
Abstract
To investigate cost affordable and robust HER and OER catalysts with significant low overpotentials, we have successfully embedded FeCoSe2 spheres on smooth surfaces of graphitic carbon nitride that demonstrated high stability and electrocatalytic activity for H2 production. We systematically analyzed the composition and morphology of FexCo1−xSe2/g-C3N4 and attributed the remarkable electrochemical performance of the catalyst to its unique structure. Fe0.2Co0.8Se2/g-C3N4 showed a superior HER activity, with quite low overpotential value (83 mV at −20 mA cm−2 in 0.5 M H2SO4) and a current density of −3.24, −7.84, −14.80, −30.12 mA cm−2 at 0 V (vs RHE) in Dulbecco’s Phosphate-Buffered Saline (DPBS), artificial sea water (ASW), 0.5 M H2SO4 and 1 M KOH, respectively. To the best of our knowledge, these are the highest reported current densities at this low potential value, showing intrinsic catalytic activity of the synthesized material. Also, the catalyst was found to deliver a high and stable current density of −1000 mA cm−2 at an overpotential of just 317 mV. Moreover, the synthesized catalyst delivered a constant current density of −30 mA cm−2 for 24 h without any noticeable change in potential at −0.2 V. These attributes confer our synthesized catalyst to be used for renewable fuel production and applications.
Similar content being viewed by others


Introduction
Significant increase in the fossil fuel consumption and associated environmental threats of greenhouse gases have urged the scientific community to explore a sustainable and everlasting source of energy1. The fuel cell technology is considered as an efficient, long-lasting and environmental friendly source of energy. Among various existing forms of fuel cells, the H2/O2 fuel cells are deemed as zero carbon discharging technology with only water as the combustion product2,3. To ensure higher cell efficiency, the supply of pure fuel is essential and water electrolysis is one of the most convenient ways of producing H2. In fact, water splitting has garnered enough spotlight because of the highly pure and plentiful supply of fuel produced during this process. Likewise, this strategy does not demand elevated temperatures and pressure reactors. The overall water electrolysis can be explained by the following two half-cell reactions: hydrogen evolution reaction (HER) at the cathode4,5 and oxygen evolution reaction (OER) at the anode6,7, investigated in acidic and alkaline conditions, respectively5,8.
The noble metals i.e., Platinum for HER9,10,11,12,13,14 and Iridium or Ruthenium for OER15,16,17,18 being expensive and scarce, must be substituted by earth abundant and cost effective non-noble metals19 or metal free catalysts such as carbon based electrocatalysts. This work is an effort to substitute the expensive noble metals with only a small amount of earth abundant and cost effective non-noble metal catalysts to achieve better HER activity than the commercial catalysts reported so far. Among the metal free substrates, Carbon Nitride (C3N4) is the best choice as it offers π-conjugated graphitic planner layers with good conductivity and large surface area20,21. The C3N4 is reported as one of the primitive synthetic polymeric materials that offers different ways to modify its reactivity without any change in composition22,23,24,25,26,27,28. The g-C3N4 is believed to possess N-bridged poly(tri-s-triazine) planar structure with high degree of condensation. The s-triazine framework makes it thermally (up to 600 °C) and chemically stable. Moreover, it is a semiconducting material of medium band gap value (2.7 eV) that finds extensive use as a photo(electro)catalyst29,30,31,32,33.
Recently, transition-metals sulfides, phosphides and selenides have been found to be efficient HER catalysts34,35. Likewise, various electrocatalysts including metal alloys36,37, metal nitrides38,39, metal phosphides40, metal carbides41, metal dichalcogenides42 and carbon-based catalysts have also attracted attention of researchers in the last few years43,44. The transition metal dichalcogenides (TMDs) have been found to be promising water splitting electrocatalysts and have thus gained significant interest in technological applications due to their unique electronic structure, different chemical compositions and diverse crystal symmetry. Several routes i.e. homogeneously dispersed electrocatalyst on conductive supports, chemical vapor and electron beam deposition have been developed to exploit the electrocatalytic properties of these materials45,46,47. However, the above-mentioned synthetic approaches are unfeasible for large scale production as control over the layers, chirality and influence of different substrates on its fabrication is still an area that is not well explored. The chemical doping is considered as an important structure-engineering technique to increase active sites and conductivity of these electrocatalysts48 which will be explored in this work.
Previously, Transition metal dichalcogenides (TMDs) such as NiS2, FeS2, and CoS2 displayed efficient overall water splitting performance49,50,51. Similarly, Iron (Fe) found in its natural form such as Nitrogenases and Hydrogenase (natural hydrogen evolution enzymatic catalysts) is the most promising material for HER. Hence, Iron doping is expected to increase the electrocatalytic performance of HER catalysts. Similarly, sulfur component of above-mentioned catalysts can be replaced with selenium. Sulfur and selenium are present in the same group and both exhibit similar properties, but selenides are expected to enhance HER performance of electrocatalysts due to low bond strength of Se–H (276 kJ/mol) than P–H (322 kJ/mol) and S–H (363 kJ/mol). Obviously, the bond strength has a great influence on the extent of adsorption/desorption of the intermediate states during catalysis. The weaker Se–H bond as compared to P–H and S–H is expected to facilitate the product desorption from catalytic sites which circumvents catalyst poisoning and thus stable and efficient performance is projected. Moreover, the Se sites can help to promote the delivery of oxygen molecules because of the localized negative charge of Se sites for OER. Likewise, there is a 3d–2p electronic repulsion amid metal and Se d-band centers32.
Herein, we have reported a facile method for synthesizing highly active and stable FeCoSe2 on graphitic carbon nitride substrate (FexCo1−xSe2/g-C3N4) as novel catalyst for overall water splitting. The catalytic activity of FexCo1−xSe2/g-C3N4 significantly improved with increased Fe percentages in the composite material. The Fe0.2Co0.8Se2/g-C3N4, has proven itself to be a promising electrocatalyst for HER and OER due to its high conductivity, unique structure and high surface area.
Experimental Section
Chemicals and materials
Iron (III) Nitrate (Fe(NO3)3·9 H2O), Cobalt (II) Chloride hexahydrate (CoCl2.6H2O), Urea (CO(NH2)2) and Selenium Powder (Se) were purchased from Sigma-Aldrich. Potassium hydroxide, Sulfuric acid and Iso-propanol were of analytical grade and were used as received without any further purification. Preparation of Indian standard artificial sea water (ASW) and Dulbecco’s Phosphate-Buffered Saline (DPBS) is detailed in Supplementary note 1.
Synthesis of graphitic carbon nitride
The graphitic carbon nitride was synthesized by using combustion method. The 10 g of urea powder was placed in a covered silica crucible and calcined at 500 °C for 4 h at a heating rate of 2 °C/min. The g-C3N4 powder was collected and exfoliated in excess of distilled water for 5 h, dried at 80 °C and stored for further use.
Synthesis of CoSe2 and FexCo1−xSe2 embedded in g-C3N4
To synthesize CoSe2 embedded in g-C3N4, a simple hydrothermal route was followed. Solution A was prepared by adding 0.8 mmol of cobalt source, 2 ml of 0.5 M aqueous solution of ethylene diamine tetra-acetic acid ligand, and 20 mg of g-C3N4. While solution B was prepared by dissolving 1.62 mmol of Se powder in 8 ml of 3.3 M NaOH. Both solutions were mixed by ultrasonication and the mixture was shifted to a Teflon autoclave and maintained at 180 °C for 18 h. While to produce FexCo1−xSe2/g-C3N4, cobalt source in the solution A was substituted by appropriate amount of iron nitrate and cobalt chloride solutions. In each case, black product was collected and washed with deionized water followed by air drying for characterizations and application of the as-synthesized product. Total quantity of metal sources (Fe + Co) was kept very low and constant (0.8 mmol). For Fe0.2Co0.8Se2/g-C3N4, the molar ratio of Fe, Co, and Se was maintained at 1: 4: 10. Finally, feeding ratios of metals were further confirmed by average results of EDX (Energy Dispersive X-ray Spectroscopy).
Fabrication of electrodes
3 mg of each sample and 20 μL of 5 wt.% Nafion were dispersed in 80 μL of iso-propanol to make a slurry. Then 5 μL of slurry was drop casted on glassy carbon electrode (GCE) of geometric surface area of 0.0707 cm2 and dried in air. These fabricated electrodes (with mass loading of 2.12 mg cm−2) were used as cathodes and anodes for respective electrochemical measurements of HER and OER respectively.
Characterization
To identify the crystalline structure of products, X-ray diffraction (XRD) was performed on Rigaku D/max 2500 with a Cu Kα1 radiation source (λ = 1.54056 Å). Morphologies of prepared samples were identified at Hitachi S-4800 Scanning Electron Microscope (SEM) at an accelerating voltage of 30 KV. Energy dispersive X-ray spectra (EDX) were observed on an Oxford Material Analysis equipped on SEM and Transmission electron microscopy (TEM). While, Fourier Transform Infra-Red (FTIR) spectroscopic measurements were carried on TENSOR-27 (Bruker) and differential scanning calorimetry/thermal gravimetric analysis (DSC/TGA) on TA SDT Q600.
Electrochemical measurements
Electrochemical studies were carried out on Autolab PGSTAT302N Metrohm, (running with NOVA 1.11 software) electrochemical workstation at room temperature. While, EIS measurement was made with a Gamry EIS 300 electrochemical impedance analyzer with frequencies ranging from 100 kHz to 0.1 Hz at a voltage amplitude of 10 mV. The electrocatalytic ability of all materials was studied in 0.5 M H2SO4, DPBS, ASW, and in 1 M KOH for HER using a three-electrode configuration at a scan rate of 20 mV/s. The FexCo1−xSe2/g-C3N4 powder on GCE was used as working, Platinum (Pt) wire as counter and Ag/AgCl (sat. KCl) as reference electrode. All potentials in this work are reported versus Reversible Hydrogen electrode (RHE) according to the Eq. 1.
Results and Discussion
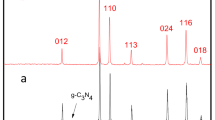
The Fe/Co ratio was controlled, and a series of samples were prepared. The effect of Fe/Co ratio on the composition and the crystalline phase was investigated by X-ray diffraction (XRD) technique and is shown in Fig. 1. The diffraction peaks at 30.6, 34.4, 35.7, 47.7 and 50.3 match well and can be indexed to the planes of powder diffraction standard database of CoSe2 (JCPDS # 53- 0449) and FeSe2 (JCPDS # 21- 0432), respectively. It is noteworthy that the diffraction peaks of g-C3N4 are not indexable in the Fe0.2Co0.8Se2/g-C3N4 partly due to their comparatively low diffraction intensity, as carbon nitride synthesized from urea shows low diffraction intensity, and partly to relatively small amount of carbon nitride present in the sample. In addition, with increase in Fe amount, the intensity of XRD signals decreased which implied low crystallinity of the Fe0.2Co0.8Se2/g-C3N4 material in comparison to the pristine ones. Furthermore, the final product (Fe0.2Co0.8Se2/g-C3N4) showed diffraction peaks corresponding to both CoSe2, and FeSe2, which indicates the coexistence of the two orthorhombic phases52. It is pertinent to mention here that owing to the small differences in ionic radii of iron and cobalt, the strongest diffraction peaks of FeCoSe2, FeSe2 and CoSe2 displayed very little interlayer spacing difference (±0.01 Å), which provided the basis for facile doping of Fe into the CoSe2 structures. Additionally, Fourier transform infrared spectroscopy (FTIR) was conducted to reveal the bonding states of FexCo1−xSe2/g-C3N4 samples (Fig. S1).

Powder X-ray diffraction pattern of g-C3N4, FeSe2/g-C3N4, CoSe2/g-C3N4, and Fe0.2Co0.8Se2/g-C3N4.
The morphologies of all samples were studied by SEM (Fig. 2). The Fe free material (CoSe2/g-C3N4) exhibited a not so well-defined nodular morphology (Figs. 2a and S2a), however, after the addition of Fe, Fe0.2Co0.8Se2/g-C3N4 showed a similar morphology to CoSe2/g-C3N4 but with the appearance of rather uniform sphere like structures embedded on to the CoSe2 smooth surface (Fig. 2b). In addition, Fig. 2b depicts a porous framework of Fe0.2Co0.8Se2/g-C3N4 electrocatalyst, which is favorable for catalytic applications. Whereas, in the case of FeSe2/g-C3N4, due to increase in Fe content in FexCo1−xSe2/g-C3N4, a completely different rod type morphology was seen (Figs. 2c and S3). This appearance of distinct morphologies of two metals (Co, Fe) with Se is as result of their different directional growths under hydrothermal conditions. Furthermore, elemental mapping of synthesized material confirmed a fairly uniform distribution of constituent elements (Fig. S2c and Table S1). The TEM image of Fe0.2Co0.8Se2/g-C3N4 (Fig. 2d) shows crystalline nature of the material where fringe analysis reveal that the lattice distances remain same throughout the sample. The spherical shape of Fe0.2Co0.8Se2/g-C3N4, corresponds to the results obtained from SEM.

SEM micrographs of (a) CoSe2/g-C3N4 (b) Fe0.2Co0.8Se2/g-C3N4 and (c) FeSe2/g-C3N4. d) TEM micrograph and elemental mapping images of Fe0.2Co0.8Se2/g-C3N4.
The as-obtained compounds were further investigated by the simultaneous application of thermogravimetry and differential scanning calorimetry (TG-DSC) to study the effect of temperature, phase transition phenomenon and the associated endothermic or exothermic effects (Figs. S4 and S5). Due to the presence of g-C3N4 substrate, the compounds are found to be stable for a wide temperature range. Among these samples, g-C3N4 was found to be the most stable one. While comparing Fe0.2Co0.8Se2 and Fe0.2Co0.8Se2/g-C3N4, it is clear that g-C3N4 has a significant effect on the thermal stability of prepared samples. DSC studies of g-C3N4 reveal that it undergoes an exothermic phase transition in the temperature range of 675–725 °C. Whereas, Fe0.2Co0.8Se2/g-C3N4 first shows an endothermic transition at 425.6 °C and then an exothermic phase transition at 580 °C. All the samples were further investigated for electrocatalytic overall water splitting performance.
Electrocatalytic water splitting performance
The HER performance of the prepared samples FexCo1−xSe2/g-C3N4 (x = 0, 0.2, 0.4, 0.6, 0.8 and 1) were investigated in 0.5 M H2SO4, DPBS, ASW and 1 M KOH. The best electrocatalytic performance was observed for Fe0.2Co0.8Se2/g-C3N4. Fig. S6a compares the HER activity of the materials of all composition without iR compensation in 0.5 M H2SO4. The Fe0.2Co0.8Se2/g-C3N4 showed best electrocatalytic activity with highest current density values of −14.8, −20, −100 mA cm−2 at potential values of 0, 83, and 209 m V respectively in 0.5 M H2SO4 solution. As HER is facilitated in acidic medium, the current density of −14.8 mA cm−2 is likely due to the formation of HSeO3− in strong acidic conditions and its subsequent reduction to H2Se and or HSe− 53. To further explore the role of acid, HER was also carried out in DPBS solution where FeCoSe2 exhibited a low current density value of −3.24 mA cm−2, confirming the role of acid in the formation and reduction of HSeO3− (Fig. S6b)5. Basu et al. have reported similar results by employing CoSe2/C3N4 catalyst which displayed a photocurrent density of −4.89 mA cm−2 54. The Fig. 3a presents the comparison of Fe0.2Co0.8Se2/g-C3N4 with CoSe2/g-C3N4, g-C3N4 substrate, and Pt after iR compensation. The poor electrocatalytic HER activity of g-C3N4 suggests that the major role for higher HER activity is not coming from the substrate itself but it helps in the dispersion of FexCo1−xSe2 onto the smooth surface of g-C3N4.

Hydrogen evolution activity of (a) bare GCE, g-C3N4, CoSe2/g-C3N4 and FexCo1−xSe2/g-C3N4 and Pt after iR correction in 0.5 M H2SO4 (b) Nyquist plots for AC impedance measurement of proton conductivity taken under HER working conditions in frequency range of 1000 kHz to 0.1 Hz at 10 mV AC voltage at open-circuit potential (c) Nyquist plot of Fe0.2Co0.8Se2/g-C3N4 at different biases and (d) Stability test of Fe0.2Co0.8Se2/g-C3N4 for 24 h in 0.5 M H2SO4 (pH 0.3).
While CoSe2/g-C3N4 showed comparatively good performance with overpotentials of 193 mV, 450 mV at −20 and −100 mA cm−2 respectively than g-C3N4, the introduction of iron further enhanced the electrocatalytic activity. Therefore, by adjusting the feed ratio, highest and stable HER performance for Fe0.2Co0.8Se2/g-C3N4 with a current density as high as −1000 mA cm−2 was achieved without significant interference from the produced H2 bubbles. The negligible interference from the H2 bubbles ensures that the modified electrode favors the fast H2 desorption from the electrode surface. The fast desorption soon after production is crucial for industrial applications of electrocatalysts (Supplementary video). However, with further increment of the Fe content after optimal ratio (20 wt%), the HER activity sharply decreased due to its probable changes in electronic configurations. In the same way, the electrocatalytic ability of the Fe0.2Co0.8Se2/g-C3N4 was also assessed in DPBS, ASW, 0.1 M H2SO4 and 1 M KOH aqueous solutions as shown in Fig. S6b. It is important to note that the relatively high value of −30.12 mA cm−2 in 1 M KOH is probably due to a reduction contribution of SeO32− species that are generated at the catalyst-electrolyte interface during testing at high pH conditions. The overall performance of Fe0.2Co0.8Se2/g-C3N4 is comparable to the benchmark HER electrocatalysts (comparison given in Table S2)10.
To further investigate the charge transfer behavior at electrode electrolyte interface, the electrochemical impedance spectroscopy (EIS) of CoSe2 and Fe0.2Co0.8Se2 embedded in g-C3N4 was performed. It can be seen from the Nyquist plots in Fig. 3b, that Fe0.2Co0.8Se2/g-C3N4 had smaller charge transfer resistance (RCT) in comparison with CoSe2/g-C3N4, as the arc diameter of FeCoSe2 is smaller than CoSe2 dispersed on substrate. Thus, the EIS of Fe0.2Co0.8Se2/g-C3N4 was further performed at different applied potentials. It can be seen from Fig. 3c that the RCT values decrease with increase in applied potential value. Therefore, further increase in applied potential i.e. −0.295 V decreases the RCT and hence enhances the HER activity. Moreover, determining the electrocatalytic active surface area (ECSA) is an important parameter since it shows the intrinsic performance of electrocatalysts. ECSA was calculated by double-layer capacitance (Cdl) value (Supplementary note 3) at the electrolyte–electrode interface using cyclic voltammetry (CV given in Fig. S7). As shown in S8, CoSe2 increased the Cdl of g-C3N4 from 0.20 and 0.52 mF cm−2 to 0.85 and 0.56 mF cm−2, respectively at different potential values. After the addition of iron, Cdl values rose to 2.73 and 1.20 mF cm−2 for Fe0.2Co0.8Se2/g-C3N4. These results indicate that the introduction of Fe favorably modulates the ECSAs of FexCo1−xSe2/g-C3N4 samples (Table S3).
To assess the stability of Fe0.2Co0.8Se2/g-C3N4, chronopotentiometric response was recorded at constant current density of −30 mA cm−2 (Fig. 3d). The results show that the composite delivers almost an unchanged potential of −0.2 V at −30 mA cm−2 for 24 hours. After long term chronopotentiometric response, the samples were subjected to SEM, EDX and elemental mapping. The analysis (Figs. S9 and 10 and Table S1), confirms that after 24 h of testing the constituent elements in the sample and morphology of the synthesized material remained intact. Further, this elemental analysis of hybrid materials was used to find minimum Turn over frequency (TOFmin), (Supplementary note 2) where TOFs of all catalysts reveal that Fe0.2Co0.8Se2/g-C3N4 shows highest TOFmin (Fig. S6c). All of these results suggest that introduction of iron is of great importance in terms of electrochemical performance as it enhances the charge transfer ability of electrocatalysts and natural hydrogen evolution enzymatic catalysts.
The catalytic activity for oxygen evolution reaction (OER) was investigated in aqueous 1 M KOH solution. Fe0.2Co0.8Se2/g-C3N4 showed moderate overpotentials of 230 and 360 mV at 10 and 50 mA cm−2, respectively. The results have been compared with recently reported transition metal chalcogenides based electrocatalysts in Table S4 for OER. Moreover, the synthesized material can deliver high current density of over 500 mA cm−2 without any interference from the O2 bubbles (Fig. 4a). To acquire a direct estimation of intrinsic OER and HER kinetics of electrocatalysts, the Tafel slope was evaluated from the polarization curves. While selecting the region, we avoided very high potentials in order to circumvent oxygen and hydrogen bubble evolution that affects mass transport, and low potentials at which the redox transition for cobalt arises. In each case (i.e., in acidic and alkaline medium), the low Tafel slope of iron incorporated material indicated extremely competitive performance of synthesized material amongst TMDs (Figs. 4b and S11). The Tafel slope values further quantify the enhanced kinetics of charge transfer in the synthesized material.

(a) OER activity of Bare GCE, g-C3N4 and FexCo1−xSe2/g-C3N4 in 1 M KOH (b) corresponding Tafel slope of CoSe2/g-C3N4 and Fe0.2Co0.8Se2/g-C3N4 taken at wider potential range in 1 M KOH solution.
Conclusions
A low cost and a facile hydrothermal route was adopted for the preparation of FeCoSe2 hybrid uniformly supported on g-C3N4 that performed efficiently as a bifunctional electrocatalyst. Introduction of iron was found to be of vital importance as it led to increase in TOFmin and electrocatalytic ability of nanohybrid material. Due to optimized Fe incorporation, presence of Se atom and a good conducting substrate, the Fe0.2Co0.8Se2/g-C3N4 ensured high ECSAs and enhanced electrocatalytic performance in the context of low overpotential, small Tafel slope, high current density, high proton conductivity, and stability. In addition, the designed catalyst also performed good OER activity with moderate onset potential and high current density values. Due to facile synthesis technique and good electronic properties, g-C3N4 can offer a perfect platform for the conversion and modification of catalysts with varied structure and composition. In this work, g-C3N4 allowed the catalyst to work in wide pH ranges which is highly desirable for industrial applications. These peculiar qualities imply that Fe0.2Co0.8Se2/g-C3N4 hybrid electrocatalyst has the potential to replace benchmark electrocatalysts for clean fuel production in industrial applications.
References
Habas, S. E., Platt, H. A. S., van Hest, M. F. A. M. & Ginley, D. S. Low-cost inorganic solar cells: from ink to printed device. Chem. Rev. 110, 6571–6594 (2010).
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Wasmus, S. & Küver, A. Methanol oxidation and direct methanol fuel cells: A selective review. J. Electroanal. Chem. 461, 14–31 (1999).
Xiao, P., Chen, W. & Wang, X. A review of phosphide-based materials for electrocatalytic hydrogen evolution. Adv. Energy Mater 5, 1500985 (2015).
Shi, Y. & Zhang, B. Recent advances in transition metal phosphide nanomaterials: Synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev. 45, 1529–1541 (2016).
Casalongue, H.S., et al Direct observation of the oxygenated species during oxygen reduction on a platinum fuel cell cathode. Nat. Commun. 4 (2013).
Gao, M. R., Xu, Y. F., Jiang, J. & Yu, S. H. Nanostructured metal chalcogenides: Synthesis, modification, and applications in energy conversion and storage devices. Chem. Soc. Rev. 42, 2986–3017 (2013).
Lu, F., Zhou, M., Zhou, Y. & Zeng, X. First-row transition metal based catalysts for the oxygen evolution reaction under alkaline conditions: Basic principles and recent advances. Small 13(45), 1701931 (2017).
Zeng, Z., Tan, C., Huang, X., Bao, S. & Zhang, H. Growth of noble metal nanoparticles on single-layer TiS2 and TaS2 nanosheets for hydrogen evolution reaction. Energy Environ. Sci. 7, 797–803 (2014).
Anantharaj, S., Karthik, P. E., Subramanian, B. & Kundu, S. Pt nanoparticle anchored molecular self-assemblies of DNA: An extremely stable and efficient HER electrocatalyst with ultralow Pt content. ACS Catal 6, 4660–4672 (2016).
Wang, J. X., Zhang, Y., Capuano, C. B. & Ayers, K. E. Ultralow charge-transfer resistance with ultralow Pt loading for hydrogen evolution and oxidation using Ru@Pt core-shell nanocatalysts. Sci. Rep 5, 12220 (2015).
Wang, C., Dekrafft, K. E. & Lin, W. Pt nanoparticles@photoactive metal-organic frameworks: Efficient hydrogen evolution via synergistic photoexcitation and electron injection. J. Am. Chem. Soc. 134, 7211–7214 (2012).
Sun, C. L. et al. Ultrafine platinum nanoparticles uniformly dispersed on arrayed CNx nanotubes with high electrochemical activity. Chem. Mater. 17, 3749–3753 (2005).
Song, H. M., Anjum, D. H., Sougrat, R., Hedhili, M. N. & Khashab, N. M. Hollow Au@Pd and Au@Pt core-shell nanoparticles as electrocatalysts for ethanol oxidation reactions. J. Mater. Chem. 22, 25003–25010 (2012).
Burke, M. S., Enman, L. J., Batchellor, A. S., Zou, S. & Boettcher, S. W. Oxygen evolution reaction electrocatalysis on transition metal oxides and (oxy)hydroxides: Activity trends and design principles. Chem. Mater. 27, 7549–7558 (2015).
Karan, H. I. et al. Catalytic activity of platinum monolayer on iridium and rhenium alloy nanoparticles for the oxygen reduction reaction. ACS Catal 2, 817–824 (2012).
Chen, L. Y., Hou, Y., Kang, J. L., Hirata, A. & Chen, M. W. Asymmetric metal oxide pseudocapacitors advanced by three-dimensional nanoporous metal electrodes. J. Mater. Chem. A 2, 8448–8455 (2014).
McCrory, C. C. L., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Yingying, G et al. Co2P–CoN Double Active Centers Confined in N‐Doped Carbon Nanotube: Heterostructural Engineering for Trifunctional Catalysis toward HER, ORR, OER, and Zn–Air Batteries Driven Water Splitting. Adv. Funct Mater. 28 (51), 1805641 (1–9) (2018).
Groenewolt, M. & Antonietti, M. Synthesis of g-C3N4 nanoparticles in mesoporous silica host matrices. Adv. Mater. 17, 1789–1792 (2005).
Wang, Y. et al. Excellent visible-light photocatalysis of fluorinated polymeric carbon nitride solids. Chem. Mater. 22, 5119–5121 (2010).
Logakis, E. et al. Structure-property relationships in isotactic polypropylene/multi-walled carbon nanotubes nanocomposites. Compos. Sci. Technol 70, 328–335 (2010).
Benard, D. J. et al. Dissociation of cyanogen azide: An alternative route to synthesis of carbon nitride. J. Phys. Chem. B 102, 6010–6019 (2002).
Khabashesku, V. N., Zimmerman, J. L. & Margrave, J. L. Powder synthesis and characterization of amorphous carbon nitride. Chem. Mater. 12, 3264–3270 (2000).
Bai, X., Cao, C. & Xu, X. Formation and characterization of flower-like carbon nitride by pyrolysis of melamine. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol 175, 95–99 (2010).
Xia, X. et al. Preparation of magnetic graphitic carbon nitride nanocomposites. Mater. Lett. 64, 2620–2623 (2010).
Jiang, G. et al. Controllable preparation of graphitic carbon nitride nanosheets via confined interlayer nanospace of layered clays. Mater. Lett. 64, 2718–2721 (2010).
Byers, J. C., Tamiasso-Martinhon, P., Deslouis, C., Pailleret, A. & Semenikhin, O. A. Atomic force microscopy studies of carbon nitride (CNx) films deposited on a conducting polymer substrate. J. Phys. Chem. C 114, 18474–18480 (2010).
Xiang, Q., Yu, J. & Jaroniec, M. Preparation and enhanced visible-light photocatalytic H2- production activity of graphene/C3N4 composites. J. Phys. Chem. C 115, 7355–7363 (2011).
Cao, S. & Yu, J. g-C3N4-based photocatalysts for hydrogen generation. J. Phys. Chem. Lett. 5, 2101–2107 (2014).
Ge, L. et al. Synthesis and efficient visible light photocatalytic hydrogen evolution of polymeric g-C3N4 coupled with CdS quantum dots. J. Phys. Chem. C 116, 13708–13714 (2012).
Zhu, T., Chong, M. N. & Chan, E. S. Nanostructured tungsten trioxide thin films synthesized for photoelectrocatalytic water oxidation: A review. ChemSusChem 7, 2974–2997 (2014).
Abdullah Khan, M., Teixeira, I. F., Li, M. M. J., Koito, Y. & Tsang, S. C. E. Graphitic carbon nitride catalysed photoacetalization of aldehydes/ketones under ambient conditions. Chem. Commun. 52, 2772–2775 (2016).
Anantharaj, S. et al. Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe, Co, and Ni: A review. ACS Catalysis 6, 8069–8097 (2016).
Long, A. et al. MoS2 nanosheets grown on nickel chalcogenides: Controllable synthesis and electrocatalytic origins for the hydrogen evolution reaction in alkaline solution. J. Mater. Chem. A (2019).
Lu, Q. et al. Highly porous non-precious bimetallic electrocatalysts for efficient hydrogen evolution. Nat. Commun. 6, 6567 (2015).
Tian, J. et al. Self-supported NiMo hollow nanorod array: An efficient 3D bifunctional catalytic electrode for overall water splitting. J. Mater. Chem. A. 3(40), 20056–20059 (2015).
Xie, J. et al. Atomically-thin molybdenum nitride nanosheets with exposed active surface sites for efficient hydrogen evolution. Chem. Sci 5, 4615–4620 (2014).
Zhou, M. et al. Construction of polarized carbon-nickel catalytic surfaces for potent, durable, and economic hydrogen evolution reactions. ACS Nano 12(5), 4148–4155 (2018).
Popczun, E. J. et al. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 135, 9267–9270 (2013).
Huang, Y. et al. Mo2C nanoparticles dispersed on hierarchical carbon microflowers for efficient electrocatalytic hydrogen evolution. ACS Nano 10, 11337–11343 (2016).
Tian, T., Huang, L., Ai, L. & Jiang, J. Surface anion-rich NiS2 hollow microspheres derived from metal-organic frameworks as a robust electrocatalyst for the hydrogen evolution reaction. J. Mater. Chem. A 5, 20985–20992 (2017).
Hu, C. & Dai, L. Multifunctional carbon-based metal-free electrocatalysts for simultaneous oxygen reduction, oxygen evolution, and hydrogen evolution. Adv. Mater. 29, 1604942 (2017).
Shi, Y. et al. Carbon-nanotube-incorporated graphene scroll-sheet conjoined aerogels for efficient hydrogen evolution reaction. ACS Sustain. Chem. Eng. 5, 6994–7002 (2017).
Yu, Y., van Aken, P. A., Maier, J., Zhu, C. & Mu, X. Single-layered ultrasmall nanoplates of MoS2 embedded in carbon nanofibers with excellent electrochemical performance for lithium and sodium storage. Angew. Chemie Int. Ed. 53, 2152–2156 (2014).
Kong, D., Cha, J. J., Wang, H., Lee, H. R. & Cui, Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 6, 3553–3558 (2013).
Kong, D., Wang, H., Lu, Z. & Cui, Y. CoSe2 nanoparticles grown on carbon fiber paper: An efficient and stable electrocatalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 136, 4897–4900 (2014).
Liang, H. et al. Hydrothermal continuous flow synthesis and exfoliation of NiCo layered double hydroxide nanosheets for enhanced oxygen evolution catalysis. Nano Lett. 15, 1421–1427 (2015).
Faber, M. S., Lukowski, M. A., Ding, Q., Kaiser, N. S. & Jin, S. Earth-abundant metal pyrites (FeS2, CoS2, NiS2, and their alloys) for highly efficient hydrogen evolution and polysulfide reduction electrocatalysis. J. Phys. Chem. C 118, 21347–21356 (2014).
Faber, M. S. et al. High-performance electrocatalysis using metallic cobalt pyrite (CoS2) micro- and nanostructures. J. Am. Chem. Soc. 136, 10053–10061 (2014).
Zhou, M. et al. In situ electrochemical formation of core-shell nickel-iron disulfide and oxyhydroxide heterostructured catalysts for a stable oxygen evolution reaction and the associated mechanisms. J. Mater. Chem. A 5, 4335–4342 (2017).
Akhtar, M., Malik, M. A., Raftery, J. & O’Brien, P. Synthesis of iron selenide nanocrystals and thin films from bis(tetraisopropyldiselenoimidodiphosphinato)iron(II) and bis(tetraphenyldiselenoimidodiphosphinato)iron(II) complexes. J. Mater. Chem. A 2, 20612–20620 (2014).
Cook, P., Kim, Y. J., Yuan, K., Marcano, M. C. & Becker, U. Electrochemical, spectroscopic, and computational investigations on redox reactions of selenium species on galena surfaces. Minerals 9, 437 (2019).
Basu, M. et al. CoSe2 embedded in C3N4: An efficient photocathode for photoelectrochemical water splitting. ACS Appl. Mater. Interfaces 8, 26690–26696 (2016).
Acknowledgements
Aamir Hassan Shah from CAS Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, China is acknowledged for his valuable suggestions to improve the manuscript.
Author information
Authors and Affiliations
Contributions
Muhammad Zulqarnain, Afzal Shah and Faiza Jan Iftikhar designed and performed electrode modification experiments, electrochemical study for its catalytic role and characterization of the synthesized catalyst such as FTIR and DSC/TGA. Muhammad Zulqarnain, Muhammad Abdullah Khan and Afzal Shah contributed in writing the manuscript. Afzal Shah and Muhammad Abdullah Khan characterized the modified electrode by XRD, EDX and SEM along with contribution in analysis of results. Jan Nisar was involved in characterization of TEM and revision of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zulqarnain, M., Shah, A., Khan, M.A. et al. FeCoSe2 Nanoparticles Embedded in g-C3N4: A Highly Active and Stable bifunctional electrocatalyst for overall water splitting. Sci Rep 10, 6328 (2020). https://doi.org/10.1038/s41598-020-63319-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63319-7
This article is cited by
-
Preparation of high-crystalline and non-metal modified g-C3N4 for improving ultrasound-accelerated white-LED-light-driven photocatalytic performances
Scientific Reports (2023)
-
In situ synthesize Bi nanostructures on copper foam toward efficient electrocatalytic hydrogen evolution reaction
Chemical Papers (2023)
-
Fabrication of g-C3N4 with Simultaneous Isotype Heterojunction and Porous Structure for Enhanced Visible-Light-Driven Photocatalytic Performance Toward Tetracycline Hydrochloride Elimination
Topics in Catalysis (2023)
-
Design and preparation of HPW-anchored magnetic carbon nitride nanosheets: an efficient and eco-friendly nanocomposite for one-pot synthesis of α-amino phosphonates
Research on Chemical Intermediates (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
