
Abstract
Early detection of breast cancer and its correct stage determination are important for prognosis and rendering appropriate personalized clinical treatment to breast cancer patients. However, despite considerable efforts and progress, there is a need to identify the specific genomic factors responsible for, or accompanying Invasive Ductal Carcinoma (IDC) progression stages, which can aid the determination of the correct cancer stages. We have developed two-class machine-learning classification models to differentiate the early and late stages of IDC. The prediction models are trained with RNA-seq gene expression profiles representing different IDC stages of 610 patients, obtained from The Cancer Genome Atlas (TCGA). Different supervised learning algorithms were trained and evaluated with an enriched model learning, facilitated by different feature selection methods. We also developed a machine-learning classifier trained on the same datasets with training sets reduced data corresponding to IDC driver genes. Based on these two classifiers, we have developed a web-server Duct-BRCA-CSP to predict early stage from late stages of IDC based on input RNA-seq gene expression profiles. The analysis conducted by us also enables deeper insights into the stage-dependent molecular events accompanying IDC progression. The server is publicly available at http://bioinfo.icgeb.res.in/duct-BRCA-CSP.
Similar content being viewed by others

Introduction
Breast cancer ranks second among all the cancer types arranged in the order of increasing death rates, also the most prevalent cancer in women1. The cancer has been categorized into three therapeutic groups: ER - ER+ patients receive endocrine therapy, HER – HER+ group is treated by therapeutic targeting of HER/ERBB2, and TNBC - lacking expression of ER, PR, HER receptors2. It has been categorized into two major histological types- Invasive Ductal Carcinoma (IDC) and Invasive Lobular Carcinoma (ILC), occurring in 47–79% and 2–15% of invasive cancers amongst women of different worldwide races, respectively3,4. These two sub-types show similarity in certain features such as tumour site, size, stage and grade, but have different metastatic patterns, characteristic histology and malignant calcifications5,4. IDC starts from ducts and spreads to the breast fatty tissue, whereas ILC is restricted to milk producing lobules6. These two sub-types are also discriminated at the molecular level with differential expression of gene encoding vimentin, cathepsin D, thrombospondin, E-cadherin, vascular endothelial growth factor, cytokeratin 8, and cyclin A4,7,8,9,10,11,12. The pathological differences between the two sub-types arises as a result of separate gene regulatory networks, which warrants further exploration for the development of appropriate diagnostic and therapeutic treatment strategy6. According to reports, 75% cases of invasive breast carcinoma cases are accounted by IDC, however, advanced treatment of IDC patients still remains a challenge due to lack of molecular targets for IDC treatment13,14. Also, there is the availability of higher number of datasets for IDC patients in TCGA-BRCA, which is favourable for development of efficient classifiers using machine learning. Hence, we implemented machine-learning and developed a web-server for efficient prediction of the correct IDC stage, which can potentially aid in designing appropriate treatment strategies and precise molecular targeting.
Early detection of breast cancer has led to a significant decrease in mortality rate. Prognostic and predictive factors used for therapy are not sufficient and we need new markers for treatment as individuals differ15. Although PET and MR imaging techniques are available for early detection of breast cancer, these techniques are based on morphological features that do not provide any clue for molecular events accompanying cancer progression. In these cases, gene expression based analyses are able to capture early stage markers and also detect molecular events and pathways for driving disease from early to late stage. Early availability of such information can lead to identification of patients who would require targeted or personalized therapy. Further, it may also shed some light on tumors for which no standard treatment is available (https://www.cancer.gov/about-cancer/treatment/types/precision-medicine/tumor-dna-sequencing). Unfortunately, imaging techniques will not be help for such treatments. Further, using ML-based methods, not only can the process be automated (thereby eliminating the need of a skilled professional to assess the images), but more information can be derived from a single procedure. Previous studies has been done to identify non coding RNA expression profile associated with early stage of invasive ductal carcinoma16. Also early stage markers of breast cancer has been identified using microarray datasets from peripheral blood cell17. Machine learning has been previously utilized to discriminate early-late stage based on gene expression profile of clear cell renal cell carcinoma patients18. However machine learning based analysis on tissue site based expression profile in invasive ductal carcinoma has not yet been performed.
The increased incidence of breast cancer and higher mortality rate has attracted significant research efforts to unravel its causes, and development of better treatment options19. Breast cancer is a heterogeneous disease with varied features, such as morphological appearances, profile, response to therapy, TNM staging, histological grade, etc.20. There is a direct correlation between mortality rate and stages of cancer, and the stage progression could be checked by early detection and appropriate treatment strategies21. Although knowledge about genomic profiling has been identified in terms of varied molecular features associated with subtypes of cancer, its molecular mechanism of progression is poorly understood22. Tumour stage is defined as the anatomic extent of cancer at the time of diagnosis, which is important for an individual patient prognosis, and determination of best treatment strategy23. Pierre Denoix and the Union of International Cancer Control (UICC) has classified tumour staging based on TNM classification23. TNM classification overlaps with breast cancer stages, where T describes the extent of a primary tumour by the size or depth of invasion mainly in stage I or II, N describes the extent of regional lymph node metastasis in mainly stage II or III, and M describes the presence of metastasis mainly in stage IV23. The incorporation of this staging system into molecular or genetic profiles can help in detecting prognostic groups that guide the disease intervention23. There is a sharp decrease in the 5-year survival rate of patients with the stage-wise progression of breast cancer21. Treatment of cancer remains a challenge because of the lack of knowledge about factors for cancer progression and metastasis23. Potential treatment options are available based on clinical and pathological prognostic factors with the histological grade being the most important predictive factor23. High throughput techniques such as Next Generation Sequencing (NGS) that capture expression of thousands of genes in a single assay can act as powerful analytical tools for capturing breast cancer prognostic signature20. We can obtain information about a large number of genes, but their intertwining relationship cannot be captured by traditional techniques like statistical and correlational analyses, hence advanced methods such as machine-learning are important to capture cryptic signatures inherent in these data19. Molecular profiling helps in finding predictive information and identifying prognostic biomarkers that can serve as therapeutic targets20. Most of the cancer research is focussed to determine for finding driver genes, which are related to chimeras or splice junctions, which do not utilize the high resolution features of RNA-seq24. Machine-learning techniques are increasingly being used for modelling the progression and treatment of cancer due to its ability to detect key features from complex datasets25. Personalized treatment strategies could be developed for patients with similar molecular sub-types based on the patterns identified from systematically collected molecular profiles of tumour samples26. In this study, we developed classification methods to analyse the genomic datasets of invasive ductal carcinoma obtained from TCGA, using supervised machine-learning algorithms and feature selection methods. We developed prediction models that could discriminate between early and late stages of IDC using RNA-seq datasets. Different feature selection methods such as RFE, RLASSO, linear modelling, linear regression and random forest were trained and evaluated using Python scikit-learn library which provides individual rankings to gene features. Based on the most comprehensive ranking of gene features by various feature selection methods the top gene features were selected for enriched classifier training that helped us efficiently classify the tumours based on the tumour stage-specific gene expression profiles.
Results
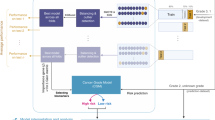
The workflow followed in our study is shown in Fig. 1. The TCGA level 3 RNA-seq datasets representing 1,093 breast cancer patients were retrieved using the TCGA2STAT R package27. The datasets represent 610 IDC patients, the distribution of samples across testing and training set by tumour stage is given in the Table 1. TCGA2STAT package merges the molecular profile information with clinical information into a data frame that is ready for supervised machine-learning. Each of the molecular profiles consists of RNA-seq gene expression data of 20,505 genes. The import dataset consists of ‘expression’ representing the gene expression profiles of patients in terms of RPKM values (described in methods), ‘clinical data’ which consists of clinical information related to patients, and ‘merged data’ in which both the information is mapped. Samples without clinical stage assignments were excluded from our study. Samples bearing clinical stages of stage I and II were pooled together as ‘early stage’, while the stages III and IV were pooled together as ‘late stage’. We generated gene expression data frames as comma separated value (CSV) format from the data retrieved using TCGA2STAT R package, with 20505 genes as column labels and 610 TCGA patient IDs as row labels. The values obtained by mapping the reads to genome generated as gene expression estimates were used as feature vectors for training the machine-learning classifier. Hence, the entire dataset consists of a gene expression data frame with a dimension of 610 * 20505. Near zero variance features and features having correlation coefficient more than 80% were removed using caret, an R package28. This led to a preliminary reduction of the number of features from 20,505 to 17,373. The training datasets were standardized using z-score normalization. It converts all the features to common scale with mean zero and standard deviation 1 (Supplementary file, Figure S1, S2). The normalized data-set was used for model generation to discriminate early versus late stages of the cancer.

Flowchart of the study to develop classification models, trained with relevant gene expression profiles to efficiently discriminate between the early and late IDC stages.
The normalized datasets were divided into two training sets, the first dataset comprises of complete gene expression datasets which were the original datasets representing expression of 17,373 genes used for feature selection. The second dataset consists of gene expression data corresponding to the driver-gene list in which the training genes were reduced to driver-genes responsible for progression of different cancers. The list of 881 driver genes was obtained from three well curated driver genes lists- Cosmic, IntoGen and Bailey29,30,31. The gene expression of the selected genes of the two datasets were further used for feature selection and classifier model generation (for details, see methods section).
The top 30 gene feature list enriched models rendered the highest accuracy for driver gene expression with a mean accuracy of 0.64 for all the machine-learning methods, hence, these features were used for training the model (Fig. 2a,b). The relevance of selected gene features was further validated by survival Kaplan-Meir estimate. Survival estimate revealed that median survival in cases with alteration 95.63 months and cases without alteration 129.6 months (Supplementary file, Figure S7). Top 20 gene feature enriched models gave the highest accuracy for the complete gene expression-based model with a mean accuracy of 0.70 for all the machine-learning methods hence, these features were used for training the models (Fig. 2c,d). The relevance of selected gene feature was further validated by survival Kaplan-Meir estimate. Survival estimate revealed that median survival in cases with alteration months 128.98 months and cases without alteration 129.6 months (Supplementary file, Figure S8). We also performed gene ontology enrichment analysis of selected gene features in biological process of cancer for both the models (Supplementary file, Table ST1). Despite using relevant features, the accuracy was low as the dataset was not balanced, i.e., there are more samples representing early stage as compared to that of late stage (469 for early stage, 141 for late stage). In order to tackle the class imbalance, Synthetic Minority Oversampling Technique (SMOTE) was employed using Python scikit-learn library. SMOTE was employed using ENN (Edited Nearest Neighbour) in which oversampling and under-sampling is performed until there is no difference with k- neighbour of majority class32. Real world datasets have higher composition of ‘normal class’ as compared to ‘abnormal class’, introducing bias in classification model. Combination of over-sampling of minority class along with under-sampling of majority class can aid in increasing the classifier performance33. To check the SMOTE resampling, models were trained on datasets where SMOTE resampling was employed (Supplementary file, Figure S5). The dataset where SMOTE was employed, the classification accuracy improved from 77% to nearly 89% on the validation set (Supplementary file, Figure S6, S11, S12). For training, validation, and testing, the samples were randomly stratified and split into 80% training-cum-validation sets (available on duct-BRCA-CSP webserver) and 20% independent testing datasets (available on duct-BRCA-CSP webserver).

(a) Feature selection methods were used to rank the gene features used in the training datasets. Top 20, 30, 40, 50, 60 and 80 features were used to train the binary classification model and their accuracy was evaluated. Based on that, top 30 gene features renders highest accuracy for all the machine learning algorithms evaluated by us. NB: Naïve Bayes, LR: Logistic Regression, RF: Random Forest, SVM: Support Vector Machine, DT: Decision Tree. X- axis: model accuracy, Y-axis: no. of features selected for model building (b). Correlation plot for top 30 gene features used in classification model building. X axis: Genes selected by feature selection Y axis: Genes selected by feature selection. (c) Feature selection methods were used to rank the gene features used in the training datasets. Top 20, 30, 40, 50, 60 and 80 features were used to train the binary classification model and their accuracy was evaluated. Based on that, top 20 gene features renders highest accuracy for all machine learning algorithms. NB: Naïve Bayes, LR: Logistic Regression, RF: Random Forest, SVM: Support Vector Machine, DT: Decision Tree. X- axis: model accuracy Y-axis: no. of features selected for model building (d). Correlation plot for top 20 driver gene features used in model building X, Y axis: Genes selected by feature selection.
Training-cum-validation
The classification accuracy of the generated prediction models ranges from 74% for SVM, to 95% for Random forest; and auROC value ranges from 0.76 for LR to 0.93 for the Random forest trained model for complete gene expression-based model. Based on the model accuracy and auROC, we inferred that the Random forest based prediction model has outperformed the other four machine-learning algorithms implemented in the study (Table 2). Random forest based model achieved the best performance with auROC of 0.93 on the training dataset, evaluated using ten-fold cross-validation for the complete gene expression-based model (Fig. 3a). The Random forest model displayed highest auROC as compared to the other models for complete gene expression-based model(Fig. 3b). The classification accuracy of the generated prediction models ranges from 72% for NB, to 92% for Random forest; and auROC value ranges from 0.72 for LR to 0.96 for Random forest for driver gene expression-based model. Based on accuracy and auROC, we inferred that Random forest based prediction model has outperformed the four other machine-learning algorithms implemented in the study. (Table 2). Random forest based model achieved maximum performance with auROC of 0.96 on training dataset when evaluated using ten-fold cross-validation for driver gene expression-based model (Fig. 4a). Random forest model exhibited the highest area under the curve as compared to the other models for driver gene expression-based model (Fig. 4b).

(a) Random forest based model achieved maximum performance with auROC of 0.93 on the training dataset when evaluated using ten-fold cross-validation, for the complete gene expression-based model. (b) Precision-recall curve is a trade of between precision and recall with high area under the curve representing low false positive and low false negative for all classifiers. Amongst all the prediction models, Random forest achieved the maximum area under precision-recall curve for complete-gene expression model.

(a) Random forest based model achieved maximum performance with auROC of 0.96 on the training dataset when evaluated using ten-fold cross-validation, for the driver gene expression-based model. (b) Precision-recall curve is a trade of between precision and recall with high area under the curve representing low false positive and low false negative for all classifiers. Amongst all the prediction models, the Random forest model achieved the maximum area under precision-recall curve for driver-gene expression-model.
Independent data-set performance
Further, we evaluated the performance of the trained models on independent datasets. The performance was re-evaluated based on accuracy, sensitivity, specificity, MCC and auROC for all the models. We observed coherence in the performance of the models between independent data testing and 10-fold cross validation based on auROC values for the complete gene expression-based model. Random forest achieved maximum auROC of 0.96 with an accuracy of 90% for testing datasets implemented in the complete gene expression-based model (Table 3, Supplementary file, Figure S9). Also, we observed coherence in the performance of the models between independent data testing and 10-fold cross validation based on auROC values for driver gene expression-based model. Random forest achieved maximum auROC of 0.99 with an accuracy of 94% for testing datasets in driver gene expression-based model (Table 3, Supplementary file, Figure S10).
External validation for a microarray dataset
We also evaluated the performance of the models developed by us for another dataset representing a microarray data, obtained from GEO. The models were able to achieve a maximum auROC of 0.47 with an accuracy of 67% for the Random forest based model (Table 4). A maximum auROC of 0.45 with accuracy 38% with Random forest based model trained on driver gene expression features (Table 4). Heatmap of differential expression analysis of microarray datasets between early and late stage for the complete gene expression-based features set (Supplementary file, Figure S3); and driver gene-based features set, showing differences in gene expression between early and late stages for the selected gene features (Supplementary file, Figure S4).
t-SNE (T-distributed stochastic neighbour embedding)
t-SNE technique was used for visualization of our gene expression datasets that displays high-dimensional data providing each data point a location in 2D or 3D space. It helps to model features into high-dimensional object to three-dimensional space such that similar objects tend to cluster together and dissimilar ones are modelled to distant points. The t-SNE analysis on our datasets segregates samples representing early and late stages, which shows that the dataset features are separable (Fig. 5).

t-SNE visualization was implemented on our gene expression data-sets to check if data-sets are segregating to early stage and late stage class labels based on selected features. This technique visualize our data-sets in 3D space in which early stage and late stage samples are segregating. X axis: X in t-SNE Y axis: Y in t-SNE.
Protein-protein interaction analysis of genes selected for model building
We performed protein-protein interaction analysis on gene features selected by our models using STRING database (Search Tool for Retrieval of Interacting Genes): the complete gene expression-based model, driver gene-based model and the combination of two. We found that as compared to the former two gene sets, more interacting partners are exhibited by STRING analysis of their combination (Fig. 6a-c). Thus, we were able to decipher major pathway that were targeted by gene sets in IDC selected by our models.

(a) Protein-protein interaction analysis using STRING of the gene set for the complete gene expression based-model. (b) STRING protein-protein interaction analysis of gene set from driver gene expression-based model. (c) STRING protein-protein interaction analysis of the combined gene sets. We found that as compared to the 11a and 11b, more interacting partners are exhibited by STRING analysis of their combination 11c which helps to decipher major pathways associated with IDC progression.
Several of these genes have been suggested to play a role in tumor progression from early to late stage. Genomic instability or DNA damage repair is the main driving factor of early stage of cancer development34. Cell adhesion and ECM pathway interaction are found to be dys-regulated in early tumorigenesis of ER+ cancer35. Whereas in later stages, patients diagnosed at stage IV, develop distant metastasis, which becomes nearly incurable. Although strategies targeting primary tumor has improved, treatment strategies for preventing metastasis is less developed which may be catered using machine learning36. Advanced stage of breast cancer is accompanied by genetic marker associated by cell division and proliferation pathway (https://ww5.komen.org/BreastCancer/RecommendedTreatmentsforMetastaticBreastCancer.html). In our analysis we have discovered several genes associated with these pathways suggesting their role in progression from early stage to late stage.
Four proteins encoded by DNAJB1, DNAJA1, CCT5 and FKBP4 are revealed to be in direct interactions, using STRING analysis. These proteins are major components of ubiquitin protein conjugation pathway by interacting with heat shock protein (Fig. 6c). This process mediate cellular processes such as protein localization, cell cycle regulation and DNA damage repair37. Ubiquitin dys-regulation can affect tumour suppressor or oncogene leading to cellular transformation and cancer38. DNAJB1 binds to mitogen-inducible gene MIG6, a tumour suppressor, which positively regulates epidermal growth factor signalling, leading to breast cancer development39.
CCT5 belongs to CCT gene family that serves as potential biomarker and display alteration in majority of breast cancer cases40. FKBP4 is found to be over-expressed in ductal carcinoma and under-expressed in lobular carcinoma by expression profiling41.
Five proteins encoded by CTTN, NCK1, CBL, PLCG1 and ERBB2IP depicts direct interaction in STRING analysis involved in RTK signalling pathway (Fig. 6c). Its aberrant expression results in enhanced cell proliferation, survival and metastasis leading to malignancy42. CTTN encodes cortactin which is a substrate for tyrosine Src nonreceptor tyrosine kinase whose amplification has been reported in primary metastatic breast carcinoma43.
NCK1 is an tyrosine kinase that regulation cell adhesion and has role in breast carcinoma cell invasion and metastasis44. CBL over-expression results in inhibition of transforming growth factor tumor suppressor activity and breast cancer prognosis45. PLCG1 is differentially regulated in breast cancer and has role in tumorigenesis of mediating intercellular signalling cascade46. ERBB2IP is a tyrosine kinase that interact with chaperon protein HSP90 and regulates breast tumor progression47.
Four proteins encoded by TRAAP, CDKN1A, CHD9 and WHSC1 depict direct interaction in STRING analysis involved DNA replication and DNA damage repair pathway (Fig. 6c). TRAP bind to proliferating cell nuclear antigen (PCNA) resulting DNA replication inhibition and cell growth inhibition and cancer48. WHSC1 is a methyl transferase that performs histone methylation affecting cell ability to undergo DNA damage repair49.
CDKN1A is a candidate breast cancer biomarker with upregulated expression in breast cancer tissue as compared to adjacent non-tumorous breast tissue50. CHD9 is a chromodomain helicase, found to be under-expressed in tumor with high Nottingham prognostic index (NPI) widely used for breast cancer prognostic in METABARIC cohort51.
Five proteins encoded by EIF6, ITPA, YBX1, UPF3B and EIF4A1 depict direct interaction in STRING analysis involved in protein translational machinery (Fig. 6c). Deregulated protein synthesis can affect several processes such as cell growth, proliferation, apoptosis at translational level and malignancy52. Dys-regulation of EIF4A1 protein results in preferred translation of gene involved in pro-oncogenic signalling53.
EIF6 is potential biomarker for cancer as it downstream modulator of cell division resulting in oncogenesis54. ITPA downregulation promotes DNA instability, suppression of cell growth and apoptosis in SKBR3 cell lines55. YBX1 is an oncoprotein that regulates tumorigenesis and malignant progression in breast cancer56. UPF3B is prolactin induced protein that regulates cell cycle progression and found to be upregulated in majority of breast cancer57.
Proteins encoded by GAS7, NUP98, MSI2, MLLT10 and PBX1 depict direct interaction in STRING analysis involved dys-regulated DNA binding transcription factor pathway (Fig. 6c). DNA binding TFs are commonly deregulated in cancer which modulates gene expression resulting IN malignancy58. MSI2 directly regulates estrogen receptor by binding to ESR1 resulting in breast cancer cell growth59.
MLLT10 is one of the breast cancer susceptibility loci identified by genome wide association studies60. NUP98 overexpression correlates with poor outcome in breast cancer61. GAS7 expression negatively correlates with p53 expression that results in early onset of breast cancer62. PBX1 is found to be up-regulated in metastatic progression ERα-positive breast cancer63.
Threshold value of expression for genes selected by feature selection
Threshold value is the expression value beyond which the sample will segregate into two groups, in our study- ‘early’ and ‘late’ stages. For example, if Z-score of CDKN1A (over-expressed in early stage) is greater than 0.32 is then it is representative of an early stage sample otherwise if it is less than 0.32 then it is representative of a late stage sample. We calculated threshold for all the genes selected by feature selection methods for the complete gene expression-based model as well as driver gene-based model (Table 5).
Gene ontology
Clusterprofiler R package was used for gene ontology enrichment analysis of the gene set selected for the complete gene expression-based model and selected gene set for the driver gene-based model. It reveals enrichment in molecular functions such as transferase and hydrolase activity for gene set for the driver gene-based model (Fig. 7a). Cathepsin D is a lysosomal hydrolase which is having increased expression in tumors that results in degradation of extracellular matrix causing metastasis64. Increased expression of glycoprotein-sialytransferase is associated with altered membrane synthesis resulting in invasiveness and neoplastic state65.

(a) Gene ontology analysis of the gene set from driver gene expression-based model for molecular function. (b) Gene ontology analysis of gene set from complete gene expression-based model for molecular function.
The selected training gene set for the complete gene expression-based model was found to be enriched in molecular functions related to oxidoreductase activity, lyase, hydrolase and transferase activity (Fig. 7b). Glutathione-dependent oxidoreductase- CLIC3 is secreted by cancer cell which contributes to tumour micro-environment by promoting angiogenesis and tumour cell invasion66. CSE (Cystathion-gamma-lyase) regulates STAT3 signalling which promotes cell proliferation in breast cancer67.
The selected training gene set for the complete gene expression-based model was found to be enriched in cellular components related to plasma membrane, endoplasmic-reticulum membrane, organelle membrane and nuclear-endoplasmic reticulum membrane (Fig. 8a). Mitochondria-associated ER-membrane responds to various stress signals including apoptotic signalling, inflammatory signalling and unfolded protein response (UPR). These pathways may be perturbed due to abnormal or uncontrolled expression of related genes resulting in cancer development68. Training Gene set for the driver gene expression-based model is enriched in cellular component such as plasma, membrane and organelle membrane (Fig. 8b).

(a) Gene ontology analysis of gene set from feature selection from complete gene expression-based model for cellular component. (b) Gene ontology analysis of gene set by from driver gene expression-based model for cellular component.
Gene set from driver gene-based model is enriched in biological processes related to transcriptional misregulation and ErbB signaling (Fig. 9a). Transcription factors are involved in tumorigenesis by altering expression profiles of their targets69. ErbB tyrosine kinase receptors are found to be activated by epidermal growth factor controlling cellular proliferation, angiogenesis and metastasis in breast cancer70. Gene features from the complete Gene expression-based models are more enriched in biological process related to immunological response such as T cell costimulation, immunoglobin response (Fig. 9b). Impaired expression of HLA-DQB1 due to change in methylation pattern of gene is associated with esophageal squamous cell carcinoma by altering immune response pattern71.

(a) Gene ontology analysis of gene set from driver gene expression-based model for biological process. (b) Gene ontology analysis of gene set by complete gene expression-based model for biological process.
Methods
Data mining
The study dataset was obtained from TCGA using TCGA2STAT R package, which automatically downloads and processes TCGA genomics and clinical data into a format convenient for statistical analyses in R environment27. The package imports and processes molecular profile from high-throughput experiments such as microarray, next generation sequencing and methylation array.
Data pre-processing and normalization
As an initial step of pre-processing, which aids in preliminary feature reduction for a feature-rich training dataset, gene features showing near zero variance across the two classes were removed. Near zero variance features are the feature which either have unique value or have few unique value relative to the number of samples. Along similar lines, the features having more than 80% correlation with each other can prove to be problematic for machine-learning. Hence, such feature pairs/groups were also removed in a way where only a single feature of the group remains. These two tasks were performed using the Caret, a R package28. We used RPKM (Reads Per Kilobase of transcript per Million mapped reads) values of the reads for supervised machine-learning analysis. RPKM is a measure of normalization of RNA-seq data with the total read length and number of sequencing reads for a given sample72. The training datasets were standardized using z-score normalization. The normalization converts all the features to common scale with mean zero and standard deviation 1. The normalized data-set was used for training models.
Feature selection
Feature selection is an advantageous step before machine-learning which reduces the dimensionality of datasets26. Given the possibly large sets of features, it helps in searching for the subset of features that has relevance in terms of a given predictor variable73. It also helps in improving the accuracy of a classifier by removing irrelevant data74. The main challenge associated with current data mining technologies is the high dimensionality of datasets combined with homogenous nature of data75.
For reducing the dimensionality of the datasets and identifying relevant features for building efficient machine-learning classifiers, we implemented various feature selection algorithms such as RFE, RLASSO, Random forest, linear modelling and linear regression, which provide individual ranking to gene features. Recursive Feature Extraction (RFE) is a method which utilizes recursion for feature extraction where smaller and smaller sets are considered as features until the desired number of features are returned. Randomized lasso is a stability selection method, which is combination of sub-sampling of high dimensional datasets and selection algorithm76. Linear regression assumes that features which are important have highest coefficient in the model, and features which have low importance have lower coefficient in the model. When there are multiple correlated features, small change in data can lead to large change in model. Regression model uses regularization method which adds an additional penalty to a model in order to minimize the sum of squared error of training model using lasso and ridge regression methods. Lasso regression methods performs L1 regularization minimizes absolute sum of the coefficient and producing sparse solution. Ridge regression performs L2 regularization minimizing squared absolute sum of the coefficients. The Least absolute shrinkage and selection operator (LASSO) does regression analysis for parameter estimation and variable selection simultaneously77. Random forest uses decision tree based strategies to rank feature based on attribute “feature importance”. All of the feature selection methods were implemented using the Python 3.6 scikit-learn library.
These feature-selection methods were used to rank the gene features of the training datasets. All the methods were implemented using the Python 3.6 scikit-learn library. All of the above-mentioned methods report individual ranking for the features. In order to get consensus ranking, we calculated the overall mean of each feature rank obtained from individual method. Subsequently, the Top 20, 30, 40, 50, 60 and 80 features were used to train and evaluate accuracy of models for binary classification of early versus late IDC, based on 5 machine-learning methods namely - RF, Naive Bayes, SVM, Logistic regression (LR) and Decision tree. Gene features list which gave the highest accuracy for all the machine-learning method were selected for model generation and evaluation. t-SNE technique was used for visualization of our gene expression datasets, returned after feature selection, in order to check if data-sets are segregating into defined class based on selected features for visualization of high dimensional data-point. t-SNE uses random walk on neighbourhood graph that allows implicit structure of data point to influence the way groups of data is present78.
Handling data imbalance
Real world datasets are imbalanced, predominately composed of “normal” example and a small percentage of “abnormal” examples79. Imbalance results in poor predictive accuracy of minority class and difficulty in assessment of performance of classifier as most new sample are classified into minority class80. Class imbalanced datasets shows biasness which can be attenuated using SMOTE resulting in class balanced datasets80. Feature space similarity between minority classes are used to generate artificial data in SMOTE resampling81. It has been proven that over sampling of minority class with under sampling of majority class results in improvement of accuracy79. This method has been used to increase predicative accuracy of model for multiclass microarray datasets82. To check the usage of SMOTE resampling, models were trained on datasets where SMOTE resampling was employed.
Training classification models
After feature selection and data processing, we trained different algorithms to generate efficient classifiers for early and late tumour stage. We used five different algorithms – Random Forest, Naive Bayes, LinSVM (Support Vector Machines with linear kernels), Logistic regression and Decision tree. Naive Bayes is based on Bayesian theorem that calculates the probability of attribute to fall in particular instance with the assumption that every attribute is independent from other attributes83. Random forest uses ensemble of decision tree by random selection of features to split node84. SVM implements Sequential Optimization Algorithm for decision function85.
Training-cum-validation
The five supervised machine-learning algorithms (Random Forest, Naive Bayes, LinSVM, Logistic regression and Decision tree) were trained on subset of features obtained from feature selection and validated by 10-fold cross validation. The training models were compared by their accuracy, auROC, precision -recall and F-measure value.
Independent data testing
We further re-evaluated the best-trained model on an independent dataset which was not used in the classifier training at all.
Calculating threshold expression values for selected gene features
We performed differential expression analysis for the selected gene features by the two models, for early-late datasets to find out the differential expression of gene features selected by our model. Each gene feature selected by our model had range of expression across all the samples. We executed machine-learning and model evaluation for every single feature selected by our classifiers with threshold set across its expression range. The value that was giving highest ROC was considered as threshold value of expression value that could discriminate between early-late stages. Threshold value is the expression value beyond which the sample will segregate into two groups, in our case ‘early’ and ‘late’ stage.
Cancer driver gene expression-based model
The available driver gene list for the cancer were also used for building model to discriminate early-late stages of breast cancer. We compiled a list of driver genes using Cosmic, intoGen and baile, which are expert curated lists of driver genes in human cancers. Cosmic stands for catalogue of somatic mutations in cancer which is an expert curated list of driver genes in human cancers, which is widely used in medical research29. IntoGen identifies somatic mutation, gene, pathways that are involved in tumorigenesis by analysis of 13 cancers30. Bailey list identifies 299 molecular cancer genes by pan-cancer and pan-software analysis of 9,423 tumour exome datasets using 26 computational tools31. We reduced the data-set to these gene features, which was then used for feature selection and model building, repeating the above-mentioned steps to generate driver gene expression-based model for the web server.
Gene ontology
GO was performed on the list of genes returned by the feature selection methods to determine which gene families play role in the progression of breast cancer. We performed enrichment analysis using clusterprofiler R package. The package makes use of the datasets from the post genomic era high throughput technologies such as RNA-seq, micro-array, etc. to examine cellular molecules at systems level86. We also performed STRING protein-protein interaction analysis to discover major pathways targeted by selected gene features.
External data-set evaluation
To further check the performance of our model, we obtained independent datasets form GEO with accession ID GSE61304 containing 60 samples of IDC with clinical stage information obtained using microarray profiling. GEOquery package helps the user to access the information stored in GEO directly using Bioconductor without any formatting or parsing problem87. Biomart was used to annotate the probe IDs of microarray datasets with gene symbols88. If a particular probe is sequenced multiple times, WGCNA R package collapserow function was used to select one single representative row of each probe ID89. Subsequently, RMA normalization was performed using GCRMA R package converting the expression in log 2 scale to make its distribution comparable to RNA-seq datasets90,91. This independent-testing dataset were segregated into driver-testing datasets and complete gene expression datasets for performance evaluation of the generated models.
Conclusion
We have successfully applied supervised machine-learning classification on gene expression profiles to develop classification models for discriminating early and late stage of invasive ductal carcinoma. The RNA-seq data obtained from TCGA had various additional information related to the samples ranging from age, survivability, TNM staging, histological subtype and pathological stage in the form of metadata or clinical data.
The data yielded 20,505 gene expression values used as training features to be considered for classification model trainings. This voluminous dimensionality was reduced using various data pre-processing and feature selection methods. After this, the classifier models were generated by applying various machine-learning algorithms. Based on best trained classifiers, we developed a web-server Duct-BRCA-CSP that could predict whether the sample represents early or late stage using patient’s gene expression profile. The expression-based gene features returned by the feature selection methods can be used to differentiate samples between early and late stage with high accuracy, also providing candidate biomarkers for improved diagnosis and treatment, subsequent to adequate experimental validations. Thus, the combined power of machine-learning and next generation sequencing can provide important insights into the progression IDC from early to late stage. Our study proves that accurate prediction models captured features of the relevant genes, that could be candidates for further experimental evaluation for therapeutic and prognostic potential in the cancer treatment.
Discussion
We developed a web-server Duct-BRCA-CSP for invasive ductal carcinoma which predicts tumour stage of a sample on the basis of RNA-seq expression profile of selected genes, rather than its tumour size, gene expression profile of all the genes, imaging or survivability. Our study is preliminary in nature, however, in the future, the availability of datasets from higher number of patients, especially those representing late stage may help in building more efficient stage specific classifiers which may be suitable for personalized treatment strategy in clinics. In addition, further inclusion of more datasets such as mutation profile, methylation data and protein isoform data may improve accuracy of classifiers. Inclusion of paired datasets can also further aid in gaining valuable insights into the progression of breast cancer. To the best of our knowledge, the webserver Duct-BRCA-CSP is a server which is first of its kind for prediction of IDC tumour stages based on gene expression profiles of selected genes.
References
Libson, S. & Lippman, M. A review of clinical aspects of breast cancer. Int. Rev. Psychiatry 26, 4–15 (2014).
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nat. 490, 61–70 (2012).
Jay R. Harris M.E.L., Morrow M. & Osborne C.K. Diseases of the Breast. Annals of Surgery, 233(4) (2001).
Zhao, H. et al. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol. Biol. Cell 15, 2523–2536 (2004).
Winchester, D. J. et al. A comparative analysis of lobular and ductal carcinoma of the breast: presentation, treatment, and outcomes. J. Am. Coll. Surg. 186, 416–422 (1998).
Ragunath, P. K. et al. Relevance of systems biological approach in the differential diagnosis of invasive lobular carcinoma & invasive ductal carcinoma. Bioinformation 8, 359–364 (2012).
Bedner, E. et al. Cathepsin D in invasive ductal NOS, medullary, lobular and mucinous breast carcinoma. An immunohistochemical study. Pol. J. Pathol. 46, 11–15 (1995).
Serre, C. M. et al. Distribution of thrombospondin and integrin alpha V in DCIS, invasive ductal and lobular human breast carcinomas. Analysis by electron microscopy. Virchows Arch. 427, 365–372 (1995).
Berx, G. et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13, 1919–1925 (1996).
Lee, A. H. et al. Invasive lobular and invasive ductal carcinoma of the breast show distinct patterns of vascular endothelial growth factor expression and angiogenesis. J. Pathol. 185, 394–401 (1998).
Lehr, H. A. et al. Cytokeratin 8 immunostaining pattern and E-cadherin expression distinguish lobular from ductal breast carcinoma. Am. J. Clin. Pathol. 114, 190–196 (2000).
Coradini, D. et al. Infiltrating ductal and lobular breast carcinomas are characterised by different interrelationships among markers related to angiogenesis and hormone dependence. Br. J. Cancer 87, 1105–1111 (2002).
Li, C. et al. Identification of the potential crucial genes in invasive ductal carcinoma using bioinformatics analysis. Oncotarget 9, 6800–6813 (2018).
Zhang, N. et al. Dose invasive apocrine adenocarcinoma has worse prognosis than invasive ductal carcinoma of breast: evidence from SEER database. Oncotarget 8, 24579–24592 (2017).
Guler, E. N. Gene Expression Profiling in Breast Cancer and Its Effect on Therapy Selection in Early-Stage Breast Cancer. Eur. J. Breast Health 13, 168–174 (2017).
Deva Magendhra Rao, A. K. et al. Identification of lncRNAs associated with early-stage breast cancer and their prognostic implications. Mol. Oncol. 13, 1342–1355 (2019).
Sharma, P. et al. Early detection of breast cancer based on gene-expression patterns in peripheral blood cells. Breast Cancer Res. 7, R634–644 (2005).
Bhalla, S. et al. Gene expression-based biomarkers for discriminating early and late stage of clear cell renal cancer. Sci. Rep. 7, 44997 (2017).
Saleh T. Atiya, D Shaker, A O. Studying Combined Breast Cancer biomarkers using Machine Learning techniques. (2016).
Rakha, E. A. et al. Breast cancer prognostic classification in the molecular era: the role of histological grade. Breast Cancer Res. 12, 207 (2010).
Palaniappan, A., Ramar, K. & Ramalingam, S. Computational Identification of Novel Stage-Specific Biomarkers in Colorectal Cancer Progression. PLoS One 11, e0156665 (2016).
Lesurf, R. et al. Molecular Features of Subtype-Specific Progression from Ductal Carcinoma In Situ to Invasive Breast Cancer. Cell Rep. 16, 1166–1179 (2016).
Brierley, J., Gospodarowicz, M. & O’Sullivan, B. The principles of cancer staging. Ecancermedicalscience 10, ed61 (2016).
Singireddy S. et al. Identifying differentially expressed transcripts associated with prostate cancer progression using RNA-Seq and machine learning techniques. In: 2015 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), p. 1–5 (2015).
Kourou, K. et al. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 13, 8–17 (2015).
Jagga, Z. & Gupta, D. Classification models for clear cell renal carcinoma stage progression, based on tumor RNAseq expression trained supervised machine learning algorithms. BMC Proc. 8, S2 (2014).
Wan, Y. W., Allen, G. I. & Liu, Z. TCGA2STAT: simple TCGA data access for integrated statistical analysis in R. Bioinforma. 32, 952–954 (2016).
Kuhn, M. Building Predictive Models in R Using the caret Package. Journal of Statistical Software; Vol 1, Issue 5 (2008).
Sondka, Z. et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Gonzalez-Perez, A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082 (2013).
Bailey, M. H. et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318 (2018).
More A. Survey of resampling techniques for improving classification performance in unbalanced datasets (2016).
N. V. Chawla KWB, L. O. Hall, W. P. Kegelmeyer. SMOTE: Synthetic Minority Over-sampling Technique. Journal of Artificial Intelligence Research; Vol 16 (2002).
Broustas, C. G. & Lieberman, H. B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 181, 111–130 (2014).
Emery, L. A. et al. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. Am. J. Pathol. 175, 1292–1302 (2009).
Redig, A. J. & McAllister, S. S. Breast cancer as a systemic disease: a view of metastasis. J. Intern. Med. 274, 113–126 (2013).
Shi, D. & Grossman, S. R. Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol. Ther. 10, 737–747 (2010).
Qi, J. & Ronai, Z. A. Dysregulation of ubiquitin ligases in cancer. Drug. Resist. Updat. 23, 1–11 (2015).
Park, S. Y. et al. DNAJB1 negatively regulates MIG6 to promote epidermal growth factor receptor signaling. Biochim. Biophys. Acta 1853, 2722–2730 (2015).
Bassiouni, R. et al. Chaperonin Containing TCP-1 Protein Level in Breast Cancer Cells Predicts Therapeutic Application of a Cytotoxic Peptide. Clin. Cancer Res. 22, 4366–4379 (2016).
Bertucci, F. et al. Lobular and ductal carcinomas of the breast have distinct genomic and expression profiles. Oncogene 27, 5359–5372 (2008).
Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 7, 1758–1784 (2015).
MacGrath, S. M. & Koleske, A. J. Cortactin in cell migration and cancer at a glance. J. Cell Sci. 125, 1621–1626 (2012).
Morris, D. C. et al. Nck deficiency is associated with delayed breast carcinoma progression and reduced metastasis. Mol. Biol. Cell 28, 3500–3516 (2017).
Kang, J. M. et al. CBL enhances breast tumor formation by inhibiting tumor suppressive activity of TGF-beta signaling. Oncogene 31, 5123–5131 (2012).
Hernandez, P. et al. Integrative analysis of a cancer somatic mutome. Mol. Cancer 6, 13 (2007).
Tao, Y. et al. Role of Erbin in ErbB2-dependent breast tumor growth. Proc. Natl Acad. Sci. USA 111, E4429–4438 (2014).
Punchihewa, C. et al. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J. Biol. Chem. 287, 14289–14300 (2012).
Shah, M. Y. et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 35, 5905–5915 (2016).
Wei, C. Y. et al. Expression of CDKN1A/p21 and TGFBR2 in breast cancer and their prognostic significance. Int. J. Clin. Exp. Pathol. 8, 14619–14629 (2015).
Chu, X. et al. Genotranscriptomic meta-analysis of the CHD family chromatin remodelers in human cancers - initial evidence of an oncogenic role for CHD7. Mol. Oncol. 11, 1348–1360 (2017).
Hagner, P. R., Schneider, A. & Gartenhaus, R. B. Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood 115, 2127–2135 (2010).
Modelska, A. et al. The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis. 6, e1603 (2015).
Zhu, W. et al. The role of eukaryotic translation initiation factor 6 in tumors. Oncol. Lett. 14, 3–9 (2017).
Charbgoo, F. et al. RNAi mediated gene silencing of ITPA using a targeted nanocarrier: Apoptosis induction in SKBR3 cancer cells. Clin. Exp. Pharmacol. Physiol. 44, 888–894 (2017).
Shibata, T. et al. Y-box binding protein YBX1 and its correlated genes as biomarkers for poor outcomes in patients with breast cancer. Oncotarget 9, 37216–37228 (2018).
Naderi, A. & Vanneste, M. Prolactin-induced protein is required for cell cycle progression in breast cancer. Neoplasia 16(329-342), e321–314. (2014).
Bhagwat, A. S. & Vakoc, C. R. Targeting Transcription Factors in Cancer. Trends Cancer 1, 53–65 (2015).
Kang, M. H. et al. Musashi RNA-binding protein 2 regulates estrogen receptor 1 function in breast cancer. Oncogene 36, 1745–1752 (2017).
Ghoussaini, M., Pharoah, P. D. P. & Easton, D. F. Inherited genetic susceptibility to breast cancer: the beginning of the end or the end of the beginning? Am. J. Pathol. 183, 1038–1051 (2013).
Mullan, P. B. et al. NUP98 - a novel predictor of response to anthracycline-based chemotherapy in triple negative breast cancer. BMC Cancer 19, 236 (2019).
Chang, J. W. et al. Wild-type p53 upregulates an early onset breast cancer-associated gene GAS7 to suppress metastasis via GAS7-CYFIP1-mediated signaling pathway. Oncogene 37, 4137–4150 (2018).
Magnani, L. et al. The pioneer factor PBX1 is a novel driver of metastatic progression in ERalpha-positive breast cancer. Oncotarget 6, 21878–21891 (2015).
Abbott, D. E. et al. Reevaluating cathepsin D as a biomarker for breast cancer: serum activity levels versus histopathology. Cancer Biol. Ther. 9, 23–30 (2010).
Bosmann, H. B. & Hall, T. C. Enzyme activity in invasive tumors of human breast and colon. Proc. Natl Acad. Sci. USA 71, 1833–1837 (1974).
Hernandez-Fernaud, J. R. et al. Secreted CLIC3 drives cancer progression through its glutathione-dependent oxidoreductase activity. Nat. Commun. 8, 14206 (2017).
You, J. et al. Cystathionine- gamma-lyase promotes process of breast cancer in association with STAT3 signaling pathway. Oncotarget 8, 65677–65686 (2017).
Kato, H. & Nishitoh, H. Stress responses from the endoplasmic reticulum in cancer. Front. Oncol. 5, 93 (2015).
Gonzalez-Perez, A. Circuits of cancer drivers revealed by convergent misregulation of transcription factor targets across tumor types. Genome Med. 8, 6 (2016).
Hardy, K. M. et al. ErbB/EGF signaling and EMT in mammary development and breast cancer. J. Mammary Gland. Biol. Neoplasia 15, 191–199 (2010).
Rodriguez, J. A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol. Lett. 14, 4415–4427 (2017).
Mortazavi, A. et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Radovic, M. et al. Minimum redundancy maximum relevance feature selection approach for temporal gene expression data. BMC Bioinforma. 18, 9 (2017).
Yu L. & Liu H. Feature Selection for High-Dimensional Data: A Fast Correlation-Based Filter Solution. In: ICML. (2003).
Ge, G. & Wong, G. W. Classification of premalignant pancreatic cancer mass-spectrometry data using decision tree ensembles. BMC Bioinforma. 9, 275 (2008).
Nicolai Meinshausen P.B. Stability selection, Journal of the Royal Statistical Society 2010/9/1;72:417-473.
Thomas, J. et al. Probing for Sparse and Fast Variable Selection with Model-Based Boosting. Comput. Math. Methods Med. 2017, 1421409 (2017).
van der Maaten, L. H. G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Alberto Fernández S.G., F. Herrera & N.V. Chawla. SMOTE for learning from imbalanced data: progress and challenges, marking the 15-year anniversary, Journal of Artificial Intelligence Research; Volume 61 (2018).
Blagus, R. & Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinforma. 14, 106 (2013).
Yu, H. et al. Mining and integrating reliable decision rules for imbalanced cancer gene expression data sets. Tsinghua Sci. Technol. 17, 666–673 (2012).
Sujataa Dash B. & Narayan R.. Sampling based hybrid algorithms for imbalanced data classification, International Journal of Hybrid Intelligent Systems 18 April 2016;volume 13.
Friedman, N., Geiger, D. & Goldszmidt, M. Bayesian Network Classifiers. Mach. Learn. 29, 131–163 (1997).
Breiman, L. Random Forests. Mach. Learn. 45, 5–32 (2001).
Platt J. Fast Training of Support Vector Machines Using Sequential Minimal Optimization, Advances in Kernel Methods - Support Vector Learning January 1998.
Yu, G. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Davis, S. & Meltzer, P. S. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinforma. 23, 1846–1847 (2007).
Durinck, S. et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Moffitt, R. A. et al. caCORRECT2: Improving the accuracy and reliability of microarray data in the presence of artifacts. BMC Bioinforma. 12, 383 (2011).
Irizarry, R. A. et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15 (2003).
Author information
Authors and Affiliations
Contributions
D.G., S.R. and R.K. Study design; D.G., S.R. and V.M. Data analysis, concept, programming and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roy, S., Kumar, R., Mittal, V. et al. Classification models for Invasive Ductal Carcinoma Progression, based on gene expression data-trained supervised machine learning. Sci Rep 10, 4113 (2020). https://doi.org/10.1038/s41598-020-60740-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60740-w
This article is cited by
-
Hierarchical graph neural network with subgraph perturbations for key gene cluster discovery in cancer staging
Complex & Intelligent Systems (2024)
-
Enhancing the prediction of IDC breast cancer staging from gene expression profiles using hybrid feature selection methods and deep learning architecture
Medical & Biological Engineering & Computing (2023)
-
Breast cancer diagnosis based on hybrid rule-based feature selection with deep learning algorithm
Research on Biomedical Engineering (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
