
Abstract
Nanopore based DNA-sequencing delivers long reads, thereby simplifying the decipherment of bacterial communities. Since its commercial appearance, this technology has been assigned several attributes, such as its error proneness, comparatively low cost, ease-of-use, and, most notably, aforementioned long reads. The technology as a whole is under continued development. As such, benchmarks are required to conceive, test and improve analysis protocols, including those related to the understanding of the composition of microbial communities. Here we present a dataset composed of twelve different prokaryotic species split into four samples differing by nucleic acid quantification technique to assess the specificity and sensitivity of the MinION nanopore sequencer in a blind study design. Taxonomic classification was performed by standard taxonomic sequence classification tools, namely Kraken, Kraken2 and Centrifuge directly on reads. This allowed taxonomic assignments of up to 99.27% on genus level and 92.78% on species level, enabling true-positive classification of strains down to 25,000 genomes per sample. Full genomic coverage is achieved for strains abundant as low as 250,000 genomes per sample under our experimental settings. In summary, we present an evaluation of nanopore sequence processing analysis with respect to microbial community composition. It provides an open protocol and the data may serve as basis for the development and benchmarking of future data processing pipelines.
Similar content being viewed by others


Introduction
Sequencing of environmental DNA has established itself as a means to overcome the limitations of cultivation and to understand the composition and dynamics of microbial communities1,2,3. Throughout the past two decades, sequencing technologies have continually experienced a decrease in cost and increase in output. As such and due to its wide availability, next-generation (also known as second-generation) DNA sequencing is currently the major technology2,4,5. Yet, a limitation of second generation DNA sequencing remains: its short reads. While first generation Sanger sequencing yields up to 1,000 basepairs (bp), second-generation methods (e.g. Illumina MiSeq) are limited to app. 300 bp. Nanopore-based sequencing is a third-generation sequencing method enabling deciphering of nucleic acids exceeding several thousand basepairs. The technology is generally applicable to a wide variety of purposes in basic and applied research in all kingdoms, as well as to clinical and life science applications6,7,8. Sequencing devices employing this technology are currently distributed through Oxford Nanopore Technologies and the technology as a whole is, as of today, under active development. This is of particular interest since nanopore sequencing, or long-read sequencing, has previously been labelled as error prone9, although more recent advances brought improvements to both chemistry and data processing (e.g. Brown, Nanopore Community Meeting Presentation 2018;10). On the other hand, single molecule sequencing using nanopores is generating long reads, which are, among other reasons, of interest in elucidating microbial diversity11. Other advantages of the first available sequencer model, the MinION, also compared to its larger siblings GridION and PromethION, are the lower initial investment and its mobility allowing for direct field studies12,13,14. Since the introduction of the MinION, several studies have been presented concerning its performance. However, those were not taking advantage of amplification-free sequencing and employed - to date - previous15,16,17,18 or other19 versions of the sequencing chemistry. Aforementioned ongoing development of the technology as a whole necessitates new and frequent revisions and updates to sequencing protocols and downstream data processing. This is also the case with the development of bioinformatics pipelines and the design of tools20. For this purpose, suitable datasets for rigorous testing are required. Recently, such datasets have been supplied for GridION and PromethION sequencers using commercially available standards21. We investigated four mixed microbial DNA samples differing by the employed DNA-quantitation technique and their composition using the MinION sequencer. The samples were composed of DNA covering up to five orders of magnitude in genome amounts from twelve bacterial species. The aim of the study includes an establishment of a suitable classification pipeline and an assessment of the accuracy of the MinION in samples with unknown microbial composition.
Results and discussion
Raw dataset description
Using the MinION DNA-sequencing platform we generated app. 809k reads in Fast5 file format, equal to an estimate of 8.15 Gbp in a single run within 36 hours (see Supplementary Fig. S1,S2). We could observe increased yield for each pore group switch (a.k.a remux), and output of constant quality on a uniform read length distribution for our sequencing run (see Supplementary Figs. S3–S5). Approximately 807k reads equal to 7.06 Gbp were successfully basecalled and demultiplexed generating an overall yield of 662k reads equivalent to 6.17 Gbp for downstream analysis. Samples one to four, corresponding to the four barcodes used, are composed of app. 142k (#1, heterogeneous sample quantified by ddPCR), 262k (#2, heterogeneous sample quantified by Qubit), 110k (#3, equimolar sample quantified by ddPCR) and 148k (#4, equimolar sample quantified by Qubit) reads, respectively. A total of only four reads were not properly demultiplexed by Porechop, i.e. assigned to a barcode not present in the library. A total of app. 140k reads were demultiplexed as “unclassified” by Porechop, i.e. not assigned to any barcode. All reads not assigned to barcodes #1, #2, #3 or #4 (corresponding to the four samples) were discarded and thus excluded from downstream analysis (Table 1).
Data classification and validation
The use of Centrifuge with nanopore read datasets has been demonstrated before22,23. The application of Kraken and Kraken2 on nanopore data has also been described, albeit within different experimental settings, such as the taxonomic classification of reads of well characterized isolates24 or the taxonomic classification of complete assemblies21. Taxonomic classification performed by either, Centrifuge, Kraken or Kraken2 allowed for the heterogeneously concentrated samples (samples #1 and #2, adjusted by ddPCR and Qubit, respectively) an initial choice of five out of twelve strains based on the available Krona plots (see Supplementary data S1). For the samples with equimolar genomic concentration (samples #3 and #4), a selection of twelve strains was immediately possible (Fig. 1). Generally, despite the differences in the underlying software and databases/indices, we could observe substantial agreement25 between the results obtained from Kraken, Kraken 2 and Centrifuge with their respective databases as tested by Fleiss Kappa (lowest 0.778, highest 0.931).

Centrifuge, Kraken and Kraken 2 classification results on genus and species level for equimolar (sample/barcode 3, adjusted by ddPCR and sample/barcode 4, adjusted by Qubit) and heterogeneously concentrated samples (sample/barcode 1, adjusted by ddPCR and sample/barcode 2, adjusted by Qubit) of 12 target strains. Theoretical values and validation by NanoOK (alignments with minimap2) are given for comparison.
Quantitation by ddPCR delivers slightly different results than quantitation by fluorometry such as Qubit26,27. This is due to e.g. different basepair compositions, staining efficiencies or denaturation of DNA prior to droplet generation. Thus, we investigated, if the slight difference between these two quantitation approaches (Qubit vs. ddPCR) were also determinable by nanopore-based DNA-sequencing. Indeed, differences in quantitation, which resulted in different volumes necessary for sample preparations, corresponded to different amount of reads for that specific organism to the same extent (see Supplementary Fig. S6).
Unblinding the ground truth to the sequencing laboratory revealed a correct, that is true positive, selection of all twelve strains in samples of equimolar genomic concentration, as well as a correct selection of five out of twelve strains in the two samples with different genomic concentration. The five strains selected from the heterogeneously concentrated samples made up 99.38% of the genomes calculated to be available in the actual samples of different genomic concentration. This corresponds to a concentration of 2.5 million to 50 million genomes per species and sample. Notably, read classification matching the ground truth on genus level was possible for up to 99.27% (Centrifuge) between all samples, whereas read classification matching the ground truth on species level was up to 92.78% (Centrifuge) across all samples (Table 2). Generally, accuracy and deviation metrics (root mean squared deviation (RMSD) and mean absolute error (MAE)) on genus level were better than on species level. Comparing Centrifuge, Kraken and Kraken2 running their precompiled databases/indices, Centrifuge was able to assign the highest fraction of reads to the theoretically expected genera and species across all samples. Also, Centrifuge performed best with respect to both measures of deviation (RMSD, MAE), whereas Kraken 2 was superior over Kraken. However, beyond the accuracy of each classifier, computational aspects need to be considered. Especially, when limited computational resources are available, such as in field applications, Kraken 2 offers superior processing speed and lower memory consumption compared to Centrifuge and Kraken28.
Precision and recall per species and genus reached generally high values on read level (see Supplementary Table S3, S4). For genera with very low abundancy, drops in precision could be observed (see Supplementary Table S3). Reads wrongly classified on species level were, e.g., attributable to close relatives, such as Bacillus species to Bacillus licheniformis, Enterobacter cloacae to Enterobacter hormaechei, et cetera, or exhibited differences in read abundancy as compared to true positive hits, which is similar to findings reported by Deshpande et al.19 despite a different sequencing and analysis approach. This is also reflected by the lower values of recall for these species on read level (see Supplementary Table S4). The necessity for accurate databases and unified nomenclature is discussed elsewhere29,30,31,32 and has been shown to affect classification of nanopore data18. These results indicate that classification is, as of yet, more reliable on genus level than on species level.
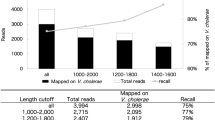
Serendipitously, rerunning the classification process after the removal of four most abundant initially selected strains from the read data allowed the additional selection and thus classification of four strains down to app. 25,000 to 500,000 genomes per sample, using Krona plots. The remaining three strains adjusted to the range of 500 to 5,000 genomes per sample could not be reliably retrieved from the two samples with heterogeneous genomic concentrations (Fig. 2). Their presence was obfuscated by the filter process, i.e. they were as abundant as falsely classified reads and, subsequently, a clear discrimination allowing selection and classification was impossible. With the experimental settings and proceeding as described here, this suggests a dynamic range of detection and viable classification between 250 and 500,000 genomes/µl of initial DNA input, corresponding to a range of 25,000 to 50 million genomes from material obtained from microbial communities of low diversity from the MinION. The range reported here is similar to the findings of Nicholls et al.21.

In silico complexity reduction of the samples with heterogeneous genomic concentration (sample/barcode 1, adjusted by ddPCR and sample/barcode 2, adjusted by Qubit) allows reliable detection of further strains down to an original genomic concentration around 25,000 genomes. Strains adjusted to the range of 500 to 5,000 genomes could not be reliably detected. Theoretical values and validation by NanoOK (alignments with minimap2) are given for comparison.
These results showed good consistency with a) the output from the NanoOK analysis by direct comparison (Table 3, see Supplementary Table S5), where at least 99.21% of all available reads could be aligned to selected references and b) the theoretical expectation. Moreover, mean coverages reported by NanoOK indicate potential for de novo genome assemblies (Fig. 3). Full genomic coverage realistically permitting de novo assembly was achieved for strains down to a concentration of 250,000 genomes per sample (see Supplementary Table S5). At comparable sequencing times, we anticipate the concentration level required to achieve full genomic coverage to be even lower for libraries that are not multiplexed.

Sequenced coverage per sample for each of the twelve identified strains in the community. Data based on NanoOK analysis.
Despite the error rates currently accompanying MinION sequencing, these results clearly illustrate the viability and possibilities of long reads for direct taxonomic classification and abundance estimation with currently available bioinformatics pipelines.
Conclusion
We present a MinION DNA sequence read dataset to facilitate the Nanopore community to improve and develop new bioinformatics pipelines aimed at the understanding of microbial diversity. Continual benchmarking using updated sequencing methods and chemistries in metagenome analyses is required32. With the presented detailed methodology, as a whole, this study follows the FAIR Guiding Principles33 for scientific data management and stewardship by contributing (F)indable and (A)ccessible data under bioproject accession PRJNA545964 and corresponding signal level data34 that is (R)eusable for the fast-paced development of third generation sequencing and downstream bioinformatics in a metagenomics context.
Based on the dataset, we present a simple and straightforward analysis pipeline to investigate the composition of microbial communities. Given our experimental approach we were able to achieve highly accurate taxonomic classification of low abundant (25,000 genomes/sample) organisms to at least genus level. Full genomic coverage was achieved for species with an abundancy of 250,000 genomes per sample and sufficient coverage for de novo assembly could be obtained.
While there is no standardized approach for the characterization of bacterial communities, molecular tools are considered powerful to gain knowledge and insight into these35,36, and nanopore sequencing is no exception to this point. In summary, the presented benchmark provides insight into nanopore data and data processing for the taxonomic classification of microbial communities. Hence, this study contributes to the toolsets and development of processing pipelines available to elucidate microbial diversity.
Material and methods
The overall experimental design is setup as follows: Bacteria cultivation, DNA extraction, quantification and creation of mock samples were performed by the Unit for Biological Agents, Federal Institute for Occupational Safety and Health (BAuA). Samples were shipped to the sequencing team (Mittweida UAS). The sequencing team performed library preparation, sequencing and downstream processing unaware of the samples’ actual respective compositions (Fig. 4).

Overall study design and process workflow. Part One (grey), mock community creation, was performed by the Unit for Biological Agents, Federal Institute for Occupational Safety and Health (BAuA, Berlin). Part Two (black), sequencing and data processing was performed by Wünschiers Group, University of Applied Sciences Mittweida (Mittweida UAS).
Sample cultivation and preparation
DNA from twelve bacterial strains was extracted to form a mock community sample (Table 4) for benchmarking the MinION sequencing platform using the following criteria: (A) Each strain is the type strain of the bacterial species and is available from the Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), the National Collection of Type Cultures (NCTC) or the American Type Culture Collection (ATCC). (B) Each strain has a reference sequence deposited at the National Center for Biological Information (NCBI). (C) Each strain has several assemblies of the same species available at the NCBI. (D) The sequencing laboratory is blind to both, the selection itself and the actual composition of the strains selected.
Bacteria were grown overnight as follows: Dickeya solaniT (Todd-Hewitt + 0,5% yeast extract (THY), 28 °C), Serratia fonticolaT (DSMZ-Medium 1, 28 °C), Bacillus licheniformisT (DSMZ-Medium 1, 37 °C), Corynebacterium glutamicumT (THY, 28 °C), Micrococcus luteusT (THY, 28 °C), Cronobacter sakazakiiT (DSMZ-Medium 1, 28 °C), Achromobacter xyloxidans subsp. xyloxidansT (DSMZ-Medium 1, 28 °C), Paenibacillus odoriferT (DSMZ-Medium 1, 28 °C), Chromobacterium violaceumT (DSMZ-Medium 1, 28 °C), Enterobacter hormaechei subsp. steigerwaltiiT (CASO, 37 °C), Staphylococcus saprophyticus subsp. saprophyticusT (DSMZ-Medium 92, 37 °C) and Xanthomonas campestrisT (DSMZ-Medium 1, 28 °C). DNA of 1 ml of the cell suspension derived from liquid culture or resuspended colonies in PBS was extracted using a modified protocol of the GenElute Plant Genomic DNA Miniprep Kit (Sigma Aldrich,37). DNA concentrations were quantified using the Qubit BR assay in a Qubit 1.0 fluorometer according to the manufacturer’s protocol. Subsequently, ddPCR targeting the 16 S rRNA-gene was conducted with app. less than 40,000 target genes according to the manufacturer’s instructions (Bio-Rad) using the ddPCR Supermix for Probes (no dUTP). Final concentrations of oligonucleotides were 0.4 pmol/µL 1055Falt (ATGGRTGTCGTCAGCT), 0.2 pmol/µL 1392 R (ACGGGCGGTGTGTAC) and 0.1 pmol/µL 1115IB (FAM-CAACGAGCG-ZEN-CAACCC-3IABkFQ) adopted from Rothrock et al.38. Droplet generation was conducted according to manufacturer’s instructions in a QX200 Droplet Generator and amplified in a T100 Thermal Cycler. PCR conditions were initial denaturation at 95 °C for 10 min, and 30 cycles of denaturation at 95 °C for 30 s, annealing at 57 °C for 45 s, extension at 72 °C for 45 s with a ramp rate of 1 °C/s, followed by a final extension at 98 °C for 10 min and cooling to 12 °C. Droplet evaluation was performed in a QX200 Droplet Reader with QuantaSoft-Software.
Based on Qubit and ddPCR quantitation, the nucleic acids were adjusted to different genomic concentrations ranging from 5 to 5*105 genomes/µl (samples #1 and #2, corresponding to sequencing library barcodes #1 and #2), or to equimolar genomic concentration of 5*104 genomes/µl (samples #3 and #4 corresponding to sequencing library barcodes #3 and #4).
Samples were shipped on ice by public postal services.
Library preparation and sequencing
A sequencing library was prepared according to manufacturer’s instructions. The Ligation Sequencing Kit (SQK-LSK108, Oxford Nanopore Technologies (ONT)) and the Native Barcoding Expansion 1–12 kit (EXP-NBD103, ONT), barcoding each of the samples (barcodes #1, #2, #3, #4), were used with the following exceptions: Shearing times were prolonged and an optional FFPE DNA repair step (M6630, New England Biolabs (NEB)) was included. The incubation times during the end-repair/dA-tailing (E7546, NEB) were extended from five to 20 minutes for both, the 20 °C and 65 °C incubation steps. Qubit checkpoint measurements were performed according to the library preparation protocol (see Supplementary Table S1). Pooling of the barcoded samples was performed ‘as is’ instead of protocol-given ‘equimolar’. Sequencing was then performed on a R9.4 flowcell (FLO-MIN106, ONT, >1200 pores, see Supplementary Table. S2) with MinKNOW (version 2.1.12, ONT) at room temperature.
Base calling and demultiplexing
Upon conclusion of sequencing, raw data in Fast5 file format were transferred to our server (4.17.2-1-ARCH, 20 cores with 2 threads each, 256 GB RAM) and basecalled using the Albacore software (version 2.0.2, ONT) with barcoding option. Subsequently, barcodes were removed from basecalled output and subsequently sorted utilizing Porechop (version 0.2.3, standard settings, https://github.com/rrwick/Porechop). Basecalled and demultiplexed sequencing data quality was assessed with NanoPack (version 1.13.0, https://github.com/wdecoster/NanoPlot)39.
Data classification and validation
Taxonomic classification was performed with standard parameters (Centrifuge “-k 1”) on native reads using Centrifuge (precompiled index: “Bacteria, Archaea (compressed), 2018-4-15”)22, as well as Kraken (precompiled database: “DustMasked MiniKraken DB 8GB”)40 and Kraken2 (precompiled database: MiniKraken2_v1_8GB)28 and the results were visualized with Krona41 and R42,43,44,45.
The interactive and intuitive Krona visualization was used to manually select up to twelve bacterial strains. The corresponding genome reference sequences were obtained from NCBI Reference Sequence Database46 (accessed on 2018-07-31).
NanoOK (version 1.34)47 was utilized for an assessment of the read dataset against the selection of NCBI genome reference sequences, using minimap2 aligner (version 2.11)48. To create the minimap2 index, the reference sequences obtained from NCBI Reference Sequence Database were concatenated into a single FastA file.
Statistics and additional visualizations were computed with R42,43,44,45,49,50. We calculated the accuracy of the classification performed by Centrifuge, Kraken and Kraken 2 on each sample the proportion of reads assigned to the known input organism at the genus and species level out of the total number reads given any assignment at that rank18. To calculate a corresponding estimate of the accompanying error, the mean absolute error, as well as root mean squared deviation of classified to theoretically present fractions on genus and species level were computed. On read level, precision and recall for genus and species identification were computed32 for Centrifuge, Kraken and Kraken 2 vs. the results obtained from the NanoOK analysis, with precision being the proportion of reads classified correctly to reads classified and recall being the proportion of reads classified correctly to the reads from the NanoOK dataset, which was used as “ground truth”. All additional bioinformatics processing was performed in the Linux Bourne Again Shell (bash), using Samtools (version 1.9)51 and seqtk (version 1.3-r106, https://github.com/lh3/seqtk).
Data availability
The data sets supporting the results of this article are available in the under bioproject accession PRJNA545964 (https://www.ncbi.nlm.nih.gov/sra/PRJNA545964) and as Zenodo deposit 3600229 (10.5281/zenodo.3600229).
References
Handelsman, J. Metagenomics: application of genomics to uncultured microorganisms. Microbiology and molecular biology reviews: MMBR 68, 669–685, https://doi.org/10.1128/MMBR.68.4.669-685.2004 (2004).
Schloss, P. D. & Handelsman, J. Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome biology 6, 229, https://doi.org/10.1186/gb-2005-6-8-229 (2005).
Tringe, S. G. & Rubin, E. M. Metagenomics: DNA sequencing of environmental samples. Nature reviews. Genetics 6, 805–814, https://doi.org/10.1038/nrg1709 (2005).
Shendure, J. & Ji, H. Next-generation DNA sequencing. Nature biotechnology 26, 1135–1145, https://doi.org/10.1038/nbt1486 (2008).
Shendure, J. & Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature biotechnology 30, 1084–1094, https://doi.org/10.1038/nbt.2421 (2012).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232, https://doi.org/10.1038/nature16996 (2016).
Jain, M. et al. Linear assembly of a human centromere on the Y chromosome. Nature biotechnology 36, 321–323, https://doi.org/10.1038/nbt.4109 (2018).
Bronzato Badial, A. et al. Nanopore Sequencing as a Surveillance Tool for Plant Pathogens in Plant and Insect Tissues. Plant disease 102, 1648–1652, https://doi.org/10.1094/PDIS-04-17-0488-RE (2018).
Laver, T. et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomolecular detection and quantification 3, 1–8, https://doi.org/10.1016/j.bdq.2015.02.001 (2015).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome biology 20, 129, https://doi.org/10.1186/s13059-019-1727-y (2019).
Bleidorn, C. Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Systematics and Biodiversity 14, 1–8, https://doi.org/10.1080/14772000.2015.1099575 (2016).
Walter, M. C. et al. MinION as part of a biomedical rapidly deployable laboratory. Journal of biotechnology 250, 16–22, https://doi.org/10.1016/j.jbiotec.2016.12.006 (2017).
Walper, S. A. et al. Detecting Biothreat Agents: From Current Diagnostics to Developing Sensor Technologies. ACS sensors 3, 1894–2024, https://doi.org/10.1021/acssensors.8b00420 (2018).
Hansen, S. et al. Serotyping of foot-and-mouth disease virus using oxford nanopore sequencing. Journal of virological methods 263, 50–53, https://doi.org/10.1016/j.jviromet.2018.10.020 (2019).
Kilianski, A. et al. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. GigaScience 4, 12, https://doi.org/10.1186/s13742-015-0051-z (2015).
Benítez-Páez, A., Portune, K. J. & Sanz, Y. Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION™ portable nanopore sequencer. GigaScience 5, 4, https://doi.org/10.1186/s13742-016-0111-z (2016).
Benítez-Páez, A. & Sanz, Y. Multi-locus and long amplicon sequencing approach to study microbial diversity at species level using the MinION™ portable nanopore sequencer. GigaScience 6, 1–12, https://doi.org/10.1093/gigascience/gix043 (2017).
Brown, B. L., Watson, M., Minot, S. S., Rivera, M. C. & Franklin, R. B. MinION™ nanopore sequencing of environmental metagenomes: a synthetic approach. GigaScience 6, 1–10, https://doi.org/10.1093/gigascience/gix007 (2017).
Deshpande, S. V. et al. Offline Next Generation Metagenomics Sequence Analysis Using MinION Detection Software (MINDS). Genes 10; https://doi.org/10.3390/genes10080578 (2019).
Sedlazeck, F. J., Lee, H., Darby, C. A. & Schatz, M. C. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nature reviews. Genetics 19, 329–346, https://doi.org/10.1038/s41576-018-0003-4 (2018).
Nicholls, S. M., Quick, J. C., Tang, S. & Loman, N. J. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience 8; https://doi.org/10.1093/gigascience/giz043 (2019).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome research 26, 1721–1729, https://doi.org/10.1101/gr.210641.116 (2016).
Sanderson, N. D. et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC genomics 19, 714, https://doi.org/10.1186/s12864-018-5094-y (2018).
Tyler, A. D. et al. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Scientific reports 8, 10931, https://doi.org/10.1038/s41598-018-29334-5 (2018).
Landis, J. R. & Koch, G. G. The measurement of observer agreement for categorical data. Biometrics 33, 159–174 (1977).
Bhat, S. et al. Comparison of methods for accurate quantification of DNA mass concentration with traceability to the international system of units. Analytical chemistry 82, 7185–7192, https://doi.org/10.1021/ac100845m (2010).
Sanders, R. et al. Evaluation of digital PCR for absolute DNA quantification. Analytical chemistry 83, 6474–6484, https://doi.org/10.1021/ac103230c (2011).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome biology 20, 257, https://doi.org/10.1186/s13059-019-1891-0 (2019).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nature reviews. Microbiology 12, 635–645, https://doi.org/10.1038/nrmicro3330 (2014).
Edgar, R. C. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 6, e4652, https://doi.org/10.7717/peerj.4652 (2018).
Lydon, K. A. & Lipp, E. K. Taxonomic annotation errors incorrectly assign the family Pseudoalteromonadaceae to the order Vibrionales in Greengenes: implications for microbial community assessments. PeerJ 6, e5248, https://doi.org/10.7717/peerj.5248 (2018).
McIntyre, A. B. R. et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome biology 18, 182, https://doi.org/10.1186/s13059-017-1299-7 (2017).
Wilkinson, M. D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Scientific data 3, 160018, https://doi.org/10.1038/sdata.2016.18 (2016).
Leidenfrost, R., Pöther, D.-C., Jäckel, U. & Wünschiers, R. Nanopore raw signal data to Benchmarking the MinION: Evaluating long reads for microbial profiling. Available at https://zenodo.org/record/3600229 (2020).
Schäfer, J., Weiß, S. & Jäckel, U. Preliminary Validation of a Method Combining Cultivation and Cloning-Based Approaches to Monitor Airborne Bacteria. Annals of work exposures and health 61, 633–642, https://doi.org/10.1093/annweh/wxx038 (2017).
Jäckel, U., Martin, E. & Schäfer, J. Heterogeneity in Cultivation-Based Monitoring of Airborne Bacterial Biodiversity in Animal Farms. Annals of work exposures and health 61, 643–655, https://doi.org/10.1093/annweh/wxx039 (2017).
Martin, E., Kämpfer, P. & Jäckel, U. Quantification and identification of culturable airborne bacteria from duck houses. The Annals of occupational hygiene 54, 217–227, https://doi.org/10.1093/annhyg/mep088 (2010).
Rothrock, M. J., Hiett, K. L., Kiepper, B. H., Ingram, K. & Hinton, A. Quantification of Zoonotic Bacterial Pathogens within Commercial Poultry Processing Water Samples Using Droplet Digital PCR. AiM 03, 403–411, https://doi.org/10.4236/aim.2013.35055 (2013).
Coster, W., de, D’Hert, S., Schultz, D. T., Cruts, M. & van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics (Oxford, England) 34, 2666–2669, https://doi.org/10.1093/bioinformatics/bty149 (2018).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology 15, R46, https://doi.org/10.1186/gb-2014-15-3-r46 (2014).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC bioinformatics 12, 385, https://doi.org/10.1186/1471-2105-12-385 (2011).
R, C. T. R: A Language and Environment for Statistical Computing (Vienna, Austria, 2014).
Wickham, H. & Sievert, C. ggplot2. Elegant Graphics for Data Analysis. 2nd ed. (Springer International Publishing, Cham, 2016).
Kassambara, A. ggpubr:’ggplot2’ Based Publication Ready Plots (2018).
Neuwirth, E. RColorBrewer: ColorBrewer Palettes (2014).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic acids research 44, D733–45, https://doi.org/10.1093/nar/gkv1189 (2016).
Leggett, R. M., Heavens, D., Caccamo, M., Clark, M. D. & Davey, R. P. NanoOK: multi-reference alignment analysis of nanopore sequencing data, quality and error profiles. Bioinformatics (Oxford, England) 32, 142–144, https://doi.org/10.1093/bioinformatics/btv540 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics (Oxford, England) 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Gamer, M., Lemon, J., Fellows, I. & P Singh. irr: Various Coefficients of Interrater Reliability and Agreement (2019).
Hamner, B. & Frasco, M. Metrics: Evaluation Metrics for Machine Learning. Available at, https://CRAN.R-project.org/package=Metrics (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Acknowledgements
RL acknowledges funding through European Social Fund (ESF) PhD Scholarship, Grant #100316182. RW received funding from the Saxon State Ministry of Science and Art and the BMBF-funded Saxony5 Initiative. The funders had no role in the design of the study, the collection, analysis and interpretation of data and the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
The conception and design of the study were done by all authors. D.P. and U.J. performed bacteria growth, DNA extraction, ddPCR based DNA quantification and sample preparation. R.L. received the samples, prepared barcoded sequencing libraries, performed sequencing and downstream sequence and statistical analyses. The latter were supervised by R.W. Result interpretation and manuscript preparation were done by all authors. All authors have edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leidenfrost, R.M., Pöther, DC., Jäckel, U. et al. Benchmarking the MinION: Evaluating long reads for microbial profiling. Sci Rep 10, 5125 (2020). https://doi.org/10.1038/s41598-020-61989-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61989-x
This article is cited by
-
MetageNN: a memory-efficient neural network taxonomic classifier robust to sequencing errors and missing genomes
BMC Bioinformatics (2024)
-
Comparative analysis of metagenomic classifiers for long-read sequencing datasets
BMC Bioinformatics (2024)
-
Benchmarking bacterial taxonomic classification using nanopore metagenomics data of several mock communities
Scientific Data (2024)
-
Long-read sequencing of metagenomes from wet deposition samples in the Western USA during an elevated precipitation in February 2019
Aerobiologia (2024)
-
Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets
BMC Bioinformatics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
