
Abstract
Kv7 channels are enriched at the axonal plasma membrane where their voltage-dependent potassium currents suppress neuronal excitability. Mutations in Kv7.2 and Kv7.3 subunits cause epileptic encephalopathy (EE), yet the underlying pathogenetic mechanism is unclear. Here, we used novel statistical algorithms and structural modeling to identify EE mutation hotspots in key functional domains of Kv7.2 including voltage sensing S4, the pore loop and S6 in the pore domain, and intracellular calmodulin-binding helix B and helix B-C linker. Characterization of selected EE mutations from these hotspots revealed that L203P at S4 induces a large depolarizing shift in voltage dependence of Kv7.2 channels and L268F at the pore decreases their current densities. While L268F severely reduces expression of heteromeric channels in hippocampal neurons without affecting internalization, K552T and R553L mutations at distal helix B decrease calmodulin-binding and axonal enrichment. Importantly, L268F, K552T, and R553L mutations disrupt current potentiation by increasing phosphatidylinositol 4,5-bisphosphate (PIP2), and our molecular dynamics simulation suggests PIP2 interaction with these residues. Together, these findings demonstrate that each EE variant causes a unique combination of defects in Kv7 channel function and neuronal expression, and suggest a critical need for both prediction algorithms and experimental interrogations to understand pathophysiology of Kv7-associated EE.
Similar content being viewed by others


Introduction
Epilepsy is the second most prominent neurological disease (www.epilepsy.com), in which excessive electrical activity within networks of neurons in the brain manifests clinically as recurrent unprovoked seizures1. Recent discoveries of epilepsy-related genes in multiple laboratories and through large consortia have revealed a diverse array of proteins that may contribute to epileptogenesis1,2. Among these proteins, neuronal KCNQ/Kv7 potassium (K+) channels have been implicated in epilepsy since mutations in the principle subunits, KCNQ2/Kv7.2 and KCNQ3/Kv7.3, cause Benign Familial Neonatal Epilepsy (BFNE [MIM: 121200]) and Epileptic Encephalopathy (EE [MIM: 613720]) (RIKEE database www.rikee.org).
Neuronal Kv7 channels are mainly composed of heterotetramers of Kv7.2 and Kv7.33, which show overlapping distribution in the hippocampus and cortex4. They generate slowly activating and non-inactivating voltage-dependent K+ currents that contribute to resting membrane potential, prevent repetitive and burst firing of action potentials (APs), and modulate AP threshold3,5,6,7.They are enriched at the plasma membrane of axonal initial segments (AIS) and distal axons8,9, where APs initiate and propagate10. Membrane phosphatidylinositol-4,5-bisphosphate (PIP2) is required for Kv7 channels to open3, although its exact binding sites in Kv7.2 and Kv7.3 are still under investigation11,12,13,14,15. They are also called ‘M-channels’ because their currents are inhibited by PIP2 depletion upon activation of Gq/11-coupled receptors such as the M1 muscarinic acetylcholine receptor14,16,17.
Nearly 200 BFNE and EE mutations in KCNQ2 and KCNQ3 genes have been identified to date (RIKEE database www.rikee.org). Dominantly inherited BFNE variants cause neonatal seizures that show spontaneous remission with benign psychomotor and intellectual outcomes18, and their effects on Kv7 channel function and excitability have been extensively studied19,20. A large number of de novo EE variants in KCNQ2 have been recently discovered since 20122,21,22,23,24,25,26. Patients with EE variants display early-onset seizures, developmental delay, neuroradiological abnormalities, and behavioral comorbidities including intellectual disability and autism21,23,24,25. Current anti-epileptic drugs are ineffective in treating many patients with KCNQ2 EE variants21,25,27, posing a critical need to understand how EE mutations disrupt Kv7 channels and lead to severe symptomatic epilepsy.
One important step is to determine if EE variants cluster at key functional domains that are critical for voltage-dependent gating and expression of Kv7 channels. Each Kv7 subunit contains 6 transmembrane segments (S1–S6)3,28. The S1-S4 segments form voltage sensing domains with S4 as a main sensor for depolarization3,28,29. The pore loop between S5 and S6 contains a highly conserved selectivity filter that controls K+ permeability and selectivity3,28,29. The C-terminal intersection of four S6 segments constitutes the main gate28,29. The intracellular C-terminal tails of Kv7.2 and Kv7.3 contain sites for PIP2-dependent modulation and ankyrin-G-dependent targeting to the AIS, and 4 helical structures (helices A-D) that mediate channel assembly through helix C and interaction with calmodulin (CaM) through helices A and B8,30. Therefore, we hypothesize that EE mutations are enriched at specific functional domains of Kv7.2 and disrupt their functions.
In this study, we test this hypothesis by developing novel statistical algorithms and modeling of Kv7.2, since computational in-silico algorithms have been shown to be useful tools for predicting pathogenicity of sequence variants31. We show that EE variants are significantly clustered at S4, the pore loop, S6, helix B, and the helix B-C linker of Kv7.2. Our investigation of selected EE variants in epilepsy mutation hotspots revealed that each mutation impaired the function of its associated protein domain. Unexpectedly, we discovered that selected EE mutations reside in the PIP2 binding regions of Kv7.2. Furthermore, selected mutations in the pore loop and helix B impaired Kv7 current enhancement upon increasing PIP2 and severely decreased surface expression of heteromeric channels in neurons. These findings emphasize the importance of both prediction algorithms and experimental interrogations to understand pathophysiology of Kv7-associated EE.
Results
MHF algorithm identifies EE mutation clusters in Kv7.2
We compiled genetic information and clinical symptoms of 194 epilepsy mutations in Kv7.2 that were reported until December 31, 2017 (Fig. 1a, Supplementary Table S1). These variants include 10 submicroscopic and partial gene deletions, 17 splice site mutations, 10 nonsense mutations, 25 frameshift mutations, 2 non-initiation mutations, 126 missense mutations that lead to single amino acid substitutions, and 4 mutations that result in single amino acid deletions (Fig. 1a). These mutations were classified into three groups according to the severities of their clinical outcomes described in the RIKEE database (Fig. 1a, Supplementary Table S1). The “mild or BFNE” mutations lead to seizures but not developmental delay in patients. The “severe or EE” mutations cause neonatal encephalopathy, seizures, and developmental delays. The “uncertain severity” mutations are associated with both benign seizures and EE or have limited clinical information. In addition, 130 silent mutations (with highest allele frequency from 0.0017% to 19%) and 25 relatively common nonpathogenic missense mutations of Kv7.2 (with allele frequency ≥ 0.01%) were identified from the Exome Aggregation Consortium (ExAC) database that collected protein-coding genetic variations from 60,706 humans (http://exac.broadinstitute.org).

Epilepsy mutations cluster at the S4, the pore, and the S6 domains of Kv7.2. (a) Pie charts showing the relative proportions of different types of epilepsy variants in human Kv7.2 (left) and of single amino acid Kv7.2 mutations that cause epilepsy with different clinical severity including “mild or BFNE”, “uncertain severity”, and “severe or epileptic encephalopathy (EE)” (right). All mutations identified up to December 31, 2017 are shown in Supplementary Table S1. (b) Single amino acid mutations except for 2 missense mutations in the primary start ATG codon (green markers) and silent mutations (purple markers) are mapped to Kv7.2 primary structure (NP_742105.1). The MHF statistical algorithm identified S4, the pore loop, and S6 as hotspots for all pathogenic epilepsy mutations (brackets, ***p < 0.005, Supplementary Table S2). (c) The MHF algorithm revealed S4, the pore loop, and S6 as hotspots for “severe or EE” mutations (brackets, **p < 0.01, ***p < 0.005, Supplementary Table S3). (d) The S1-S6 domains of tetrameric human Kv7.2 were modeled based on the cryoEM structure of Xenopus Kv7.1 (Protein Data Bank: 5VMS)28. At left, two opposing subunits are viewed from the plane of the plasma membrane. At right, the model is viewed from the extracellular space, with two opposing subunits shown as ribbons and their neighbors shown as transparent surfaces. Sites of pathogenic mutations are highlighted with colored spheres on the C-alpha atoms of one subunit: mild or BFNE (blue), uncertain severity (purple), severe or EE (red). Where more than one mutation occurs at a single position, the residue is colored by the most severe phenotype. (e) Location of amino acids mutated in mild or BFNE (blue), uncertain severity (purple), or severe or EE (red) in S4, the pore loop and S6 of Kv7.2.
In contrast to the evenly distributed silent mutations, pathogenic single amino acid mutations are concentrated at transmembrane segments S1 to S6, the pore loop, and intracellular helices A and B of Kv7.2 (Fig. 1b). To test if this trend was statistically significant, we developed a resampling algorithm titled Mutation Hotspot Finder (MHF). This algorithm was applied under the null hypothesis that pathogenic mutations are equally observed at every residue of a functional domain in the full-length Kv7.2 protein when there is no further association between the mutations and the domains. Because our MHF examines the association between the pathogenic variants and the functional domains, we used 130 single amino acid mutations and excluded nonsense and frameshift mutations that truncate one or more functional domains in Kv7.2. This analysis revealed that epilepsy mutations are significantly clustered at the voltage sensing S4, the pore loop, and S6 of Kv7.2 (p < 0.005), whereas silent and nonpathogenic mutations did not cluster at any of the functional domains (Fig. 1b, Supplementary Table S2). Importantly, epilepsy mutations of Kv7.2 were significantly associated with the “severe or EE” group (p < 0.001) and not the “mild or BFNE” and “uncertain severity” groups (Fig. 1a–e, Supplementary Table S3).
Our MHF analysis also revealed that helix B and helix B-C linker have significantly more pathogenic mutations (p < 0.01) than other domains within the Kv7.2 C-terminal tail due to the clustering of “severe or EE” mutations (p < 0.05)(Fig. 2a–d, Supplementary Tables S2–3. Since Kv7.2 binds to CaM through helices A and B (Fig. 2e)28,32, we next tested if the clinical severity of epilepsy mutations is associated with the extent to which Kv7.2 variants bind to CaM. Both “mild or BFNE” and “severe or EE” mutations located at helices A and B decreased the CaM binding energy of Kv7.2 (Fig. 2e,f). Furthermore, EE mutations occur at the positively charged residues in the distal portion of helix B and the helix B-C linker away from the CaM contact site in the modeled Kv7.2 structure (Fig. 2a,g). These results suggest that disruption of CaM binding alone cannot explain why “severe or EE” mutations were selectively enriched at helix B and the helix B-C linker.

Epilepsy mutations cluster at CaM-binding helix B and the helix B-C linker of Kv7.2. (a) Tetrameric human Kv7.2 (ribbons) in complex with four CaM subunits (transparent green surfaces) was modeled based on the structure of Xenopus Kv7.1 (Protein Data Bank: 5VMS)28. Sites of pathogenic mutations in Kv7.2 C-terminal tail are highlighted with colored spheres on the C-alpha atoms of one subunit: mild or BFNE (blue), uncertain severity (purple), severe or EE (red). In addition to epilepsy mutation hotspots (S4, the pore loop, and S6), “severe or EE” mutations cluster near the inner leaflet of the plasma membrane and the C-terminus of S6 at the base of the gate. (b,c) The MHF statistical algorithm on the intracellular C-terminal tail of Kv7.2 identified helix B and the helix B-C linker as hotspots for all pathogenic epilepsy mutations (brackets, **p < 0.01, ***p < 0.005, Supplementary Table S2) (b) and EE mutations (brackets, *p < 0.05, ***p < 0.005, Supplementary Table S3) (c). (d) Location of amino acids mutated in mild or BFNE (blue), uncertain severity (purple), or severe or EE (red) in helix A containing a consensus IQ motif for binding CaM (underlined), helix B, and the helix B-C linker of Kv7.2. (e) Mutated amino acids are highlighted on a model of Kv7.2 helix A and B (grey) bound to Ca2+-CaM (green), which was modeled after the crystal structure of chimeric Kv7.3 helix A - Kv7.2 helix B protein in complex with Ca2+-CaM (Protein Data Bank: 5J03)32. Side chains for residues with pathogenic mutations are colored spheres: mild or BFNE (blue), uncertain severity (purple), severe or EE (red). (f) Predicted changes in Ca2+-CaM binding energy of pathogenic Kv7.2 missense mutations within helices A and B. The higher the energy the weaker the predicted affinity for Ca2+-CaM. (g) Positively charged basic residues at proximal helix A, distal helix B and the helix B-C linker (purple) are located close to basic residues from the S2–3 linker (yellow), S4, S6 (blue), and CaM (green).
Voltage-dependent activation of homomeric Kv7.2 channel is disrupted by selected EE mutations in epilepsy mutation hotspots
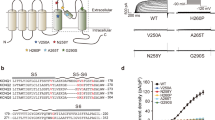
To test if EE variants within the mutation hotspots disrupt key functional protein domains of Kv7.2, we selected four EE mutations which have not been previously characterized: L203P at the voltage-sensing S423, L268F at the pore loop26, and K552T and R553L at helix B22,24 (Fig. 3a,b). To determine their effects on voltage-dependent activation of homomeric Kv7.2 channels, we performed whole-cell patch clamp recording in Chinese hamster ovary (CHOhm1) cells, which display very low expression of endogenous K+ channels and depolarized resting membrane potential of −10 mV12,33. Application of depolarizing voltage steps from −100 to +20 mV in GFP-transfected CHOhm1 cells produces very little voltage-dependent currents that reverse around −26 mV12,34. In contrast, the same voltage steps in cells transfected with GFP and Kv7.2 wild-type (WT) generated slowly activating voltage-dependent outward K+ currents that reached peak current densities of 17.3 ± 1.1 pA/pF at +20 mV (Fig. 3d,e, Supplementary Fig. S1). The average V1/2 of WT channels (−26.8 ± 2.1 mV) was similar to the previously published value of −25 ± 1.9 mV35. Consistent with increased outward K+ current, cell expressing Kv7.2 displayed hyperpolarized resting membrane potential (−35.5 ± 1.1 mV) and reversal potential (−38.8 ± 1.9 mV) (Supplementary Tables S4–5.

All selected EE mutations variably alter voltage-dependent activation of homomeric Kv7.2 channels and disrupt their current enhancement upon diC8-PIP2 inclusion. (a) Sites of selected EE mutations (L203P, L268F, K552T, and R553L) characterized in this study. These mutations are highlighted with red spheres on the C-alpha atoms of one subunit on the modeled tetrameric human Kv7.2 structure (ribbons) in complex with four CaM subunits (transparent green surfaces). (b) Localization of selected EE mutations are shown in red in the amino acid sequence of Kv7.2 (NP_742105.1). The EE mutations are shown in bold. The critical residues in S4 and the selectivity filter in the pore are underlined. (c–h) Whole cell voltage clamp recordings of macroscopic K+ currents in CHO hm1 cells transfected with GFP and Kv7.2 WT or EE mutants. Cells were held at -80 mV. Currents were evoked by depolarization for 1.5 s from −100 mV to +20 mV in 10 mV increments, followed by a step to 0 mV for 300 ms. To examine PIP2 sensitivity of Kv7.2 channels, the recording was repeated with internal patch pipette solution containing diC8-PIP2 (100 μM) which also contained EGTA to sequester free Ca2+. The raw current traces and data are shown in Supplementary Figs. S1–S2. (c) Immunoblot analyses of CHOhm1 cells reveal both monomeric bands (around 90 kD) and multimeric bands (around 180 and 270 kD) of Kv7.2 proteins. For clarity, cropped gel images are shown. Full-length gels can be found in Supplementary Fig. S7,a. (d) Representative recordings after subtraction of leak currents. Leak current was defined as non-voltage-dependent current from GFP-transfected cells. (e) Average peak current densities at all voltage steps. *p < 0.05, ***p < 0.005 based on one-way ANOVA Fisher’s test. (f) Average peak current densities at -20 mV (left) and + 20 mV (right). p values are computed from one-way ANOVA Tukey test. (g) Normalized conductance (G/Gmax) at all voltage steps. (h) Activation time constant (τ) at + 20 mV. The number of GFP-cotransfected cells that were recorded without diC8-PIP2: Kv7.2 WT (n = 12), L203P (n = 17), L268F (n = 17), K552T (n = 13), or R553L (n = 13). The number of GFP-cotransfected cells that were recorded with diC8-PIP2: Kv7.2 WT (n = 11), L203P (n = 14), L268F (n = 13), K552T (n = 11), or R553L (n = 11). Data shown represent the Ave ± SEM.
Cells expressing GFP and Kv7.2-L203P produced K+ currents with a large depolarizing shift in their voltage dependence and V1/2 and increased activation time constants, decreasing peak current densities at voltage steps up to 0 mV. These cells also displayed depolarizing resting membrane potential (−26.8 ± 2.0 mV) and reversal potential (−22.1 ± 1.2 mV) (Fig. 3d–f, Supplementary Fig. S1, Supplementary Tables S4–5. Surprisingly, their peak current density at +20 mV was larger (27.6 ± 1.88 pA/pF) than that of WT channels despite their slower activation kinetics (Fig. 3c–e, Supplementary Fig. S1). The L268F mutation in the pore loop decreased outward K+ currents through Kv7.2 channels but not their protein level (Fig. 3c–f, Supplementary Fig. S1). While the R553L mutation in distal helix B had no effect on Kv7.2 channels, the K552T mutation reduced both protein and current expression (Fig. 3c–f, Supplementary Fig. S1). The L268F and K552T mutations did not alter voltage-dependence, activation kinetics, and reversal potential of Kv7.2 currents (Fig. 3g,h, Supplementary Tables S4–5).
PIP2-induced potentiation of Kv7.2 current is blocked by selected EE variants
PIP2 is a critical cofactor required for the opening of Kv7 channels14,16,17,36 and is proposed to bind to the intracellular side of S4, the S2-S3 and S4-S5 linkers, and intracellular region from pre-helix A to the helix B-C linker11,12,13,14,28,36,37,38. Therefore, we next tested if selected EE mutations alter gating modulation of Kv7 channels by PIP2. Previous studies have shown that the activation of Kv7 channels is far from saturated by the endogenous membrane level of PIP239 and that supplying exogeneous PIP2 can enhance single-channel open probability and whole-cell current densities of homomeric Kv7.2 channels12,14,37,40.
Indeed, inclusion of diC8-PIP2 (100 μM) in the intracellular pipette solution increased K+ currents through Kv7.2 WT channels by 2-fold and caused a modest left shift in voltage-dependence (Fig. 3d–f, Supplementary Figs. S1–2) as previously shown12,14,37. Surprisingly, all selected EE mutations abolished diC8-PIP2-induced potentiation of Kv7.2 channels and hyperpolarizing shift in their voltage dependence, resulting in a significant reduction in their current densities compared to WT channels in the presence of diC8-PIP2 (Fig. 3d–f, Supplementary Figs. S1–2, Supplementary Table S5).
To increase cellular PIP2 levels, we transfected phosphatidylinositol-4-phosphate 5- kinase (PIP5K), which catalyzes the formation of PIP2 via the phosphorylation of phosphatidylinositol-4-phosphate41. Consistent with previous reports11,40,42, co-expression of PIP5K increased K+ currents through Kv7.2 WT channels with a hyperpolarizing shift in their voltage dependence. Consistent with the recording with diC8-PIP2 inclusion (Fig. 3), this effect was absent in Kv7.2 channels containing L268F, K552T, and R553L variants (Supplementary Fig. S3), indicating that these mutations abolished current potentiation of Kv7.2 channels upon increasing cellular PIP2 levels.
Modeled Kv7.2 structure and molecular dynamics simulation suggest that selected EE mutations reside in PIP2 binding regions of Kv7.2
To investigate if selected EE mutations are located in PIP2-binding regions, we compared our modeled Kv7.2 structure bound to CaM and the published structure of TRPV1 channel embedded in lipid nanodiscs with phosphatidylinositol bound (PDB: 5irz) (Fig. 4a)43. In the modeled Kv7.2 structure, the voltage-sensor (S1-S4) and the pore domain of Kv7.2 form the hydrophobic cavity where L203 and L268 are located (Fig. 4a). Similar to the binding of phosphatidylinositol to TRPV1 channel (Fig. 4a)43, the fatty acid tails of amphiphilic PIP2 are most likely embedded in this hydrophobic cavity of Kv7.2 where L203P and L268F mutations reside. Furthermore, the bottom of the voltage-sensor (S1-S4) together with pre-helix A, helix B and the helix B-C linker of Kv7.2 form a highly basic environment favorable for binding the phosphate headgroup of PIP2 (Fig. 4a), consistent with previous studies in Kv7.128.

Modeled Kv7.2 structure and molecular dynamics simulation suggest that selected EE mutations reside in PIP2 binding regions of Kv7.2. (a) Homology structure of Kv7.2 from Fig. 3a and its surface charge representation were compared side-by-side with the cartoon representation of the TRPV1 structure in nanodiscs (PDB: 5irz)43. The cross-section view of Kv7.2 with electrostatic surface showing basic surface in blue, acidic surface in red and nonpolar surface in white. The surface charge representation reveals the nonpolar cavities between the voltage-sensor and the pore domain and indicates that parts of the voltage-sensor, S6, pre-helix A, helix B and helix B-C linker form two basic pockets. (b) The open-state conformation of Kv7.2 channel was embedded in the lipid bilayer. An R553 residue from the helix B of each subunit is represented as a purple sphere. Each subunit is labeled in green, blue, light grey or orange. (c) Sample trajectory of PIP2 in the modeled Kv7.2 structure viewed from the bottom (cytoplasmic) side in Fig. 4b. The initial locations of PIP2 at the beginning of the molecular dynamics (MD) simulations are indicated in red, and the final positions of PIP2 are labeled in blue. The arrow shows the direction of PIP2 diffusion in MD simulations. (d) The distance between two phosphate groups of PIP2 (P4 and P5) and the R553 residue in helix B of Kv7.2 resulted from two simulations. In the first stimulation, 2 out of 4 Kv7.2 monomers (monomers 2 and 3) displayed close contact with P4 and P5 of PIP2. In the second simulation, 3 out of 4 monomers (monomers 1, 3, and 4) interacted with P4 and P5 of PIP2. (e) Representative snapshot of the PIP2 interacting with K552-R553-K554 in distal helix B of Kv7.2 (Supplementary Video 1). The confirmation of non-equilibrium simulation and the stability of the modeled open-conformation Kv7.2 channel structure is shown in Supplementary Fig. 4.
To test if PIP2 interacts with K552 and R553 in distal helix B, we performed molecular dynamics (MD) simulation. We constructed a homology model of the CaM-bound closed state conformation of Kv7.2 using the structure of Kv7.1 (PDB: 5VMS)28 as a template, and employed targeted MD to model the open-state conformation of Kv7.2 in the explicit lipid bilayers containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and PIP2 lipids. To extensively sample the lipid-protein interactions, we constructed two independent simulation systems, each containing seven PIP2 molecules randomly placed around Kv7.2 without CaM in both outer and inner membrane leaflets (~2.2% PIP2) (Fig. 4b). Within the time frame of the simulations, PIP2 molecules diffused from the periphery of the Kv7.2 structure towards its central region (Fig. 4c).
To examine PIP2 binding to helix B, we measured the distance between the center of mass of R553 from each monomer and that of the phosphate groups at position 4 and 5 of PIP2 (Fig. 4d). Binding of PIP2 molecules towards R553 was observed in 2 out of 4 monomers in the first simulation and in 3 out of 4 monomers in the second simulation (Fig. 4d). In both simulations, PIP2 molecules interacted with K552-R553-K554 within 100 ns and remained stably bound throughout the simulations (Fig. 4e, Supplementary Video 1, Supplementary Fig. S4), consistent with previous in vitro biochemical studies and molecular docking simulations that demonstrated PIP2 binding to the corresponding residues in the C-terminal helix A-B fragments of Kv7.137. These findings suggest that K552T and R553L mutations are located in helix B of Kv7.2 that interacts with the phosphate head group of PIP2.
To test if selected EE mutations alter PIP2 affinity, we examined the Kv7.2 current decay upon PIP2 depletion induced by activation of voltage-sensitive phosphatase (VSP)11,42,44. In CHOhm1 cells coexpressing danio rerio VSP11, the 10s-depolarization step at voltages from +40 mV decreased peak currents of Kv7.2 channels, reaching the maximal decay of 53.2 ± 4.0% at +100 mV (Supplementary Fig. S5). Current decay of Kv7.2-K552T was greater than that of WT at +40 mV but was comparable to that of WT from +60 to +100 mV (Supplementary Fig. S5), suggesting that the K552T mutation modestly decreased PIP2 affinity to Kv7.2. Interestingly, the same depolarization steps delayed current decay of Kv7.2 channels containing L203P, L268F, and R553L mutations (Supplementary Fig. S5), indicating their reduced sensitivity to PIP2 depletion.
Selected EE variants decrease current expression of heteromeric Kv7 channels and their current potentiation by diC8-PIP2
Since KCNQ2-associated EE is an autosomal dominant epileptic syndrome, we repeated voltage-clamp recording in CHOhm1 cells transfected with plasmids for Kv7.3, wild-type Kv7.2, and mutant Kv7.2 at a 2:1:1 ratio as described35 (Fig. 5, Supplementary Figs. S6–7). Although the L203P variant induced a large depolarizing shift in voltage-dependence of homomeric channels (Fig. 3d–g), heteromeric L203P mutant channels were indistinguishable from WT channels (Fig. 5a–d). Similar to homomeric channels (Fig. 3), heteromeric channels containing mutations L268F and K552T but not R553L produced significantly less current than WT channels without changing their voltage dependence (Fig. 5a–d, Supplementary Table S5). The L268F variant also increased their activation kinetics (Fig. 5e). None of the tested mutations affected total protein expression of Kv7.2 and Kv7.3 (Fig. 5f).

The L268F and K552T mutations decreased current expression of heteromeric Kv7 channels whereas all selected EE mutations disrupted their current potentiation by diC8-PIP2 inclusion. Whole cell patch clamp recordings were measured using the voltage clamp protocol described in Fig. 3 from GFP-positive CHOhm1 cells cotransfected with Kv7.3 and Kv7.2 WT (1:1 ratio) or Kv7.3, Kv7.2 WT, Kv7.2 mutant (2:1:1 ratio). The raw current traces and data are shown in Supplementary Figs. S6 and S7. (a) Representative leak-subtracted current traces. (b) Average leak-subtracted peak current densities at all voltage steps. *p < 0.05, ***p < 0.005 based on one-way ANOVA Fisher’s test. (c) Average leak-subtracted peak current densities at -20 mV (top) and at + 20 mV (bottom). p values are computed from one-way ANOVA Tukey test. (d) Normalized conductance (G/Gmax) at all voltage steps. (e) Activation time constant (τ) at + 20 mV. The number of GFP-cotransfected cells that were recorded without diC8-PIP2: Kv7.2 WT (n = 14), L203P (n = 15), L268F (n = 18), K552T (n = 15), or R553L (n = 15). The number of GFP-cotransfected cells that were recorded with diC8-PIP2: Kv7.2 WT (n = 12), L203P (n = 12), L268F (n = 16), K552T (n = 14), or R553L (n = 15). (f) Immunoblot analyses of CHOhm1 cells co-transfected with Kv7.2 wild-type or mutant and Kv7.3 reveal both monomeric bands (around 90 kD) and multimeric bands (around 180 kD, 270 kD, and 370 kD) of Kv7.2 proteins. For clarity, cropped gel images are shown. Full-length gels can be found in Supplementary Fig. S8,b. Data represent the Ave ± SEM. ***p < 0.005.
When diC8-PIP2 was added in the intracellular pipette, all tested EE mutations significantly decreased current densities of heteromeric channels at +20 mV compared to WT without altering their voltage-dependence (Fig. 5a–d, Supplementary Table S5) and their activation time constant was increased by L203P and K552T mutations (Fig. 5e). Importantly, all tested EE variants abolished PIP2-induced current potentiation of heteromeric channels (Fig. 5a–c, Supplementary Figs. S6–7).
Selected EE variants variably decrease axonal surface expression of heteromeric Kv7 channels
The physiologically relevant current through Kv7 channels is controlled by both their function and expression at the neuronal plasma membrane. Given that Kv7.2 interaction with CaM and Kv7.3 are critical for axonal surface expression of Kv7 channels9,45, we next tested if selected EE variants of Kv7.2 affect interaction with CaM and Kv7.3 and axonal targeting of Kv7 channels (Figs. 6–7, Supplementary Figs. S9–11). Coimmunoprecipitation assay in HEK293T cell lysate12,45 revealed that the K552T and R553L mutations in helix B decreased Kv7.2 binding to YFP-tagged CaM whereas the mutations including L203P in S4 and L268F in the pore loop had no effect (Fig. 6a,b). None of the tested mutations affected Kv7.2 interaction with Kv7.3 (Fig. 6c,d). Total Kv7.2 expression was also reduced by the L203P and K552T variants in cells co-expressing CaM but not Kv7.3 (Fig. 6).

The K552T and R553L mutations reduced CaM binding to Kv7.2 whereas none of the tested EE mutations affected Kv7.3 interaction with Kv7.2. (a,b) Co-immunoprecipitation of YFP-CaM with wild-type Kv7.2 (WT) or Kv7.2 containing EE mutations in the presence of EGTA. (a) Representative immunoblots of HEK293T cells expressing Kv7.2 and YFP-CaM. For clarity, cropped gel images are shown. Full-length gels can be found in Supplementary Fig. S9. (b) Quantification of immunoblots: untransfected/None (n = 6), YFP-CaM (n = 6), YFP-CaM cotransfection with Kv7.2 WT (n = 6), L203P (n = 3), L268F (n = 3), K552T (n = 3), or R553L (n = 3). (c,d) Co-immunoprecipitation of HA-Kv7.3 with wild-type Kv7.2 (WT) or Kv7.2 containing EE mutations in the presence of EGTA. (c) Representative immunoblots of HEK293T cells expressing Kv7.2 and Kv7.3. For clarity, cropped gel images are shown. Full-length gels can be found in Supplementary Figs. S9–10. (d) Quantification of immunoblots: untransfected cells (None: n = 3), or cells transfected with HA-Kv7.3 (n = 4), HA-Kv7.3 and Kv7.2 WT (n = 4), L203P (n = 3), L268F (n = 3), K552T (n = 4), or R553L (n = 3). GAPDH served as a loading control. Both monomeric Kv7.2 bands (around 90kD, arrows) and multimeric Kv7.2 bands (around 180 kD, 270 kD, and 370 kD) are observed in the IP samples and lysate in (a,c). Data represent the Ave ± SEM (*p < 0.05, ***p < 0.005 against Kv7.2 WT).

L268F, K552T, and R553L mutations decreased enrichment of heteromeric Kv7 channels at the axonal surface in cultured hippocampal neurons. Immunostaining of surface Kv7.3 and total Kv7.2 containing an extracellular hemagglutinin epitope (HA-Kv7.3) in healthy hippocampal neurons cotransfected with Kv7.2 WT or Kv7.2 with EE mutations L203P, L268F (a–e), K552T, and R553L (f–j). (a,f) Representative images of surface HA-Kv7.3 (Upper) as pseudo-color that display differences in the surface HA intensities from high (red) to low (blue). Total Kv7.2 (Middle-inverted gray) and the AIS identified by antibodies for phospho IκBα-Ser32 (14D4) (Lower-fluorescence) are shown in same neurons. Arrows mark the AIS. Scale bars: 25 μm. (b,g) Normalized background-subtracted mean intensities of surface HA fluorescence from neurons expressing WT and EE mutant HA-Kv7.3/Kv7.2 channels. The number of transfected neurons that were analyzed in Fig. 7b: WT (n = 41), L203P (n = 39), L268F (n = 34), untransfected (UT) (n = 20). The number of transfected neurons that were analyzed in Fig. 7g: WT (n = 42), K552T (n = 34), R553L (n = 21), untransfected (UT) (n = 19). The raw data from 3 independent experiments are shown in Supplementary Fig. S11. (c,h) Surface HA intensity ratio at distal axon over dendrite. (d,i) Background-subtracted mean intensities of total Kv7.2 fluorescence in Kv7.2-transfected neurons and untransfected neurons (UT, blue dotted lines). The number of transfected neurons that were analyzed in Fig. 7d: Kv7.2 WT (n = 14), L203P (n = 15), L268F (n = 14), UT (n = 14). The number of transfected neurons that were analyzed in Fig. 7i: Kv7.2 WT (n = 17), K552T (n = 23), R553L (n = 14), UT (n = 13). (e,j) Background-subtracted mean intensities of surface HA fluorescence from the transfected neurons treated with vehicle (CTL) control or dynamin inhibitory peptide (DIP). The number of transfected neurons that were analyzed in Fig. 7e: WT + CTL (n = 14), WT + DIP (n = 13), L268F + CTL (n = 8), L268F + DIP (n = 8). The number of transfected neurons that were analyzed in Fig. 7j: WT + CTL (n = 8), WT + DIP (n = 13), L268F + CTL (n = 6), L268F + DIP (n = 7). Data represent the Ave ± SEM (*p < 0.05, **p < 0.01, ***p < 0.005 against WT channels).
To test if selected EE mutations of Kv7.2 affect surface density of Kv7 channels, we transfected rat dissociated hippocampal cultured neurons with Kv7.3 containing an extracellular HA epitope (HA-Kv7.3) and performed surface immunostaining of HA-Kv7.3 as described9,12,45 (Fig. 7, Supplementary Fig. S11). In cultured neurons, transfection of HA-Kv7.3 alone yields negligible surface expression of HA-Kv7.39. However, co-transfection of Kv7.2 WT results in robust HA-Kv7.3 expression on the plasma membrane of the AIS and distal axons compared to the soma and dendrites (Fig. 7a,b)9,12,45, resulting in a surface fluorescence “Axon/Dendrite” ratio of 3.9 ± 0.49 (Fig. 7c).
Although the L203P mutation in S4 did not affect surface and total expression of HA-Kv7.3/ Kv7.2 channels (Fig. 7a–d), the L268F mutation in the pore loop abolished their preferential enrichment at the axonal surface by severely decreasing their axonal surface density (surface Axon/Dendrite ratio = 0.85 ± 0.10, Fig. 7a–c) and also reduced intracellular Kv7.2 expression in the axon (Fig. 7d). The K552T and R553L mutations in helix B significantly reduced surface expression of heteromeric channels in both distal axon and dendrites (Fig. 7f–i), resulting in similar surface Axon/Dendrite ratios as the WT channels (Fig. 7h).
Disruption of CaM binding to Kv7.2 has been shown to impair axonal enrichment of Kv7 channels by inhibiting their trafficking from the endoplasmic reticulum (ER)45. The ability of the L268F mutation to impair axonal Kv7 surface expression without affecting Kv7.2 binding to CaM or Kv7.3 (Figs. 6, 7a–d) suggests a different mechanism. To test if the L268F mutation reduces axonal enrichment of Kv7 channels by increasing their endocytosis, we used dynamin inhibitory peptide (DIP, 50 μM) which blocks dynamin-dependent endocytosis in cultured hippocampal neurons46. The DIP treatment for 45 min induced a small increase in surface HA-Kv7.3/ Kv7.2 WT and L268F mutant channels in the soma and dendrites but not axons (Fig. 7e,j), indicating their basal endocytosis in somatodendritic membrane. Although the DIP treatment had no effect on K552T mutant channels, the same treatment modestly increased axonal surface expression of L268F mutant channels (Fig. 7e,j). However, this increase did not reach the axonal level of WT channels (Fig. 7e), suggesting that increased endocytosis is not the main cause for reduced axonal surface expression of L268F mutant channels.
Discussion
In this study, we investigated the pathogenetic mechanisms underlying de novo EE mutations of Kv7.2. Visual inspection in Kv7.2 primary sequence has suggested the enrichment of EE variants at S4, the pore domain from S5 to S6, and helices A and B12,25,47. Clustering of epilepsy mutations in the ion transport domain of Kv7.2 has also been detected by identifying its variation-intolerant genic sub-regions48. Our novel MHF statistical algorithm interpreted in the context of modeled Kv7.2 atomic structure (Figs. 1–2) supports these earlier observations. We discovered that “severe or EE” missense variants cluster at S4, the pore loop that contains the selectivity filter, S6, helix B, and the helix B-C linker of Kv7.2 (Fig. 1). A recent study by Goto et al., reported that the EE missense variants cluster at the pore domain, S6, and pre-helix A of Kv7.249. The regional differences in mutation clusters between our study and Goto et al., could be attributed to the use of different algorithms and databases (ExAC and GnomAD) as sources for non-pathogenic mutations. Nonetheless, both studies identified the pore domain and S6 as hotspots of EE variants, supporting the functional importance of these regions.
However, sequence variant interpretation from the prediction algorithms should be used carefully. The presence of both gain-of-function and loss-of-function EE variants in S447,50,51,52 suggest that it is not straight forward to predict the genotype-phenotype correlation of EE. In addition, both EE and BFNE variants exist in each of the identified hotspots and even at the same codon18,49, suggesting that different amino acid substitutions at the same residue may cause diverse effects on Kv7 channels and the clinical severity of epilepsy. Although this challenge has been addressed recently by Percent Accepted Mutation (PAM)30 algorithm based on amino acid substitution in evolution49, the in vivo impact of a mutation is difficult to predict in patients due to their variable exposures to genetic and environmental factors. Thus, the use of multiple in-silico tools and comprehensive experimental analyses of epilepsy variants are needed to understand their effects on Kv7 channels ex vivo and in vivo.
Our functional characterization of de novo EE variants selected from the mutation hotspots revealed that each mutation impaired the function of its associated protein domain within Kv7.2. The L203P mutation in the main voltage sensor S4 induced a large depolarizing shift in voltage-dependence and slowed activation kinetics of homomeric Kv7.2 channels (Fig. 3) but had no effect on heteromeric channels (Fig. 5). In contrast, the L268F mutation in the pore loop decreased current densities of both homomeric and heteromeric channels without affecting their voltage dependence (Figs. 3, 5). K552T and R553L mutations in CaM-binding helix B decreased the interaction between CaM and Kv7.2 (Fig. 6), which is shown to play critical roles in M-current expression and inhibition of hippocampal neuronal excitability53. Current suppression of homomeric channels is a common feature of EE variants of KCNQ254. Given the overlapping distribution of Kv7.2 and Kv7.3 throughout the hippocampus and cortex4, the dominant negative functional effects of L268F and K552T variants on heteromeric channels (Figs. 3, 5) are expected to induce neuronal hyperexcitability and may underlie severe symptomatic EE with drug-resistant seizures, psychomotor delay, and profound intellectual disability22,26.
Interestingly, our modeled Kv7.2 structure revealed that the distal helix B and helix B-C linker come together with pre-helix A to form a positively charged surface close to the voltage sensor S1-S4 and the base of S6 (Fig. 2). Mutations of basic amino acid residues including H328C, R325G, and R333W at pre-helix A and R560W at the helix B-C linker of Kv7.2 have been shown to impair regulation of Kv7.2 currents by PIP211,12,14, which couples voltage sensor activation to the opening of the gate28,36. Our MD simulations revealed that K552 and R553 in distal helix B bind to the negatively charged head group of PIP2 (Fig. 4). Importantly, K552T and R553L mutations impaired current enhancement of both homomeric and heteromeric channels upon acute or tonic increase in PIP2 (Figs. 3, 5, Supplementary Fig. S3), suggesting that these mutant channels cannot respond to the changes in cellular PIP2. Since stable binding of CaM to Kv7.2 is crucial for PIP2 modulation of neuronal Kv7 channels33, a decrease in CaM binding (Fig. 6) may also contribute to the loss of PIP2-induced current enhancement of K552T and R553L mutant channels (Figs. 3, 5).
The impairment of PIP2-induced current enhancement of L268F mutant channels was unexpected (Figs. 3, 5–7) because it is unlikely for the hydrophobic L268 to bind a negatively charged head group of PIP2. A comparison between modeled Kv7.2 structure and TRPV1 structure (Fig. 4a) suggests that the amphiphilic chains of PIP2 may extend to the hydrophobic cavity created by the voltage-sensors S1-S4 and the pore domain of Kv7.2. We speculate that the L268F mutation at this hydrophobic interface impair Kv7.2 interaction with PIP2. Furthermore, analogous residue for L268 in the bacterial KcsA structure can secure the proper opening size of the pore55. Therefore, it is also possible that the L268F mutation may disrupt PIP2-dependent coupling to the pore opening36.
Several studies including our own have investigated PIP2 affinity of Kv7 function by inclusion of diC8-PIP2 in the intracellular pipette solution12,14,33,37. However, caution must be exercised in interpreting their results. Potentiation of Kv7.2-L203P current by tonic elevation of cellular PIP2 upon PIP5K expression but not acute application of diC8-PIP2 (Fig. 3, Supplementary Fig. S3), suggest that diC8-PIP2 inclusion may not readily potentiate the mutant channels that displayed very slow activation kinetics (Fig. 3). Furthermore, the loss of diC8-PIP2-induced current potentiation can be either due to decreased PIP2 affinity or saturated level of interaction with PIP2 at low PIP2 concentration. We found that the K552T mutation modestly weakens PIP2 affinity, whereas other mutant channels were resistant to PIP2 depletion (Supplementary Fig. S5). Considering multiple proposed PIP2 binding sites in Kv7.2 including S2-S3 and S4-S5 linkers, pre-helix A, helix B, and helix A-B and helix B-C linkers11,12,13,14,15, selected EE mutations may cause conformational change that weakens or enhances PIP2 affinity to other regions within Kv7.2. As Suh and Hille (2008) pointed out56, it is not straight forward to determine PIP2 affinity of mutant channels by assessing their currents after manipulation of PIP2 level. Nonetheless, the lack of current potentiation upon increasing cellular or exogenous PIP2 (Fig. 3, Supplementary Fig. S3) and the location of the EE mutated residues in a region of Kv7.2 that binds to fatty acid tails or polar headgroups of PIP2 (Fig. 4) strongly suggest that there are multiple ways by which the selected EE variants may influence PIP2 interaction with Kv7 channels and reduce their currents.
Our investigation of selected EE variants on neuronal expression of Kv7 channels revealed that K552T and R553L mutations in helix B reduced enrichment of Kv7 channels at the axonal surface (Fig. 7), supporting previous observations that the degree of CaM interaction with Kv7.2 correlates with the overall amount of Kv7 channels at the axonal surface12,45. Unexpectedly, the L268F mutation at the pore loop severely decreased both surface and intracellular expression of heteromeric channels in axons without affecting Kv7.2 binding to CaM or Kv7.3 (Figs. 6–7), demonstrating the importance of studying Kv7 expression in neurons. Decreased axonal expression of Kv7.2-L268F and minor effects of endocytosis inhibition (Fig. 7) suggest that a severe reduction of L268F mutant channels at the axonal surface is caused by a CaM- and endocytosis-independent mechanism. Given that misfolded membrane proteins are retained in the ER for chaperone-assisted refolding57, the L268F mutation may cause a folding defect that facilitates ER retention and disrupts forward trafficking of heteromeric channels to the axon.
Taken together, we identified EE mutation hotspots in Kv7.2 and discovered that each variant selected from these hotspots impairs the function of its associated protein domain and displays a combination of defects in voltage- and PIP2-dependent activation and axonal expression of Kv7 channels (Fig. 8). Such combinations of defects may decrease Kv7 current and its ability to inhibit neuronal excitability in neonatal brain5,12, as conditional deletion of Kv7.2 during embryonic development results in hippocampal and cortical hyperexcitability and spontaneous seizures in mice7. Continued optimization of prediction algorithms and experimental interrogations to understand pathophysiology of Kv7-associated EE will aid the development of better therapeutic strategies for this disease.

A summary for pathogenetic mechanisms of epilepsy mutation hotspots in Kv7.2. Our functional characterization of EE mutations selected from the epilepsy mutation hotspots of Kv7.2 show that they not only disrupt the functions of key protein domains they reside in, but also reduce PIP2-dependent current potentiation and axonal surface expression of Kv7 channels. The missense EE mutations are highlighted with red spheres on the C-alpha atoms of one subunit on the modeled tetrameric human Kv7.2 structure (ribbons) in complex with four CaM subunits (transparent green surfaces). The list of these EE variants can be found in Supplementary Table S7.
Materials and Methods
The resampling statistical algorithm
A resampling algorithm titled Mutation Hotspot Finder (MHF) was developed to search for mutation clusters that localize to the functional domains in human Kv7.2 (GenBank: NP_742105.1). The complete MHF algorithm can be found in GitHub repository (https://github.com/jerrycchen/MutationHotspotFinder). The functional domains were annotated based on multiple published sources3,28,30,32 and the RIKEE database (www.rikee.org). Briefly, the MHF algorithm compares the observed numbers of mutations against the expected numbers of mutations, and computes corresponding statistical significance through bootstrapping within each pre-specified protein functional domains. The following sections explain the MHF algorithm in detail.
The \(S\) denote the set of all observed single amino acid mutations for the whole sequence of protein \(X\) (e.g. the whole sequence of Kv7.2), or the subset of the sequence of protein \(X\) (e.g. the intracellular C-terminal tail of Kv7.2). \(|S|=length(S)\) indicates the number of unique mutations in \(S.\)
The \({D}_{j},\,j\in \{1,\,2,\ldots ,J\},\) denote the number of mutations in \(S\) that fall into the functional domain \(J\) of protein sequence \(X\). Among 14 functional domains in Kv7.2 (Supplementary Table S2), \({D}_{1}\) is the number of mutations in the S1, and \({D}_{2}\) is the number of mutations in the S2, and etc. Among 9 functional domains in intracellular Kv7.2 C-terminal tail (Supplementary Table S2), \({D}_{1}\) is the number of mutations in the pre-Helix A, and \({D}_{2}\) is the number of mutations in the Helix A, and etc.
The MHF algorithm assumes that mutations are equally observed at each amino acid position within functional domains when there is no further association between the mutations and the domains. Due to this null hypothesis58, the application of MHF algorithm is restricted to single amino acid mutations, and is not suitable for mutations outside of the coding sequence as well as nonsense or frameshift mutations that delete one or more function domains. Under such assumption, we can randomly draw samples (i.e. bootstrap), with size = \(|S|\), from the sequence \(X\) to construct the bootstrapped “mutation sets” in order to simulate the distribution of mutations.
For iteration \(\,k\) of bootstrapping, the \(\tilde{S}(k)\) denote the bootstrapped mutation set where \(k\in \{1,\,2,\ldots ,K\}\). In the context of this paper, we ran 10,000 iterations of bootstrapping (i.e. \(K=10,000\)). The \({\tilde{D}}_{j}(k)\) is defined as the number of mutations in \(\tilde{S}(k)\) that fall into the functional domain \(j\) of protein sequence \(X\). The empirical expected number of mutations within each domain \(j\) was constructed from
The empirical P-values (\({\hat{P}}_{j}\)) were computed from a right-tailed test to measure the level of statistical significance on the proportion of the bootstrapped mutation sets that had more mutations than the observed mutation set \(S\) at each individual protein functional domain.
where \(I(\cdot )\) is the Indicator function.
The computed P-values were adjusted for multiple comparisons (\(J\) times) using Bonferroni’s correction. Mutations were visualized and mapped to Kv7.2 and Kv7.3 primary structures with MutationMapper (http://www.cbioportal.org/mutation_mapper.jsp Mapper). Fisher’s Exact Test was implemented using the standard fisher.test() function in R (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/fisher.test.html).
Structure modeling and visualization
The S1-S6 sequence of Kv7.2 (R75-Q326) was threaded to the cryo-EM structure of Xenopus laevis Kv7.1 bound to CaM (PDB: 5VMS)28. The loops of Kv7.2 (E86-W91 and K255-T263) were rebuilt in FoldIt (https://fold.it/portal). The structure was relaxed in Rosetta software (https://www.rosettacommons.org/software) using two rounds of rotamer sampling followed by side chain and backbone minimization, ending with minimization of all degrees of freedom while maintaining C4 symmetry. The lowest scoring decoy with Root mean square deviation (RMSD) < 2.0 Å was chosen as the final model. The amino acid residues mutated in BFNE and EE are indicated in the Rosetta-based model.
To model the interaction between CaM and Kv7.2 helices A and B, the helix A sequence of Kv7.2 (E322-V367) was threaded to the crystal structure of chimeric Kv7.3 helix A - Kv7.2 helix B in complex with Ca2+-bound CaM (PDB: 5J03)32. The structure was relaxed with Rosetta using two rounds of sequential rotamer, side chain and backbone minimization, followed by rigid body minimization. Mutations were made to the model in Rosetta followed by sequential rotamer, side chain, backbone, and rigid body minimization. The binding energy was calculated from 20 simulations. Structures were visualized using PyMOL 2.0 (Schrödinger, LLC).
DNA Constructs and mutagenesis
EYFP-hCaM was a gift from Dr. Emanuel Strehler (Addgene plasmid # 47603). The plasmid pIRES-dsRed-PIPKIγ90 was a gift from Dr. Anastasios. Tzingounis (University of Connecticut) and was previously described42. Plasmids pcDNA3 with KCNQ3 cDNA (GenBank: NM004519) encoding Kv7.3 (GenBank: NP_004510.1), HA-Kv7.3, and KCNQ2 cDNA (GenBank: Y15065.1) encoding Kv7.2 (GenBank: CAA 75348.1) have been previously described9,12,45. Compared to the reference sequence of Kv7.2 (GenBank: NP_742105.1), this shorter isoform lacks 2 exons which do not harbor pathogenic variants to date. However, the amino acid numbering in the manuscript conforms to the reference sequence of Kv7.2 for clarity. Epileptic encephalopathy mutations (L203P, L268F, K552T, R553L) were generated using the Quik Change II XL Site-Directed Mutagenesis Kit (Agilent).
Electrophysiology
Whole cell patch clamp recordings in Chinese hamster ovary (CHO hm1) was performed as described12. To express homomeric Kv7.2 channels, cells were transfected with pEGFPN1 (0.2 μg) and pcDNA3-Kv7.2 WT or mutant (0.8 μg). To express Kv7.2 channels and PIP5K, cells were transfected with pEGFPN1 (0.2 μg), pIRES-dsRed-PIPKIγ90 (0.45 ug, a kind gift from Dr. A. Tzingounis, U. Conn42), pcDNA3-Kv7.2 WT or mutant (0.45 μg). For the negative control for the PIP5K experiment, the cells were transfected with pEGFPN1 (0.65 μg) and pcDNA3-Kv7.2 WT or mutant (0.45 μg). To express heteromeric channels, cells were transfected with pEGFPN1 (0.4 μg), pcDNA3-Kv7.3 (0.8 μg), pcDNA3-Kv7.2 WT (0.4 μg), or pcDNA3-Kv7.2 WT or mutant (0.4 μg). Leak-subtracted current densities (pA/pF), normalized conductance (G/Gmax), and channel biophysical properties were computed as described12,35 with the exception that V1/2 and the slope factor k were calculated as described35,59 by fitting the plotted points of G/Gmax with a Boltzmann equation G/Gmax = 1/ {1 + exp (V0.5 – Vc) / k}.
To examine the decline of Kv7.2 current upon activation of Dr-VSP, CHO hm1 cells were transfected with pDrVSP-IRES2-EGFP (0.5 μg) and pcDNA3-Kv7.2 WT or mutant (0.5 μg). The pDrVSP-IRES2-EGFP plasmid was a gift from Yasushi Okamura (Addgene plasmid # 80333). Voltage-clamp recording of Kv7.2 current upon depolarization-induced Dr-VSP activation was performed as described60 with an external solution containing 144 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose and 10 mM HEPES (pH 7.4). Patch pipettes (3 – 4 MΩ) were filled with intracellular solution containing 135 mM potassium aspartate, 2 mM MgCl2, 1 mM EGTA, 0.1 mM CaCl2, 4 mM ATP, 0.1 mM GTP and 10 mM HEPES (pH 7.2). Cells were held at -70 mV and 10 s step depolarizations were applied in 20 mV steps from -20 to +100 mV with 2 min inter-step intervals to allow PIP2 regeneration. The extent of Kv7.2 current decay upon Dr-VSP activation during 10 s depolarization was measured as the ratio of current at 10 s over peak current at each voltage step.
Molecular dynamics simulation
For modeling of open and closed states of Kv7.2, the closed-state conformation of KCNQ2 in calmodulin-bound form was modeled based on the recent cryo-EM structure of Kv7.1 (PDB code 5VMS)28. Multiple sequence alignment of the template and KCNQ2 sequence was performed by using TCoffee web server (https://www.ebi.ac.uk/Tools/msa/tcoffee/). After the alignment, the homology model of closed-state conformation was built with MODELLER61. The stability of the closed-state conformation of Kv7.2 was tested by performing all-atom molecular dynamics (MD) simulations in explicit lipid bilayer.
In order to model the open-state conformation of Kv7.2, we performed non-equilibrium MD simulations. Using our stable closed-state conformation of Kv7.2, we performed 20-ns of Targeted MD (TMD)62 simulations in an explicit lipid bilayer. TMD has been shown to drive the conformational changes by gradually minimizing the RMSD of S4-S5 and S6 helices of the closed-state conformation and the target structure which is Kv1.2/Kv2.1 in open conformation (PDB: 2R9R)63. As major structural changes occur in the pore region of the channel, we applied a restraint (force constant = 250 kcal/mol/Å) on the S4-S5 and S6 helices of each monomer to drive it towards the target state which was defined by a highly homologous Kv1.2/Kv2.1 channel in open-state conformation (PDB code 2R9R)63. The success of TMD was gauged by measuring the backbone RMSD of S4-S5 and S6 helices with respect to the target (Supplementary Fig. S6). Upon completion of TMD, all the structural restraints were released and the stability of the obtained open-state conformation of Kv7.2 was tested by performing MD simulations in explicit lipid bilayer (Supplementary Fig. S6).
For MD simulation, the modeled Kv7.2 without calmodulin was embedded in the lipid bilayer, containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylinositol 4,5-bisphosphate (PIP2) generated using CHARMM-GUI membrane builder64. The initial position of PIP2 was at least 15 Å away from the protein surface. The membrane/protein systems were then solvated with TIP3P water and neutralized with 150 mM KCl.
All the MD simulations were performed with NAMD2.1265 using CHARMM36m force field for lipid/protein66 and a timestep of 2 fs. Long range electrostatic interactions were evaluated with particle mesh Ewald (PME)67 and periodic boundary conditions were used throughout the simulations. Non-bonded forces were calculated with a cutoff of 12 Å and switching distance of 10 Å. During the simulation, temperature (T = 310 K) and pressure (P = 1 atm) (NPT ensemble) was maintained by Nosé-Hoover Langevin piston method68. During pressure control, the simulation box was allowed to fluctuate in all the dimensions with constant ratio in the x-y (lipid bilayer) plane.
Immunoblot analysis
At 48 h post transfection, the CHOhm1 cells were washed with 1X PBS, and harvested in ice-cold lysis buffer containing (in mM): 50 Tris, 150 NaCl, 2 EGTA, 1 EDTA, 1% Triton, 0.5% deoxycholic acid, 0.1% SDS (pH 7.4) supplemented with Halt protease inhibitors (Thermo Fisher Scientific) as described12,45. After 15 min incubation, the cells in lysis buffer were centrifugated at 14,000 x g for 15 min at 4 °C. The lysates were mixed with SDS sample buffer in 1:5 dilution (in mM): containing 75 Tris, 10% SDS, 50 TCEP, 12.5% glycerol, 0.50 EDTA, 0.50 mg/mL Bromophenol Blue. After heating at 75 °C for 30 min, the samples were run on 12% non-gradient and 4–20% gradient SDS-PAGE gels (Bio-Rad), transferred to a polyvinyl difluoride (PVDF) membrane (Immobilon, Millipore), and analyzed by immunoblotting12,45. Briefly, the membranes were blocked in blocking buffer (5% milk, 0.1% Tween-20 in TBS), and incubated with mouse anti-Kv7.2 (1:200 dilution), rabbit anti-Kv7.3 (1:500 dilution) or anti-GAPDH antibody (1:1000 dilution) in wash buffer (1% milk, 0.1% Tween-20 in TBS) overnight at 4 °C. After incubating with horse radish peroxidase-conjugated secondary antibodies in wash buffer for 1 hr, the blots were washed, and treated with Pierce ECL or SuperSignal Pico Plus substrate (Thermo Fisher Scientific #32106, #34577). The immunoblot membranes were immediately imaged with the iBright CL1000 imaging system (Thermo Fisher Scientific). ImageJ software (NIH, http://rsb.info.nih.gov/ij) was used to measure background-subtracted immunoblot band intensities of Kv7.2 and Kv7.3 (monomers, dimers, multimers) and GAPDH as previously decreased12,45. The ratio of Kv7.2/GAPDH and Kv7.3/GAPDH from Kv7.2 WT samples were taken as 100% and the ratio of EE mutant samples were normalized to the ratio of WT samples to obtain % Kv7.2 WT. Antibodies used in immunoblotting include anti-Kv7.2 (Neuromab, N26A/23), rabbit anti-Kv7.2 (Alomone, APC-050), rabbit anti-Kv7.3 (Alomone, APC-051), anti-GAPDH antibodies (Cell Signaling, 2118), donkey anti-rabbit and anti-mouse HRP secondary antibodies (The Jackson Laboratory, 711-035-152, 715-035-150).
Immunoprecipitation
HEK293T cells were plated on 100 mm cell culture dishes (BD Biosciences, 2 × 106 cells per dish) and maintained in Minimal Essential Medium containing 10% Fetal Bovine Serum, 2 mM glutamine, 100 U/mL penicillin and 100 U/mL streptomycin at 37 °C and 5% CO2. At 24 hr post plating, the cells were transfected with plasmids (total 1.6 μg) containing Kv7.2 and EYFP-hCaM (1:1 ratio), using FuGENE6 transfection reagent (Promega). For coimmunoprecipitation studies of Kv7.2 and Kv7.3, the cells were transfected with Kv7.2 and Kv7.3 containing an extracellular hemagglutinin epitope (HA-Kv7.3) (1:1 ratio). At 48 h post transfection, the cells were washed with ice-cold PBS and lysed in ice-cold immunoprecipitation (IP) buffer containing (in mM): 20 Tris-HCl, 100 NaCl, 2 EDTA, 5 EGTA, 1% Triton X-100 (pH 7.4) supplemented with Halt protease inhibitors (Thermo Fisher Scientific). The lysate containing equal amount of proteins were first precleared with Protein A/G agarose beads (100 μL, Santa Cruz) for 1 hr at 4 °C, and then incubated overnight at 4 °C with Protein A/G-agarose beads (100 μL) and rabbit anti-Kv7.2 antibody (2 μg). This amount of anti-Kv7.2 antibody allowed us to immunoprecipitate the equal amount of Kv7.2 proteins and analyze the effects of mutations on the amount of co-immunoprecipitated EYFP-hCaM and HA-Kv7.3. After washing with IP buffer, the immunoprecipitates were eluted with SDS sample buffer by incubating at 75 °C for 10–15 min, and analyzed by western blot analysis with mouse anti-GFP (1:500 dilution), mouse anti-Kv7.2 (1:200 dilution), mouse anti-HA antibodies (1:500 dilution), and rabbit anti-GAPDH antibodies (1:1000 dilution). Antibodies used in coimmunoprecipitation and immunoblotting include anti-Kv7.2 (Neuromab, N26A/23), rabbit anti-Kv7.2 (Alomone, APC-050), anti-GFP, anti-HA, anti-GAPDH antibodies (Cell Signaling, 2955, 2367, 2118), rabbit anti-Kv7.3 (Alomone, APC-051), donkey anti-rabbit and anti-mouse HRP secondary antibodies (The Jackson Laboratory, 711–035–152, 715-035-150).
Immunocytochemistry
All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Illinois Urbana-Champaign and conducted in accordance with the guidelines of the U.S National Institute of Health (NIH). Primary rat dissociated hippocampal cultured neurons prepared from 18-day old embryonic rats were plated on 12 mm glass coverslips (Warner Instruments, 105 cells per coverslip) coated with poly L-lysine (0.1 mg/mL). These neurons were maintained in neurobasal medium supplemented with B27 extract, 200 mM L-glutamine, and 100 U/mL penicillin and streptomycin in a cell culture incubator (37 °C, 5% CO2). At 5 days in vitro (DIV), neurons were transfected with plasmids (total 0.8 μg) containing Kv7.3 with an extracellular hemagglutinin epitope (HA-Kv7.3) and wild-type or mutant Kv7.2 using lipofectamine LTX as described12,45.
Immunostaining for surface HA-Kv7.3 and total Kv7.2 subunits were performed at 48 h post transfection as described12,45. In brief, neurons were washed once with artificial cerebral spinal fluid (ACSF) solution containing (in mM): 10 HEPES, 150 NaCl, 3 KCl, 2 CaCl2, 10 Dextrose (pH 7.4). Neurons were fixed in 4% paraformaldehyde / 4% sucrose in Phosphate buffered saline (PBS) for 8 min, washed with PBS, blocked with 10% normal donkey serum (NDS) in PBS for 1 hr. To label surface HA-Kv7.3, neurons were incubated with rabbit anti-HA antibody (1:500 dilution) in 3% NDS in PBS overnight at 4 °C without permeabilization, followed by incubation with donkey anti-rabbit Alexa488-conjugated secondary antibodies (1:200-1:300 dilution). To label total Kv7.2 and AIS marker, neurons were fixed for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 30 min, and incubated with goat anti-Kv7.2 antibody (1:200 dilution) and rabbit anti-phospho IκBα Ser32 (14D4) antibody (1:500 dilution) or mouse anti-Ankyrin G antibody (1:500) in 3% NDS in PBS at 4 °C overnight. After the PBS wash, the neurons were incubated with donkey anti-goat Alexa594-conjugated secondary antibodies (1:200-1:300 dilution) and anti-rabbit Alexa680-conjugated secondary antibodies (1:200-1:300 dilution) for 2 hr. The coverslips were mounted using Fluorogel anti-fade mounting medium (Electron Microscopy Sciences).
Cell permeable dynamin inhibitory peptide (DIP, Tocris Bioscience Cat. No. 1775) and diluted to 50 μM by artificial cerebrospinal fluid (ACSF). Transfected coverslips were incubated in the DIP or vehicle for 45 minutes before immunostaining. Antibodies used in immunofluorescence staining include anti-HA (Cell Signaling, 3724), anti-Kv7.2 (Santa-Cruz, sc-7793), anti-ankyrin G (Neuromab, 75–146), anti-phospho IκBα Ser32 (14D4) (Cell Signaling, 2859) and Alexa Fluor secondary antibodies (Invitrogen, A10043, A21206, A10038, A11058).
Fluorescence and phase contrast images of transfected neurons were viewed using a Zeiss Axio Observer inverted microscope High-resolution gray scale images of healthy transfected neurons were acquired using a 20X objective with a Zeiss AxioCam 702 mono Camera and ZEN Blue 2.6 software and saved as 16-bit CZI and TIFF files. To compare the fluorescence intensity of the neurons transfected with different constructs, the images were acquired using the same exposure time within one experiment.
The image analyses were performed from the healthy transfected neurons using ImageJ Software as described12,45 and excluded the transfected neurons with broken neurites or soma as well as regions where fasciculation or overlapping processes occurred. The axon was identified as a process that were labeled for the AIS marker 14D4, whereas the dendrites were identified as the processes that were absent for 14D4 in the transfected neurons. ImageJ software was used to trace the all major primary dendrites, the AIS (defined as the first 0–30 μm segment of the axon), and distal axon (defined as the segment between 50 and 80 μm from the beginning of the axon) as 1 pixel-wide line segments, and obtain their mean fluorescent intensities. The perimeter of the neuronal soma was also traced to obtain background-subtracted mean fluorescent intensities of the soma.
Statistical analyses
All analyses are reported as mean ± SEM. Using Origin 9.1 (Origin Lab), the Student t test and one-way ANOVA with post-ANOVA Tukey and Fisher’s multiple comparison tests were performed to identify the statistically significant difference with a priori value (p) < 0.05. The number of separate transfected cells for immunostaining and electrophysiology was reported as the sample size n.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Noebels, J. Pathway-driven discovery of epilepsy genes. Nature neuroscience 18, 344–350, https://doi.org/10.1038/nn.3933 (2015).
Epi, K. C. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221, https://doi.org/10.1038/nature12439 (2013).
Brown, D. A. & Passmore, G. M. Neural KCNQ (Kv7) channels. British journal of pharmacology 156, 1185–1195, https://doi.org/10.1111/j.1476-5381.2009.00111.x (2009).
Devaux, J. J., Kleopa, K. A., Cooper, E. C. & Scherer, S. S. KCNQ2 is a nodal K+ channel. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 1236–1244, https://doi.org/10.1523/JNEUROSCI.4512-03.2004 (2004).
Shah, M. M., Migliore, M., Valencia, I., Cooper, E. C. & Brown, D. A. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America 105, 7869–7874, https://doi.org/10.1073/pnas.0802805105 (2008).
Yue, C. & Yaari, Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 4614–4624, https://doi.org/10.1523/JNEUROSCI.0765-04.2004 (2004).
Soh, H., Pant, R., LoTurco, J. J. & Tzingounis, A. V. Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. The Journal of neuroscience: the official journal of the Society for Neuroscience 34, 5311–5321, https://doi.org/10.1523/JNEUROSCI.3919-13.2014 (2014).
Pan, Z. et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. The Journal of neuroscience: the official journal of the Society for Neuroscience 26, 2599–2613, https://doi.org/10.1523/JNEUROSCI.4314-05.2006 (2006).
Chung, H. J., Jan, Y. N. & Jan, L. Y. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proceedings of the National Academy of Sciences of the United States of America 103, 8870–8875, https://doi.org/10.1073/pnas.0603376103 (2006).
Clark, B. D., Goldberg, E. M. & Rudy, B. Electrogenic tuning of the axon initial segment. Neuroscientist 15, 651–668, https://doi.org/10.1177/1073858409341973 (2009).
Soldovieri, M. V. et al. Early-onset epileptic encephalopathy caused by a reduced sensitivity of Kv7.2 potassium channels to phosphatidylinositol 4,5-bisphosphate. Scientific reports 6, 38167, https://doi.org/10.1038/srep38167 (2016).
Kim, E. C. et al. Reduced axonal surface expression and phosphoinositide sensitivity in Kv7 channels disrupts their function to inhibit neuronal excitability in Kcnq2 epileptic encephalopathy. Neurobiol. Dis. 118, 76–93, https://doi.org/10.1016/j.nbd.2018.07.004 (2018).
Hernandez, C. C., Zaika, O. & Shapiro, M. S. A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361–381, https://doi.org/10.1085/jgp.200810007 (2008).
Zhang, H. et al. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975 (2003).
Zhang, Q. et al. Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proceedings of the National Academy of Sciences of the United States of America 110, 20093–20098, https://doi.org/10.1073/pnas.1312483110 (2013).
Suh, B. C. & Hille, B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520 (2002).
Winks, J. S. et al. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. The Journal of neuroscience: the official journal of the Society for Neuroscience 25, 3400–3413, https://doi.org/10.1523/JNEUROSCI.3231-04.2005 (2005).
Maljevic, S. & Lerche, H. Potassium channel genes and benign familial neonatal epilepsy. Prog. Brain. Res. 213, 17–53, https://doi.org/10.1016/B978-0-444-63326-2.00002-8 (2014).
Ihara, Y. et al. Retigabine, a Kv7.2/Kv7.3-Channel Opener, Attenuates Drug-Induced Seizures in Knock-In Mice Harboring Kcnq2 Mutations. PloS one 11, e0150095, https://doi.org/10.1371/journal.pone.0150095 (2016).
Singh, N. A. et al. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. The Journal of physiology 586, 3405–3423, https://doi.org/10.1113/jphysiol.2008.154971 (2008).
Weckhuysen, S. et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Annals of neurology 71, 15–25, https://doi.org/10.1002/ana.22644 (2012).
Weckhuysen, S. et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 81, 1697–1703, https://doi.org/10.1212/01.wnl.0000435296.72400.a1 (2013).
Milh, M. et al. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet journal of rare diseases 8, 80, https://doi.org/10.1186/1750-1172-8-80 (2013).
Kato, M. et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287, https://doi.org/10.1111/epi.12200 (2013).
Millichap, J. J. et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurology. Genetics 2, e96, https://doi.org/10.1212/NXG.0000000000000096 (2016).
Pisano, T. et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 56, 685–691, https://doi.org/10.1111/epi.12984 (2015).
Numis, A. L. et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology 82, 368–370, https://doi.org/10.1212/WNL.0000000000000060 (2014).
Sun, J. & MacKinnon, R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 169, 1042–1050 e1049, https://doi.org/10.1016/j.cell.2017.05.019 (2017).
Cui, J. Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys. J. 110, 14–25, https://doi.org/10.1016/j.bpj.2015.11.023 (2016).
Haitin, Y. & Attali, B. The C-terminus of Kv7 channels: a multifunctional module. The Journal of physiology 586, 1803–1810, https://doi.org/10.1113/jphysiol.2007.149187 (2008).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Strulovich, R., Tobelaim, W. S., Attali, B. & Hirsch, J. A. Structural Insights into the M-Channel Proximal C-Terminus/Calmodulin Complex. Biochemistry 55, 5353–5365, https://doi.org/10.1021/acs.biochem.6b00477 (2016).
Kosenko, A. et al. Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. The EMBO journal 31, 3147–3156, https://doi.org/10.1038/emboj.2012.156 (2012).
Gamper, N., Stockand, J. D. & Shapiro, M. S. The use of Chinese hamster ovary (CHO) cells in the study of ion channels. J Pharmacol Toxicol Methods 51, 177–185, https://doi.org/10.1016/j.vascn.2004.08.008 (2005).
Devaux, J. et al. A Kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances Kv7/M channel activity. Epilepsia 57, e87–93, https://doi.org/10.1111/epi.13366 (2016).
Zaydman, M. A. et al. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proceedings of the National Academy of Sciences of the United States of America 110, 13180–13185, https://doi.org/10.1073/pnas.1305167110 (2013).
Tobelaim, W. S. et al. Competition of calcified calmodulin N lobe and PIP2 to an LQT mutation site in Kv7.1 channel. Proceedings of the National Academy of Sciences of the United States of America 114, E869–E878, https://doi.org/10.1073/pnas.1612622114 (2017).
Sun, J. & MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 180, 340–347 e349, https://doi.org/10.1016/j.cell.2019.12.003 (2020).
Li, Y. et al. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proceedings of the National Academy of Sciences of the United States of America 108, 9095–9100, https://doi.org/10.1073/pnas.1100872108 (2011).
Li, Y., Gamper, N., Hilgemann, D. W. & Shapiro, M. S. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. The Journal of neuroscience: the official journal of the Society for Neuroscience 25, 9825–9835, https://doi.org/10.1523/JNEUROSCI.2597-05.2005 (2005).
van den Bout, I. & Divecha, N. PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. Journal of cell science 122, 3837–3850, https://doi.org/10.1242/jcs.056127 (2009).
Kim, K. S., Duignan, K. M., Hawryluk, J. M., Soh, H. & Tzingounis, A. V. The Voltage Activation of Cortical KCNQ Channels Depends on Global PIP2 Levels. Biophys J 110, 1089–1098, https://doi.org/10.1016/j.bpj.2016.01.006 (2016).
Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351, https://doi.org/10.1038/nature17964 (2016).
Choveau, F. S., De la Rosa, V., Bierbower, S. M., Hernandez, C. C. & Shapiro, M. S. Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates KCNQ3 K(+) channels by interacting with four cytoplasmic channel domains. J. Biol. Chem. 293, 19411–19428, https://doi.org/10.1074/jbc.RA118.005401 (2018).
Cavaretta, J. P. et al. Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PloS one 9, e103655, https://doi.org/10.1371/journal.pone.0103655 (2014).
Kelly, B. L. & Ferreira, A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience 147, 60–70, https://doi.org/10.1016/j.neuroscience.2007.03.047 (2007).
Orhan, G. et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Annals of neurology 75, 382–394, https://doi.org/10.1002/ana.24080 (2014).
Traynelis, J. et al. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome research 27, 1715–1729, https://doi.org/10.1101/gr.226589.117 (2017).
Goto, A. et al. Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia 60, 1870–1880, https://doi.org/10.1111/epi.16314 (2019).
Niday, Z. & Tzingounis, A. V. Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox. Neuroscientist 24, 368–380, https://doi.org/10.1177/1073858418763752 (2018).
Miceli, F. et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proceedings of the National Academy of Sciences of the United States of America 110, 4386–4391, https://doi.org/10.1073/pnas.1216867110 (2013).
Miceli, F. et al. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. The Journal of neuroscience: the official journal of the Society for Neuroscience 35, 3782–3793, https://doi.org/10.1523/JNEUROSCI.4423-14.2015 (2015).
Shahidullah, M., Santarelli, L. C., Wen, H. & Levitan, I. B. Expression of a calmodulin-binding KCNQ2 potassium channel fragment modulates neuronal M-current and membrane excitability. Proceedings of the National Academy of Sciences of the United States of America 102, 16454–16459, https://doi.org/10.1073/pnas.0503966102 (2005).
Gomis-Perez, C. et al. Homomeric Kv7.2 current suppression is a common feature in KCNQ2 epileptic encephalopathy. Epilepsia 60, 139–148, https://doi.org/10.1111/epi.14609 (2019).
Doyle, D. A. et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 (1998).
Suh, B. C. & Hille, B. PIP2 is a necessary cofactor for ion channel function: how and why? Annual review of biophysics 37, 175–195, https://doi.org/10.1146/annurev.biophys.37.032807.125859 (2008).
Tyedmers, J., Mogk, A. & Bukau, B. Cellular strategies for controlling protein aggregation. Nature reviews. Molecular cell biology 11, 777–788, https://doi.org/10.1038/nrm2993 (2010).
Ye, J., Pavlicek, A., Lunney, E. A., Rejto, P. A. & Teng, C. H. Statistical method on nonrandom clustering with application to somatic mutations in cancer. BMC Bioinformatics 11, 11, https://doi.org/10.1186/1471-2105-11-11 (2010).
Abidi, A. et al. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis 80, 80–92, https://doi.org/10.1016/j.nbd.2015.04.017 (2015).
Kosenko, A. & Hoshi, N. A change in configuration of the calmodulin-KCNQ channel complex underlies Ca2+-dependent modulation of KCNQ channel activity. PloS one 8, e82290, https://doi.org/10.1371/journal.pone.0082290 (2013).
Webb, B. & Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics 54, 5 6 1–5 6 37, https://doi.org/10.1002/cpbi.3 (2016).
Schlitter, J., Engels, M., Kruger, P., Jacoby, E. & Wollmer, A. Targeted Molecular-Dynamics Simulation of Conformational Change - Application to the T[–]R Transition in Insulin. Mol. Simulat 10, 291–& (1993).
Long, S. B., Tao, X., Campbell, E. B. & MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382, https://doi.org/10.1038/nature06265 (2007).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865, https://doi.org/10.1002/jcc.20945 (2008).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802, https://doi.org/10.1002/jcc.20289 (2005).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8, 3257–3273, doi:10.1021/ct300400x (2012).
Essmann, U. et al. A smooth particle mesh Ewald method. The Journal of Chemical Physics 103, 8577–8593, https://doi.org/10.1063/1.470117 (1995).
Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. The Journal of Chemical Physics 101, 4177–4189, https://doi.org/10.1063/1.467468 (1994).
Acknowledgements
We thank Dr. Claudio Grosman (University of Illinois at Urbana-Champaign) for comments on the manuscript and discussion on the results for Dr-VSP-mediated Kv7 current suppression. We also acknowledge computing resources provided by PRAC allocation at Blue Waters of National Center for Supercomputing Applications (ACI-1713784 to E.T.), and Extreme Science and Engineering Discovery Environment (grant TG-MCA06N060 to E.T.). We would also like to acknowledge Beckman Institute Graduate Fellowship for supporting S.P. This research was supported by the Research Project Grant #R01NS083402 from the NIH National institute of Neurological Disorders and Stroke (PI: Chung), Carver Young Investigator Grant Award #11–38870 from Roy J. Carver Charitable Trust (PI: Chung), Targeted Research Initiative for Severe Symptomatic Epilepsies Grant #C4107 from Epilepsy Foundation (PI: Chung). National Institute of General Medical Sciences of the National Institutes of Health under awards P41-GM104601 (PI: Tajkhorshid) and R01-GM123455 (PI: Tajkhorshid).
Author information
Authors and Affiliations
Contributions
H.J.C. conceived of the study and participated in its design and coordination. H.J.C., E.B., J.Z. and C.C. drafted the manuscript. J.Z., E.C.K., J.P., R.C. carried out the experiments and statistical analyses. C.C. developed the statistical algorithms. E.B. contributed to the analyses and manuscript preparation. J.Z., J.P., R.C., M.H., D.J. and H.J.C. performed literature mining. E.P. performed structural modeling. S.P., K.L., and E.T. performed MD simulation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Kim, E.C., Chen, C. et al. Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy. Sci Rep 10, 4756 (2020). https://doi.org/10.1038/s41598-020-61697-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61697-6
This article is cited by
-
Targeting lipid–protein interaction to treat Syk-mediated acute myeloid leukemia
Nature Chemical Biology (2023)
-
Functional characterization and in vitro pharmacological rescue of KCNQ2 pore mutations associated with epileptic encephalopathy
Acta Pharmacologica Sinica (2023)
-
Kv7/KCNQ potassium channels in cortical hyperexcitability and juvenile seizure-related death in Ank2-mutant mice
Nature Communications (2023)
-
Nine patients with KCNQ2-related neonatal seizures and functional studies of two missense variants
Scientific Reports (2023)
-
Early initial video-electro-encephalography combined with variant location predict prognosis of KCNQ2-related disorder
BMC Pediatrics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
