CDH6 (β = 0.638, P = 3.33 × 10−4, FDR = 0.030) and HAGH (β = 0.481, P = 7.20 × 10−4, FDR = 0.033) were significantly increased in AD patients who carry the APOE ε4 allele (see Table 1). Both CDH6 (β = 0.624, P = 5.52 × 10−4, FDR = 0.030) and HAGH (β = 0.491, P = 6.62 × 10−4, FDR = 0.030) proteins remained significantly associated with AD even after adjusting for other covariates in model 2 (Supplementary Table 1). In the replication analysis in the BioFINDER study (Table 2), plasma levels of CDH6 and HAGH were significantly associated with AD in the overall sample (CDH6: β = 1.212, P = 5.18 × 10−4; HAGH: β = 0.631, P = 7.56 × 10−15) as well as in APOE ε4 carriers (CDH6: β = 1.365, P = 3.97 × 10−3; HAGH: β = 0.506, P = 9.31 × 10−7) but not in APOE ε2 carriers. Plasma levels of HAGH protein were also associated with AD in APOE ε33 carriers (β = 0.739, P = 3.76 × 10−7) but in this subgroup no association was seen with CDH6.

Volcano plots representing the association of plasma protein levels with Alzheimer’s disease (AD) in (a) overall analysis; (b) APOE4 stratum; (c) APOE33 stratum and (d) APOE2 stratum. Each dot represents a protein with regression coefficient (β) of association plotted on x-axis and -log10 of P-values on y-axis. Proteins showing nominal association (P-value < 0.05) are annotated in overall and stratified analysis. Light blue color of dot indicates decreased protein levels with β −0.0 to −0.184 and dark blue to indicate with β < −0.184 while pink color indicates increased protein levels with β ranging from 0.0 to 0.184 and red color shows β > 0.184. Black dots are used for proteins which pass the multiple testing (false discovery rate <0.05).
Figure 2 shows that the APOE genotype modifies the association between proteins and AD based on nominal statistical significance. In discovery analysis, eight additional proteins (TN-R, Beta-NGF, MDGA1, JAM-B, CD200, LXN, PDGF-R-alpha, and LAIR-2) were also positively associated with AD APOE ε4 carriers (β > 0.107, P < 0.05), but they did not survive multiple testing. In the APOE2 stratum, the levels of two proteins including SMPD1 and VWC2 were reduced in AD cases compared to the APOE genotype matched controls (Supplementary Fig. 1). In APOE33 stratum, PRTG, CNTN5 and NCAN proteins, that do not emerge in the APOE4 or APOE2 stratum (see Fig. 2), showed suggestive associations but did not survive multiple testing (β > 0.193, P < 2.57 × 10−2). Both CDH6 (β = 0.202, P = 1.79 × 10−1) and HAGH (β = −0.018, P = 8.42 × 10−1) did not show association with AD in APOE33 carriers while HAGH showed nominal association in APOE ε2 carriers (β = 0.482, P = 4.25 × 10−2) (See Table 1).

Venn diagram showing the overlap of proteins identified in the association analysis results of overall and APOE stratified discovery analysis.
Sensitivity analyses
Sensitivity analyses were performed in the Rotterdam Study to test the robustness of our findings. In the first sensitivity analysis (Supplementary Table 2), we adjusted for follow-up time, taking into account that some cases or controls may die of other diseases. This analysis showed that levels of both HAGH (β = 0.477, P = 6.47 × 10−4) and CDH6 (β = 0.661, P = 1.48 × 10−4) proteins were significantly increased in AD patients compared to controls in APOE ε4 carriers. In the second sensitivity analysis, we only analyzed protein that were assessed directly (non-imputed data). Similarly, the association of HAGH and CDH6 proteins with AD remained significant in APOE ε4 carriers when analyzing non-imputed proteomics data (Supplementary Table 3). Last but not least, we performed a formal interaction test to evaluate the interaction of APOE with each of the 91 proteins (Supplementary Table 4). Only three of 91 proteins showed interaction with APOE (P-value < 0.05) including HAGH (βinteraction = 0.414, Pinteraction = 1.70 × 10−2), G-CSF (βinteraction = 0.276, Pinteraction = 2.78 × 10−2) and CRTAM (βinteraction = −0.221, Pinteraction = 3.77 × 10−2). Except, HAGH other two proteins (G-CSF, CRTAM) did not show association with AD in any of the analyzed APOE stratum (P-value > 0.05). For CDH6, the test for interaction was not significant (βinteraction = 0.078, Pinteraction = 7.23 × 10−1).
Association of CDH6 and HAGH protein levels with Aβ−42, p-tau, and t-tau in CSF
Among the two proteins that were associated to the future risk of AD, CDH6 and HAGH, the latter was not detected in the CSF in >90% of the subjects in ADC cohort. CSF CDH6 protein levels were not associated with AD (β = 0.329, SE = 0.220, P = 0.136) in the overall as well as in APOE stratified analysis (P > 0.114; see Supplementary Table 5). However, multiple regression analysis adjusted for age and sex revealed a significant association of CDH6 CSF levels with both p-tau (β = 23.2, SE = 3.4, P = 3.48 × 10−11) and t-tau (β = 207.4, SE = 36.4, P = 2.40 × 10−8) when pooling AD patients and controls (Table 3 and Fig. 3). In the APOE stratified analysis, levels of CDH6 were significantly associated with p-tau and t-tau levels but not with Aβ−42 levels in CSF in three APOE strata (see Table 3). When stratifying by case-control status (Supplementary Table 6), CDH6 levels were significantly associated with p-tau and t-tau levels in both cases and controls. In controls, also Aβ−42 was positively associated with CDH6 (P < 1 × 10−3; see Supplementary Table 6).

Correlation plot between cerebrospinal fluid (CSF) levels of CDH6 with Aβ−42, p-tau and t-tau.
Association of proteins with APOE
Next, we associated the APOE genotype to the protein levels significantly associated with AD (CDH6 and HAGH). Results of the association of protein levels with APOE genotypes are provided in Supplementary Table 7 and Supplementary Fig. 2. In the overall sample, CDH6 protein levels were increased in the APOE ε4 carriers compared to APOE ε2 carriers (β = 0.163, P = 3.79 × 10−3). In controls, levels of CDH6 protein were decreased (β = −0.131, P = 0.026) in APOE ε2 carriers compared to APOE 33 carriers. In the controls, levels of HAGH were decreased in the APOE ε4 (β = −0.192, P = 0.028) and APOE ε2 carriers (β = −0.214, P = 0.042) compared to APOE 33 carriers.
Genome-wide association study (GWAS)
The GWAS was conducted to determine the genetic drivers of the CDH6 and HAGH protein levels (Supplementary Figs. 3a and 4 respectively). We identified 13 genome-wide significant cis protein quantitative trait loci (pQTLs) located at 5p13.3 locus of chromosome 5 for CDH6 protein levels. All genome-wide significant pQTLs are located in the intergenic region at 5′ UTR region of the CDH6 gene. Among the 13 identified pQTLs, rs111283466 was the lead pQTL with the effect estimate (β) of 1.068 and P-value 1.92 × 10−9 (Supplementary Table 8). Q-Q plot (Supplementary Fig. 5a) indicates that the results are well adjusted for population stratification (λ = 1.0056). Further lookups in the GTEx database showed that the lead pQTL (rs111283466) also affects the expression of CDH6 gene in various body tissues. GWAS analysis of HAGH protein levels did not identify any genome-wide significant pQTLs. Manhattan plot and Q-Q plot for GWAS results of HAGH protein levels are provided in the Supplementary materials (Supplementary Figs. 4 and 5b).
Discussion
In our study, plasma levels of CDH6 and HAGH proteins are significantly increased in presymptomatic AD patients compared to controls in the APOE4 stratum. In the replication analysis, both CDH6 and HAGH proteins showed significant association with AD in the BioFINDER study in APOE ε4 carriers. CDH6 protein levels were significantly correlated with p-tau and t-tau measurements in CSF of the ADC. In GWAS analysis, we have also identified a genome-wide significant pQTL for CDH6 protein levels in the blood (rs111283466), which also affects the expression levels of CDH6 transcripts in several tissues.
We observed a significant increase of CDH6 protein levels in the plasma of presymptomatic AD cases carrying the APOE ε4 allele which was also replicated in the BioFINDER study. When comparing our findings to the other studies19,20,21,22, we do not have an overlap in understudy proteins. However, like previous studies we do find an effect of the APOE gene on plasma level of prorteins21,22. In the APOE4 stratum, we see that the volcano plot (Fig. 1) is clearly asymmetric suggesting increased levels of most neuronal proteins in AD patients carrying this allele before the clinical onset of disease. This might be explained by an increase in the blood-brain barrier permeability in APOE ε4 carriers27, which may lead to increased levels of CDH6 in the blood as a result of higher levels of CDH6 in the brain. We found that CDH6 levels in the blood are driven by a genetic variant (rs111283466) in the cis-regulatory region. This may determine the CDH6 levels in both brain and blood cells, leaving the possibility open that elevated CDH6 has a blood-derived origin. Yet, such a mechanism does not explain why elevated levels in the blood are only seen in patients carrying the APOE ε4 allele. It is of note that the CDH6 coding gene is part of a larger cluster of cadherin (CDH) genes including CDH9, CDH10, CDH12 and CDH18. As all of the CDH genes are paralogues and share homology, it is crucial to exclude cross-reactions of the antibodies28 used by Olink across CDH proteins. Our GWAS benchmarks that the protein assessed in our plasma is indeed CDH6, as we found that the most important driver of the protein is in the promoter region of CDH6. None of the recently published GWAS of proteins reported significant pQTLs for CDH6 protein based on SomaLogic29,30. The aptomere based measurement of the SomaLogic yields a different protein spectrum than that of the antibody-based method of Olink31. Our identified pQTLs did not show any association with AD (P-value < 0.05) in the largest AD GWAS11. However, we find in our study that levels of CDH6 were increased in APOE ε4 carriers compared to APOE ε2 carriers. We do find that the region is associated to postcentral gyri in GWAS32 and a study has reported reduced volume of postcentral gyri in dementia patients33.
Interestingly, we found significant positive associations of CSF levels of CDH6 protein with p-tau and t-tau levels in overall as well as in AD and controls only analyses, which are considered as biomarkers of neuronal injury and tau pathology34,35. The upregulation of CDH6 protein in cerebral cortices of AD mice models (APP/PS1) compared to wild type has been reported by Lu et al.36, which is in line with our observation of positive correlation between CDH6 levels and AD pathological markers in CSF. Yet, we did not observe an association of the CDH6 protein with AD in CSF. We also observed positive association between CDH6 levels with amyloid-beta 42 in CSF of the controls, which might indicate disturbance in the amyloid-beta 42 metabolism which precedes decades before the buildup of Aβ in the brain37. Alternatively, it may point towards a similar mechanism of production of the Aβ-42 and CDH6 proteins in a healthy state38. Increased levels of phosphorylated CDH6 protein levels were reported upon the addition of amyloid-beta in cortical neuronal cells39, which adds evidence to the role of CDH6 in AD pathology. Taken together, these findings suggest that CSF levels of CDH6 protein may be associated with neuronal and axonal cell injury and neurofibrillary tangles in AD.
CDH6 is a cell surface glycoprotein that belongs to type II cadherin’s40. Cadherins are highly expressed in the brain and other tissues. They strongly interact with other molecules to perform molecular processes including synaptic functions41,42,43, synaptogenesis44, TGF-B signaling45, neural crest differentiation46, presenilin-mediated signaling and integrity of blood-brain barrier47. Although it is not possible to infer whether the correlation of AD pathology with CDH6 in plasma and CSF, are cause or consequence of the disease, several pieces of evidence favor the role of CDH6 in the pathogenesis of AD39,48. A recent study showed that the ADAM10 enzyme, whose coding gene is associated with AD11, is involved in proteolytic cleavage of the CDH6 protein, resulting in the formation of C-terminal fragment49, in a similar manner as it cleaves the amyloid precursor protein (APP)50,51. The transmembrane N-cadherin (CDH2), a paralogue of CDH6 and functionally related to CDH643, is also known to be cleaved by ADAM10 into N-cadherin C-terminal fragment 1 (NcadCTF1). Andreyeva et al.52, have demonstrated that NcadCTF1 leads to accelerated amyloid-β-induced synaptic impairment, a process that characterizes an early stage event in AD53,54. Increased levels of NcadCTF1 were also found in postmortem AD brain tissues compared with controls, suggesting that cadherins might induce synaptic dysfunction in a synergistic manner52.
In addition to CDH6, increased plasma levels of HAGH (Hydroxyacylglutathione hydrolase, mitochondrial) protein also showed significant association with AD in those who carry the APOE ε4 variant and suggestive association in overall and in APOE ε2 carriers. This finding is in line with the recently published findings of the BioFINDER study25 and further supported by the APOE stratified analysis in the BioFINDER study that was conducted for the present study. In the replication analysis, plasma levels of HAGH showed significant association in both APOE ε4 and APOE ε33 carriers while in the discovery analysis in the Rotterdam Study HAGH only showed significant association in APOE ε4 carriers which may be due to the lack of power. The HAGH protein is also known as glyoxalase-2, an enzyme, which is involved in the glyoxalase system along with glyoxalase-1 and its cofactor glutathione, a key player is oxidative stress control55,56. Overall, the glyoxalase system is involved in the detoxification of glycolysis by-products particularly cytotoxic metabolite methylglyoxal57. Levels of methylglyoxal in plasma are elevated during various disease conditions including hyperglycemia, which leads to the formation of reactive oxygen species (ROS) and causes oxidative stress. Moreover, methylglyoxal is also the precursor of glycation end products (AGEs) which are implicated in neurodegeneration and AD58,59. The most compelling evidence for the role of the glyoxalase-2 protein in AD is that the AGEs and glyoxalase system is implicated in the regulation of amyloid precursor protein (APP) expression60,61. Although glyoxalase system attributes protection against methylglyoxal mediated oxidative stress, earlier studies have also observed increased levels of glyoxalase-1 enzyme (involved in the first step of methylglyoxal detoxification) in early AD stages62,63. Increased levels of glyoxalase-2 (involved in the second step of methylglyoxal detoxification) in plasma might be a compensatory mechanism to increased levels of methylglyoxal during the early phase of disease or a general stress response55. The growing number of studies have suggested the involvement of oxidative stress during the prodromal stage of AD64,65,66, which is in line with our finding of increased levels of glyoxalase-2 observed before the onset of AD.
The strength of the current study includes that it is conducted in the prospective population-based RS cohort, where samples were selected with mean 6.9 years of follow-up preceding the diagnosis of AD. It allowed us to study the plasma proteomics changes prior to the development of AD clinical symptoms. As AD is a disorder of the brain, we have validated that CSF levels of CDH6 are also associated with biomarkers of AD in CSF in an independent cohort. Further, we used the Olink neurology proteomic panel of 91 proteins for the quantification of proteins in the plasma, which estimates targeted proteins expressed in the brain from different pathways. One of the major limitations of our study is the limited sample size, including a small number of APOE ε4 carrier controls in the stratified analysis.
In conclusion, we observed elevated protein levels of CDH6 in plasma of AD patients carrying APOE ε4 allele in the discovery and replication analysis, a protein that plays a role in synaptogenesis. Positive correlation of CSF CDH6 levels with p-tau and t-tau may also indicate the association of CDH6 with neurodegeneration. We further found the association of the plasma levels of HAGH protein to AD in those carrying the APOE ε4 allele. Association of HAGH with AD further suggest the involvement of the glyoxalase and oxidative stress pathways in the pathogenesis of AD.
Methods
Study populations
Rotterdam study
The Rotterdam Study (RS) is a prospective population-based study comprising of 14,926 middle and older aged (≥45 years) individuals from the Ommoord district of Rotterdam. The RS consists of three cohorts including RS-I (started in 1990, N = 7983 participants), RS-II (started in 2000, N = 3011) and RS-III (started in 2006, N = 3932)67. Study participants were extensively interviewed and physically examined at baseline and after every 3 to 4 years. For each participant fasting blood was collected at a dedicated center, centrifuged (Speed = 3500 g for 20 min at 4 °C) within 4 hours of venipuncture to collect plasma and stored at −80 °C. The study has been approved by the Medical Ethical Committee of Erasmus Medical Center and by the Ministry of Health, Welfare and Sport of the Netherlands. Written informed consent was obtained from each study participant to participate and to collect information from their treating physicians. All methods were performed in accordance with the relevant guidelines and regulations. In current nested case-control proteomics analysis, we chose 161 incident AD cases and 155 controls match with respect to their age and sex, from the fifth visit of RS-I (RS-I-5) cohort. Table 4 shows the baseline characteristics of the selected sample. There were no significant differences in age, sex and body mass index (BMI). AD patients were more often carriers of the APOE ε4 variant and less often of the APOE ε2 variant. Blood for the proteome profiling was collected on average 6.9 years (standard deviation [SD] = 1.7) before the onset of clinical dementia in patients and mean 8.7 years (SD = 3.2) before the latest follow-up in controls.
Dementia diagnosis
Over time, all participants were screened for dementia using the Mini-Mental State Examination (MMSE)68 and Geriatric Mental Schedule (GMS)69 organic level for all participants. Screen-positive subjects (MMSE < 26 or GMS organic level > 0) underwent the Cambridge examination for mental disorders of the elderly (CAMDEX)70 and participants suspected of having dementia were extensively examined with neuropsychological testing and neuroimaging biomarkers when available. Patients were further ascertained by linking them with their medical records from general practitioners, the regional institute for outpatient mental health care and municipality. Dementia of all patients was diagnosed based on the internationally accepted Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R) criteria and AD using the National Institute of Neurological Disorders and Stroke–Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINCDS-ADRDA)71 criteria for possible, probable and definite AD. NINCDS-ADRDA criteria were also used to diagnose vascular dementia. The final diagnosis was confirmed by a panel of neurologists, neurophysiologists, and research physicians72. AD diagnosis in RS is also provided in more detail in earlier publications72.
Proteome profiling
Proteomics profiling of the 316 plasma samples was performed using neurology panel of OLINK’s Proximity Extension Assay (ProSeek, OLINK AB, Uppsala, Sweden), which includes 91 proteins involved in various pathways including axon development, axon guidance, cell adhesion, cell death, cell differentiation, cell growth, cellular metabolic process, immune response, MAPK cascade, neurogenesis, proteolysis, signal transduction and synapse assembly (https://www.olink.com/products/neurology/). This method uses affinity-based assay, in which a pair of oligonucleotide-labeled antibody probes bind to a target protein. Proximity-dependent DNA polymerization event forms a polymerase chain reaction (PCR) target sequence between two probes bound in close proximity. The generated PCR target sequence is detected and quantified using real-time PCR method. The resultant protein abundance is provided as NPX (Normalized Protein Expression), which is an arbitrary unit on log2 scale. Lower limit of detection is estimated based on negative controls inserted in each run and measurements below this limited were treated as missing. None of the detected markers in our dataset reach missingness more than 10 percent. Protein markers with missing values less than 10% were imputed with the lowest detected limit for further analysis. More detailed information about detection limits, assay performance and validation methods are available from the service provider (www.olink.com)73.
APOE genotyping
In the RS APOE genotyping was performed using Polymerase chain reaction (PCR) and amplified PCR product was digested with HhaI enzyme. Restriction fragments of enzyme products were visualized by silver staining after getting them separated with precast ExcelGel gels (Pharmacia Biotech, Uppsala, Sweden). Genotype results were examined by three independent persons. In the case of non-agreement APOE genotype was repeated74,75.
Genotyping and imputations
In the RS participant’s blood was collected during baseline and follow-up visit. DNA genotyping was performed for all the participants with proper DNA quality with the 550 K, 550 K duo, or 610 K Illumina arrays. In genotyping quality control, genetic variants exclusion criteria include, call rate <95%, Hardy-Weinberg equilibrium P < 1.0 × 10−6 and Minor Allele Frequency (MAF) < 1%. Sample exclusion criteria include excess autosomal heterozygosity (0.336), call rate <97.5%, duplicate or family relationships and ethnic outliers identified by the identity-by-state clustering analysis (having identity-by-state probability <97% or>3 standard deviation from population mean)76. Further, genetic variants were imputed with the Haplotype Reference Consortium (HRC) reference panel (version 1.0)77, using the Michigan imputation server78. The server uses SHAPEIT2 (v2.r790)79 to phase the genotype data and performs imputation with Minimac 3 software80. Genotyping information was available for 281 among 316 participants included in the current study.
BioFINDER study
In the current study, replication analysis was performed in 671 participants (AD patients = 186, Controls = 485) of the BioFINDER (Biomarkers For Identifying Neurodegenerative Disorders Early and Reliably) study. Characteristics of the BioFINDER study participants included in the replication analysis are provided in Supplementary Table 9. The BioFINDER study includes participants from southern Sweden recruited between 2009 and 2014 (www.biofinder.se). The study participants were assessed by experienced physicians including the neurological, psychiatric and cognitive assessments81. The NINCDS-ADRDA criteria were used to classify Alzheimer’s disease dementia patients for probable Alzheimer’s disease patients. All dementia due to Alzheimer’s disease patients had pathological CSF Aβ42/Aβ40 ratio of <0.1. The inclusion criteria for the cognitively normal elderly participants included (i) aged 60–80 years, (ii) MMSE scores ranging between 28–30 at their baseline screening visit, (iii) no cognitive impairment symptoms assessed by a physician, and (iv) not fulfilling the criteria for mild cognitive impairment or dementia. Exclusion criteria included (i) refused lumbar puncture, (ii) significant neurological or psychiatric disease, (iii) current alcohol or substance misuse, or (iv) systematic illness preventing them from participating in the study25,81. Written Informed consents were collected from each study participant and the study has been approved by the Regional Ethics Committee in Lund, Sweden.
Protein profiling
During the baseline visit of the BioFINDER study, plasma and lumbar CSF samples were collected from non-fasting participants. Standardized protocol was followed to analyze the plasma and CSF samples. All samples were centrifuged at 2000 g (+4 °C for 10 min), and aliquoted into 1 ml polypropylene tubes (Sarstedt AG & Co., Nümbrecht, Germany), and stored at −80 °C. Before the proteomics profiling, plasma and CSF samples underwent one cycle of freeze-thaw, and further aliquoted into 200 L Lobind tubes (Eppendorf Nordic A/S, Denmark). Protein concentrations were quantified using the ProSeek multiplex immunoassay, developed by Olink Proteomics (Uppsala, Sweden)25.
Amsterdam dementia cohort (ADC)
In the validation analysis of most interesting proteins, we used 441 participants from the ADC cohort whose CSF samples were already profiled for neurology related proteins using the OLINK’s Proximity Extension Assay (ProSeek, OLINK AB, Uppsala, Sweden). Information about characteristics of patients included in current analysis as a part of the validation dataset is listed in Supplementary Table 10.
The ADC is a prospective memory-clinic cohort that was established in September 2000 at the Alzheimer Center Amsterdam of Amsterdam UMC. The cohort has included 6000 individuals until September 201782,83. All participants underwent standardized cognitive screening including neurological and cognitive examination, blood sampling, a lumbar puncture to collect CSF and brain magnetic resonance imaging. All CSF samples were stored in agreement with the JPND-BIOMARKAPD guidelines84. All subjects provided written informed consent for use of biomaterial and clinical data for research and the study was approved by the local medical ethical review board. All methods were performed in accordance with the relevant guidelines and regulations. A sample of 441 participants selected for our validation analysis consists of 242 AD and 199 cognitively normal controls who were presented at the memory clinic with subjective cognitive decline (i.e., Criteria for mild cognitive impairment and dementia not fulfilled)). As additional inclusion criteria, controls were required to have normal AD CSF biomarkers profile: low CSF β-amyloid 1–42 (Aβ42) and high p- or t-tau level (applying local laboratory cut-offs) and to remain cognitively stable for 2 years. All participants underwent standard neurological and cognitive assessments and the diagnosis was assigned according to consensus AD criteria83. Global Mini-Mental State Examination (MMSE) was used to examine global cognition. The levels of CSF AD-related biomarkers (Aβ42, total and phosphorylated tau [t-Tau and p-Tau181]) were analyzed at Amsterdam UMC as part of the routine diagnostic procedure using commercially available kits (Innotest Aβ(1-42), total Tau, phospho-Tau(181 P); Fujirebio, Ghent, Belgium)10,24.
Statistical analysis
Plasma protein association with AD
To identify AD-associated proteins, plasma levels of 91 proteins were compared between incident AD cases and non-demented controls using logistic regression, adjusted for age and sex in the first model. In the second model, we additionally adjusted for body mass index (BMI), smoking, educational status and medication use (lipid-lowering medications, antihypertensive and anti-inflammatory medication). To identify APOE specific associations of proteins with AD, we performed stratified association analysis based on APOE genotype carrier status. All participants were divided into APOE4 stratum (APOE 44/34/24), APOE3 stratum (APOE 33) and APOE2 stratum (APOE 22/23). Participants with APOE 24 genotypes were pooled within the APOE4 stratum because an earlier study has demonstrated that the risk profiles of APOE 24 genotype to AD and dementia is similar to those with APOE 34 genotype carriers5. The association results were corrected for multiple testing using false discovery rate (FDR) by Benjamini and Hochberg method separately for the overall analysis, and in each APOE stratum85 and association tests with FDR < 0.05 were considered significant. All analyses were performed using R software (https://www.r-project.org).
Sensitivity analyses
Moreover, we performed sensitivity analyses. In the first sensitivity analysis, we repeated the overall and APOE stratified regression analysis (Model 1: age and sex) additionally adjusting for the follow-up time (the time between blood collection and onset of AD or last follow-up for controls). In the second sensitivity analysis, to assess the differential bias due to missingness, we performed the overall and APOE stratified association analysis in the non-imputed proteomics data adjusting for age and sex. We also tested the interaction of APOE genotype (ε4 carriers and non-carriers) and proteins levels using logistic regression model adjusting for age and sex.
Additional analysis of proteins showing association with AD
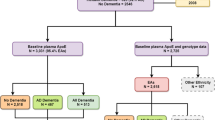
A detailed flowchart of the analysis is provided in Fig. 4 about the discovery, replication and validation analysis. Proteins that appeared significantly altered in overall or APOE stratified analysis were further tested for association with APOE genotypes; second GWAS was performed to identify pQTLs, regulating the levels of protein in blood.

Flowchart of the analyses. Rotterdam Study was used as discovery cohort in plasma-based proteomics analysis. Altered proteins in plasma analysis were replicated in the BioFINDER study and further validated in Amsterdam Dementia Cohort participants. Abbreviations; AD: Alzheimer’s disease; APOE: apolipoprotein E; GWAS: genome-wide association study; CSF: cerebrospinal fluid.
Replication analysis
Replication analysis of two proteins was performed in an independent BioFINDER study. We performed association of plasma levels of proteins with AD versus controls (AD cases = 186, controls = 485) in the overall sample and stratified by APOE genotype: APOE4 stratum (APOE 44/34), APOE33 stratum (APOE 33) and APOE2 stratum (APOE 22/23). We used logistic regression analysis adjusted for age, sex and date of sample collection.
Validation analysis: Association of CSF protein levels with Aβ-42, p-tau, and t-tau
In the validation analysis of specific proteins in an independent ADC cohort (N = 441), we performed association of CSF protein levels with AD versus control group and with Aβ-42, p-tau and t-tau levels in CSF. All the validation analyses were performed in the overall sample and stratified by APOE genotype: APOE4 stratum (APOE 44/34), APOE33 stratum (APOE 33) and APOE2 stratum (APOE 22/23). We used linear regression analysis adjusted for age and sex to evaluate the association of proteins measured in CSF with AD brain pathology biomarkers in the overall sample and stratified by clinical diagnosis (AD and controls).
Association of plasma protein levels with APOE genotype
To further evaluate the association of proteins with APOE genotypes, we compared protein levels, among APOE genotype groups (APOE 44/34/24 = 1 versus APOE 33 = 0, APOE 44/34 = 1 versus APOE 22/23 = 0 and APOE 22/23 = 1 versus APOE 33 = 0) in the overall study sample, in AD patients, and in control groups separately. Linear regression analysis was performed using protein levels as outcome and APOE status as predictor, adjusted for age and sex.
Genome-wide association study
Further, we performed the genome-wide association study (GWAS) to identify protein quantitative trait loci (pQTLs) for candidate proteins. We regressed out protein levels against age, sex and principal components to calculate residuals. To normalize the calculated residuals we applied Rank-inverse transformation on residuals. Principal components derived from genotypes were used in the association analysis to adjust for population stratification. GWAS of rank-inverse normalized residuals was performed using score test option in RVTEST software86. Variants with low imputation quality R-squared <0.3 and minor allele count less than five were excluded from the results. Manhattan and quantile-quantile (Q-Q) plots for GWAS results were generated with web-based utility Functional mapping and annotation of genetic associations (FUMA)87 and regional association plots using LocusZoom (http://locuszoom.org). pQTLs with a P-value < 2.5 × 10−8 (5 × 10−8/2 tested proteins) were considered genome-wide significant. To check the overlap of identified pQTL with expression quantitative loci (eQTLs) we used GTEx data base88.
Data availability
Current study used data from RS and ADC, where sharing of participants data is not allowed publicly due to legal and ethical permissions. Informed consents collected for both studies do not allow to share individual participants data in public repository. Data access can be made available for interested researchers upon request to corresponding author Cornelia M. van Duijn (Cornelia.vanDuijn@ndph.ox.ac.uk).
References
Au, R., Piers, R. J. & Lancashire, L. Back to the future: Alzheimer’s disease heterogeneity revisited. Alzheimers Dement (Amst) 1, 368–370 (2015).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–3 (1993).
Corder, E. H. et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7, 180–4 (1994).
Seshadri, S., Drachman, D. A. & Lippa, C. F. Apolipoprotein E epsilon 4 allele and the lifetime risk of Alzheimer’s disease. What physicians know, and what they should know. Arch Neurol 52, 1074–9 (1995).
van der Lee, S. J. et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol 17, 434–444 (2018).
Fernandez, C. G., Hamby, M. E., McReynolds, M. L. & Ray, W. J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Frontiers in aging neuroscience 11, 14 (2019).
Sunderland, T. et al. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry 56, 670–6 (2004).
de la Torre, J. C. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 3, 184–90 (2004).
Larsson, S. C. et al. Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ 359, j5375 (2017).
Henstridge, C.M., Hyman, B.T. & Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nature Reviews Neuroscience 1 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430 (2019).
Colonna, M. TREMs in the immune system and beyond. Nat Rev Immunol 3, 445–53 (2003).
Zlokovic, B. V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature Reviews Neuroscience 12, 723 (2011).
Katusic, Z. S. & Austin, S. A. Endothelial nitric oxide: protector of a healthy mind. European heart journal 35, 888–894 (2013).
Koizumi, K., Wang, G. & Park, L. Endothelial dysfunction and amyloid-β-induced neurovascular alterations. Cellular and molecular neurobiology 36, 155–165 (2016).
Han, B. H. et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-β peptide, partial restoration via γ-secretase inhibition. Journal of Neuroscience 28, 13542–13550 (2008).
Tong, X.-K., Lecrux, C. & Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. Journal of neuroscience 32, 4705–4715 (2012).
Park, L. et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Abeta Peptides. Circ Res 121, 258–269 (2017).
Hye, A. et al. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 129, 3042–50 (2006).
Ray, S. et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nature medicine 13, 1359 (2007).
Soares, H. D. et al. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Archives of neurology 69, 1310–1317 (2012).
Ringman, J. M. et al. Plasma signaling proteins in persons at genetic risk for Alzheimer disease: influence of APOE genotype. Archives of neurology 69, 757–764 (2012).
Hu, W. T. et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 79, 897–905 (2012).
Llano, D. A., Devanarayan, V. & Simon, A. J., Alzheimer’s Disease Neuroimaging, I. Evaluation of plasma proteomic data for Alzheimer disease state classification and for the prediction of progression from mild cognitive impairment to Alzheimer disease. Alzheimer Disease & Associated Disorders 27, 233–243 (2013).
Whelan, C. D. et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta neuropathologica communications 7, 1–14 (2019).
Blankenburg, M., Haberland, L., Elvers, H. D., Tannert, C. & Jandrig, B. High-Throughput Omics Technologies: Potential Tools for the Investigation of Influences of EMF on Biological Systems. Curr Genomics 10, 86–92 (2009).
Bell, R. D. et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–6 (2012).
Phizicky, E., Bastiaens, P. I., Zhu, H., Snyder, M. & Fields, S. Protein analysis on a proteomic scale. Nature 422, 208–15 (2003).
Emilsson, V. et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 361, 769–773 (2018).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. Nature Precedings, 1-1 (2010).
Zhao, B. et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nature genetics 51, 1637–1644 (2019).
Harper, L. et al. Patterns of atrophy in pathologically confirmed dementias: a voxelwise analysis. Journal of Neurology, Neurosurgery & Psychiatry 88, 908–916 (2017).
Sutphen, C. L. et al. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimers Dement 14, 869–879 (2018).
Hesse, C. et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neuroscience letters 297, 187–190 (2001).
Lv, J. et al. Quantitative proteomics reveals that PEA15 regulates astroglial Abeta phagocytosis in an Alzheimer’s disease mouse model. J Proteomics 110, 45–58 (2014).
Ye, L. et al. Abeta seeding potency peaks in the early stages of cerebral beta-amyloidosis. EMBO Rep 18, 1536–1544 (2017).
Asada‐Utsugi, M. et al. N‐cadherin enhances APP dimerization at the extracellular domain and modulates Aβ production. Journal of neurochemistry 119, 354–363 (2011).
Henriques, A. G. et al. Altered protein phosphorylation as a resource for potential AD biomarkers. Sci Rep 6, 30319 (2016).
Gul, I. S., Hulpiau, P., Saeys, Y. & van Roy, F. Evolution and diversity of cadherins and catenins. Exp Cell Res 358, 3–9 (2017).
Fannon, A. M. & Colman, D. R. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17, 423–34 (1996).
Obst-Pernberg, K. & Redies, C. Cadherins and synaptic specificity. J Neurosci Res 58, 130–8 (1999).
Yamagata, M., Duan, X. & Sanes, J. R. Cadherins Interact With Synaptic Organizers to Promote Synaptic Differentiation. Front Mol Neurosci 11, 142 (2018).
Arikkath, J. & Reichardt, L. F. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 31, 487–94 (2008).
Sancisi, V. et al. Cadherin 6 is a new RUNX2 target in TGF-beta signalling pathway. PLoS One 8, e75489 (2013).
Clay, M. R. & Halloran, M. C. Cadherin 6 promotes neural crest cell detachment via F-actin regulation and influences active Rho distribution during epithelial-to-mesenchymal transition. Development 141, 2506–15 (2014).
Montagne, A. et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015).
Inoue, T., Tanaka, T., Suzuki, S. C. & Takeichi, M. Cadherin-6 in the developing mouse brain: expression along restricted connection systems and synaptic localization suggest a potential role in neuronal circuitry. Dev Dyn 211, 338–51 (1998).
Kuhn, P.H. et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 5 (2016).
Malinverno, M. et al. Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. Journal of Neuroscience 30, 16343–16355 (2010).
Marambaud, P. et al. A CBP binding transcriptional repressor produced by the PS1/ϵ-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114, 635–645 (2003).
Andreyeva, A. et al. C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-beta. Brain 135, 2140–54 (2012).
Hu, N. W. et al. Extracellular Forms of Abeta and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep 23, 1932–1938 (2018).
Togashi, H. et al. Cadherin regulates dendritic spine morphogenesis. Neuron 35, 77–89 (2002).
Chen, F. et al. Role for glyoxalase I in Alzheimer’s disease. Proc Natl Acad Sci USA 101, 7687–92 (2004).
Thornalley, P. J. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J 269, 1–11 (1990).
Xu, Y. & Chen, X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J Biol Chem 281, 26702–13 (2006).
Desai, K. M. et al. Oxidative stress and aging: is methylglyoxal the hidden enemy? Can J Physiol Pharmacol 88, 273–84 (2010).
Takeuchi, M. et al. Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr Alzheimer Res 1, 39–46 (2004).
Ko, S. Y., Lin, Y. P., Lin, Y. S. & Chang, S. S. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic Biol Med 49, 474–80 (2010).
Wang, J. Z., Wang, Z. H. & Tian, Q. Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci Bull 30, 359–66 (2014).
More, S. S., Vartak, A. P. & Vince, R. Restoration of glyoxalase enzyme activity precludes cognitive dysfunction in a mouse model of Alzheimer’s disease. ACS Chem Neurosci 4, 330–8 (2013).
Kuhla, B. et al. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol Aging 28, 29–41 (2007).
Shea, T. B., Rogers, E., Ashline, D., Ortiz, D. & Sheu, M. S. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic Biol Med 33, 1115–20 (2002).
Baldeiras, I. et al. Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J Alzheimers Dis 15, 117–28 (2008).
Guidi, I. et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 27, 262–9 (2006).
Hofman, A. et al. The Rotterdam Study: 2016 objectives and design update. Eur J Epidemiol 30, 661–708 (2015).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12, 189–98 (1975).
Copeland, J. R. et al. A semi-structured clinical interview for the assessment of diagnosis and mental state in the elderly: the Geriatric Mental State Schedule. I. Development and reliability. Psychol Med 6, 439–49 (1976).
Roth, M. et al. CAMDEX. A standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry 149, 698–709 (1986).
McKhann, G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–44 (1984).
Ott, A., Breteler, M. M., van Harskamp, F., Stijnen, T. & Hofman, A. Incidence and risk of dementia. The Rotterdam Study. Am J Epidemiol 147, 574–80 (1998).
Brann, E. et al. Inflammatory markers in late pregnancy in association with postpartum depression-A nested case-control study. Psychoneuroendocrinology 79, 146–159 (2017).
Slooter, A. J. et al. Apolipoprotein E genotype and progression of Alzheimer’s disease: the Rotterdam Study. J Neurol 246, 304–8 (1999).
Slooter, A. J. et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol 55, 964–8 (1998).
Roshchupkin, G. V. et al. Fine-mapping the effects of Alzheimer’s disease risk loci on brain morphology. Neurobiol Aging 48, 204–211 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–83 (2016).
Das, S. et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–7 (2016).
Delaneau, O. & Marchini, J. Genomes Project, C. & Genomes Project, C. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun 5, 3934 (2014).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44, 955–9 (2012).
Palmqvist, S. et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42: a cross-validation study against amyloid positron emission tomography. JAMA Neurol 71, 1282–9 (2014).
van der Flier, W. M. & Scheltens, P. Amsterdam Dementia Cohort: Performing Research to Optimize Care. J Alzheimers Dis 62, 1091–1111 (2018).
van der Flier, W. M. et al. Optimizing patient care and research: the Amsterdam Dementia Cohort. Journal of Alzheimer’s disease 41, 313–327 (2014).
Reijs, B. L. R. et al. The Central Biobank and Virtual Biobank of BIOMARKAPD: a resource for studies on neurodegenerative diseases. Frontiers in neurology 6, 216 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society. Series B (Methodological), 289–300 (1995).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–6 (2016).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–5 (2013).
Acknowledgements
This study was funded by the ADAPTED: Alzheimer’s Disease Apolipoprotein Pathology for Treatment Elucidation and Development consortium which has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under Grant Agreement No 115975. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and the European Federation of Pharmaceutical Industries and Associations. This study was also funded by the PERADES Program (Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s disease, using multiple powerful cohorts, focused Epigenetics and Stem cell metabolomics), Project number 733051021 and Memorabel supported by ZonMW (Project number733050814). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. The generation and management of GWAS genotype data for the Rotterdam Study (RS-I, RS-II, RS-III) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. The GWAS datasets are supported by the Netherlands Organization of Scientific Research NWO Investments (Project number 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project number 050-060-810. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera and Marjolein Peters, MSc, and Carolina Medina-Gomez, MSc, for their help in creating the GWAS database, and Karol Estrada, PhD, Yurii Aulchenko, PhD, and Carolina Medina-Gomez, MSc, for the creation and analysis of imputed data. The Amsterdam Dementia Cohort: The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. The clinical database structure was developed with funding from Stichting Dioraphte. CSF analysis were supported by the Weston Brain Institute and Stichting Alzheimer Nederland.
Author information
Authors and Affiliations
Contributions
Study concept and design: S.A., A.D., N.A., C.M.D. Draft of the manuscript: S.A. Performed statistical analysis: S.A., M.C.M. A.M., Interpretation of data: S.A., M.C.M., O.H., A.D., R.A., M.E.S., N.G., A.C., M.B., A.R., T.H., E.S., N.M., P.S., W.F., M.A.I., A.M., C.E.T., N.A., C.M.D. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.A., M.C.M., A.D., R.A., M.E.S., A.R., T.H., E.S., N.M., P.S., M.A.I., N.A., C.M.D. declare no competing interests. O.H. has acquired research support (for the institution) from Roche, Pfizer, GE Healthcare, Biogen, AVID Radiopharmaceuticals and Euroimmun. In the past 2 years, O.H. has received consultancy/speaker fees (paid to the institution) from Biogen and Roche. Dr. Nikolaos Giagtzoglou was an employee at Biogen Idec, USA. Dr. Alfredo Cabrera-Socorro is an employee of Janssen Pharmaceutical NV, Turnhoutseweg 30, 2340 Beerse, Belgium. Dr. Margot H.M. Bakker is a full time employee of AbbVie GmbH & Co KG and owns AbbVie stock. AbbVie contributed to the study design, research, and interpretation of data, reviewing, and approving the publication. Prof. dr. Teunissen has functioned in advisory boards of Roche, received non-financial support in the form of research consumables from ADxNeurosciences and Euroimmun, performed contract research or received grants from Probiodrug, Biogen, Esai, Toyama, Janssen prevention center, Boehringer, AxonNeurosciences, EIP farma, PeopleBio, Roche.Research programs of Prof. dr. Wiesje van der Flier have been funded by ZonMW, NWO, EU-FP7, Alzheimer Nederland, CardioVascular Onderzoek Nederland, stichting Dioraphte, Gieskes-Strijbis fonds, Pasman stichting, Biogen MA Inc, Boehringer Ingelheim, Piramal Neuroimaging, Roche BV, Janssen Stellar, Combinostics. WF holds the Pasman chair. WF has performed contract research for Biogen MA Inc and Boehringer Ingelheim. WF has been an invited speaker at Boehringer Ingelheim and Biogen MA Inc. All funding is paid to her institution.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmad, S., Milan, M.d.C., Hansson, O. et al. CDH6 and HAGH protein levels in plasma associate with Alzheimer’s disease in APOE ε4 carriers. Sci Rep 10, 8233 (2020). https://doi.org/10.1038/s41598-020-65038-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65038-5
This article is cited by
-
Plasma levels of neurology-related proteins are associated with cognitive performance in an older population with overweight/obesity and metabolic syndrome
GeroScience (2023)
-
ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies
Molecular Neurodegeneration (2022)
-
Cross-tissue analysis of blood and brain epigenome-wide association studies in Alzheimer’s disease
Nature Communications (2022)
-
Investigation of 91 proteins implicated in neurobiological processes identifies multiple candidate plasma biomarkers of stroke outcome
Scientific Reports (2022)
-
Investigation of Mitochondrial Related Variants in a Cerebral Small Vessel Disease Cohort
Molecular Neurobiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
