
Abstract
The family Trichomycteridae is one of the most diverse groups of freshwater catfishes in South and Central America with eight subfamilies, 41 genera and more than 300 valid species. Its members are widely distributed throughout South America, reaching Costa Rica in Central America and are recognized by extraordinary anatomical specializations and trophic diversity. In order to assess the phylogenetic relationships of Trichomycteridae, we collected sequence data from ultraconserved elements (UCEs) of the genome from 141 specimens of Trichomycteridae and 12 outgroup species. We used a concatenated matrix to assess the phylogenetic relationships by Bayesian inference (BI) and maximum likelihood (ML) searches and a coalescent analysis of species trees. The results show a highly resolved phylogeny with broad agreement among the three distinct analyses, providing overwhelming support for the monophyletic status of subfamily Trichomycterinae including Ituglanis and Scleronema. Previous relationship hypotheses among subfamilies are strongly corroborated, such as the sister relationship between Copionodontinae and Trichogeninae forming a sister clade to the remaining trichomycterids and the intrafamilial clade TSVSG (Tridentinae-Stegophilinae-Vandelliinae-Sarcoglanidinae-Glanapteryginae). Monophyly of Glanapteryginae and Sarcoglanidinae was not supported and the enigmatic Potamoglanis is placed outside Tridentinae.
Similar content being viewed by others

Introduction
Unraveling the relationships of major sections of the Tree of Life is one of the most daunting challenges of the evolutionary biology. Massively parallel DNA sequencing (so-called Next-gen sequencing) is a promising tool that is helping to resolve the interrelationships of longstanding problematic taxa1,2,3,4. One of the most common classes of phylogenomic methods involves the sequence capture of nuclear regions in the flanks and cores of the ultraconserved elements (UCEs)2. The more variable flanking UCE regions allow a better resolution of nodes across a broad range of evolutionary timescales in a given phylogeny2. As variation in the flanks increases with distance from the core UCE, this combined approach displays a balance between having a high enough substitution rate while minimizing saturation, thus providing information for estimating phylogenies at multiple evolutionary timescales2,3. Recent studies of actinopterygians5, flatfishes6, cichlids7, ostariophysan8, acanthomorphs9, Loricariidae10, knifefishes11, among other vertebrates groups3,12, have shown that UCEs are excellent markers for phylogenetic studies because of their ubiquity among taxonomic groups13, low degrees of paralogy14, and low saturation3. According to Gilbert et al.15, the phylogenetic informativeness of the combined flank and core regions of UCEs outperfoms protein-coding genes used in multilocus studies. Additionally, phylogenomic approaches are characterized by their potential to collect data from at least one order of magnitude more loci than the traditional sequencing techniques applied to protein-coding legacy marker’s.
The present survey is the first to employ a new bait set to ostariophysans16 and high-throughput sequencing to address evolutionary relationships in the large catfish family Trichomycteridae (pencil and parasitic catfishes). The family contains 319 valid species17 characterized by a highly modified opercular system, with opercular and interopercular bones usually armed with distinct patches of sharp odontodes (integumentary teeth). Trichomycterids have one of the broadest ranges of trophic strategies known within a single catfish family, including insectivory, omnivory, carnivory, necrophagy, mucophagy, lepidophagy, and hematophagy18,19,20,21,22,23,24,25. The family has a wide distribution mostly in the Neotropical freshwater basins of Central and South America26,27 from Costa Rica to Chilean Patagonia, occurring on both versants of the Andes, and even in a few insular27,28 and caves environments with stygobiotic species.
Eight trichomycterid subfamilies are currently recognized: Copionodontinae, Glanapteryginae, Sarcoglanidinae, Stegophilinae, Trichogeninae, Trichomycterinae, Tridentinae, and Vandelliinae20,29. Only two papers have used explicit cladistic analyses to test the interrelationships among all eight subfamilies, one based on morphological data29 and the other based on nuclear and mitochondrial genes30. In spite of such recent advances, the descriptions of new species and clades based on labile morphological characters31 and hurried changes in subfamilial classification32 present obstacles to reconstructing the evolutionary history of Trichomycteridae. Here we present a phylogenomic analysis of a dataset of ultraconserved DNA elements (UCEs) and their flanking regions representing over 902 loci from 139 trichomycterid taxa (about 47% of species diversity of the family) to infer a new well -supported hypothesis of phylogenetic relationships discuss recent taxonomic changes in Trichomycteridae and provides an evolutionary framework to explore macroevolutionary and biogeographic processes that modeled the exceptional diversity in the Neotropics.
Results
Phylogenetic relationships within trichomycteridae
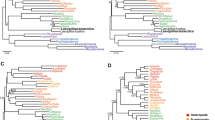
The DNA sequencing yielded a total of 323 million reads with an average of 2.1 million reads per sample (range = 40,059–6.4 million). These reads were assembled into an average of 8,752 contigs per sample (95CI, min = 176, max = 36,085), having an average length of 597 bp (Supplementary Table 2). An average (per sample) of 1,321 of those contigs matched the UCE loci from the target capture probes used and the average length of UCE-matching contigs was 598 bp (range = 164–971). The size of each matrix according to their completeness level was 1,379 (50%), 902 (75%) and 432 (90%) loci. ML and Bayesian trees inferred from each locus alignment showed identical topologies. Using a method of species-tree analysis (ASTRAL) in which a species tree history is estimated from independent gene histories, we recovered species trees partially concordant with the concatenated analysis. The ASTRAL species trees for each matrix (50%, 75%, 90% completeness) were much less resolved and had lower support values than either the Bayesian or the ML tree estimated from the concatenated dataset. In a comparison of the species tree for the 75% matrix with the Bayesian tree, approximately 25 nodes of 153 species-tree nodes showed bootstrap values < 90%; however, the species tree recovered most relationships of the gene trees. Pairwise comparisons among all nine trees obtained by the distinct inference methods (ML, Bayesian, and Species tree) with varied levels of data completeness (50%, 75%, 90%) indicates that the Bayesian tree with 75% complete matrix (Fig. 1a) exhibits the highest values of global topological similarity. This is indicated by the highest average (Fig. 1b) and lowest standard deviation (Fig. 1c) of the element-based comparison score of Bremm et al.33. The Bayesian tree with 75% complete matrix is accordingly chosen as the reference tree for the present discussion of trichomycterid relationships. Figure 1a depicts the average conservation of the nodes of the reference tree against all other trees (Supplementary Figs. 1–8), indicating high and low levels of similarity with shades of blue and red, respectively. The detailed topology of the Bayesian tree with 75% complete matrix is represented in Figs. 2–5.

Pairwise comparison among all nine trees obtained by the distinct inference methods (ML, Bayesian and Species tree) with varied levels of data completeness (50, 75 and 90%). (a) highest values of global topological similarity (b) highest average and (c) lowest standard deviation of the element-based comparison score of Bremm et al. (2011).

Phylogenetic hypothesis of Trichomycteridae from 902 ultraconserved loci using Bayesian analysis of concatenated data, highlighting the phylogenetic relationships for Copionodontinae -Trichogeninae. All nodes supported Bayesian posterior probabilities >0.99.

Phylogenetic hypothesis of Trichomycteridae from 902 ultraconserved loci using Bayesian analysis of concatenated data, highlighting the phylogenetic relationships for Trichomycterus lineage. All nodes supported Bayesian posterior probabilities >0.99.

Phylogenetic hypothesis of Trichomycteridae from 902 ultraconserved loci using Bayesian analysis of concatenated data, highlighting the phylogenetic relationships for Eremophilus lineage. All nodes supported Bayesian posterior probabilities >0.99 and blue nodes with posterior probabilities <0.99.

Phylogenetic hypothesis of Trichomycteridae from 902 ultraconserved loci using Bayesian analysis of concatenated data, highlighting the phylogenetic relationships for TSVSG clade. All nodes supported Bayesian posterior probabilities >0.99 and blue nodes with posterior probabilities <0.99.
The 75% complete matrix contains 284,349 characters (including indels), 107,270 parsimony informative sites and 30.81% of missing data. The Bayesian (Figs. 2–5) and ML (Supplementary Fig. 4) analyses for this matrix returned identical topologies. Of the 152 nodes, 150 (98.68%) were highly supported in the Bayesian tree (>0.99PP), whereas a subset of 134 of those nodes (89.79%) were also highly supported in the ML tree (>75% bootstrap score). Two nodes (1.36%) showed low support in ML trees with 50% and 8% bootstrap values, respectively, but in the Bayesian tree just one node had a low posterior probability (p = 0.5999, in the relationships of Ituglanis goya, Ituglanis ramiroi and Ituglanis sp. (Tapajos) (Fig. 4).
The phylogeny supports with highest confidence (100% bootstrap; PP = 1) the monophyly of Trichomycteridae and the subfamilies Copionodontinae (100% of the genera sampled), Stegophilinae (81.8% of the genera sampled), Trichomycterinae sensu Datovo & Bockmann29,30, Tridentinae sensu Baskin34, (75% of the genera sampled) and Vandelliinae (50% of the genera sampled) (Figs. 2–5). Our analysis provides strong evidence for the monophyly of the Clade B sensu Datovo & Bockmann29, composed of Copionodontinae and Trichogeninae (100% bootstrap; PP = 1.0; Fig. 2). This clade is herein named the Trichogeninae-group and its placement as the sister group to all remaining trichomycterids is equally well-supported. The Copionodontinae and the genus Copionodon (C. pecten, C. orthiocarinathus and Copionodon sp.) are both supported as monophyletic.
Clade C was recovered with a basal dichotomy between the TSVSG clade (Tridentinae-Stegophilinae-Vandelliinae-Sarcoglanidinae-Glanapteryginae) and Trichomycterinae (Fig. 2). The phylogenomic data provide strong evidence for the monophyly of Trichomycterinae. Our definition of the subfamily includes Ituglanis and Scleronema along with the traditional trichomycterine genera, but excludes Potamoglanis. Our study confirms the non-monophyly of Trichomycterus sensu lato, the largest trichomycterid genus has 54% of the family’s diversity (Figs. 3 and 4). In our topology, an undescribed trichomycterine from Paria on the Caribbean coast of Venezuela (Trichomycterus sp. (Paria), is placed sister to all remaining trichomycterines (Fig. 3) which group into two major clades termed the Trichomycterus and Eremophilus lineages.
In the Trichomycterus lineage, two clades diverge early from the remaining taxa. The first one to diverge is the Punctulatus Clade composed of T. cf. knerii (Orinoco) and two species from Pacific coastal rivers draining the Central Andes, T. punctulatus and T. cf. taenia. The second one to diverge is the Oroyae Clade composed of T. cf. oroyae and T. quechuorum, two Andean species from the upper Amazonas Basin. Remaining taxa form a large clade that includes Cambeva, Scleronema, and all Brazilian species of Trichomycterus. The Minutum Clade contains Scleronema minutum and several species formerly placed in Trichomycterus and recently classified into the new genus Cambeva (C. balios, C. cubataonis, C. davisi, C. iheringi, C. pascuali, C. perkos, C. poikilos, C. stawiarski and C. zonatus). The Brazilian species of Trichomycterus are grouped into three main successive subclades. The first one includes (Itatiayae Clade) includes T. itatiaye, T. pauciradiatus, T. piratymbara, T. reinhardti, T. septemradiatus, T. cf. septemradiatus and Trichomycterus. sp. (Grotão). The second (Immaculatus Clade) includes T. albinotatus, T. alternatus, T. cf. auroguttatus, T. immaculatus, T. mimosensis and T. pradensis. The third one (Brasiliensis Clade) includes T. brasiliensis, T. cf. brasiliensis, T. candidus, T. nigroauratus, T. pirabitira, plus four undescribed species identified by sampling localities, Trichomycterus sp. (Grande), Trichomycterus. sp. (São Francisco), Trichomycterus sp. (Bonito), and Trichomycterus sp. (Turvo) (Brasiliensis Clade).
The second major clade in Trichomycterine lineage (Eremophilus lineage) exhibits four successive subclades at its base (Fig. 4). The first to split (Mutisii Clade) includes T. cachiraensis, T. sandovali, and the monotypic Eremophilus mutisii, all from the Magdalena basin. The next one (Guianensis Clade) joins T. guianensis and T. cf. guianensis from the Essequibo basin. The third (Chapmani Clade) includes T. chapmani, T. aff. spilosoma, and T. transandianus, each from separate trans-Andean drainages in Colombia. The fourth (Banneaui Clade) includes T. banneaui, T. aff. striatus, T. ruitoquensis, one undetermined species from Rancheria River, Trichomycterus sp. (Rancheria), and three undetermined species from the Magdalena basin, Trichomycterus sp. 4 (Riecito), Trichomycterus sp. (Manco) and Trichomycterus sp. (Samaná Norte). A group of five undescribed species of Trichomycterus from the Paraná-Paraguay basin forms a monophyletic group that is sister group to the Areolatus Clade, composed of Bullockia maldonadoi, T. areolatus, and T. chiltoni. Finally, the monophyletic Ituglanis occupies the apex of this major trichomycterine lineage. The species sampled grouped into two main clades. One contains the Parahybae Clade (I. boitata, I. parahybae, and I. amphipotamus) and Ramiroi Clade composed of I. goya, I. cf. goya, and I. ramiroi and two undescribed Amazonian species, Ituglanis sp. (Das Brancas) and Ituglanis sp. (Tapajos). The second one (Amazonicus clade) combines species from the Amazon and La Plata systems, with a subclade composed of I. amazonicus, I. herberti, I. cf. parkoi, Ituglanis. sp. (Xingu) and I. parkoi sister to a second subclade including I. cf. amazonicus, I. eichhorniarum, I. cf. eichhorniarum plus three undescribed species Ituglanis. sp. (Araguaia), Ituglanis. sp. (Cuiabá) and Ituglanis. sp. (João Dias).
The TSVSG Clade includes the subfamilies Tridentinae, Stegophilinae, Vandelliinae, Sarcoglanidinae, and Glanapteryginae (Fig. 5). We analyzed five representatives of three currently recognized glanapterygine genera, including the most generalized Listrura (L. camposi and L. picinguabae) and the highly derived psammophilic Pygidianops (Pygidianops sp. 1, and Pygidianops sp. 2) and Typhlobelus (T. guacamaya). The only glanapterygine genus not analyzed was Glanapteryx which contains two nominal species. Half of the sarcoglanidine genera were analyzed (Microcambeva barbata, Sarcoglanis simplex and Stauroglanis gouldingi); missing were Ammoglanis, Malacoglanis, and Stenolicmus. An undescribed taxon apparently belonging to the Glanapteryginae-group, Trichomycteridae n. gen. (de Pinna & Datovo; pers. comm.) also was incorporated into our study.
The resulting hypothesis for the TSVSG Clade did not support the monophyly of Glanapteryginae and Sarcoglanidinae, respectively, or the sister group relationship between those subfamilies. Rather, representatives of both subfamilies from Atlantic coastal drainages grouped into one clade (Southeastern Clade), while those from the Amazon and Orinoco basins (Northwestern Clade) and the enigmatic Potamoglanis from the Amazon and Paraguay basins formed a second clade more closely related to other TSVSG taxa. The Southeastern Clade is at the base of the TSVSG clade and includes the undescribed genus of de Pinna & Datovo, Microcambeva barbata (Sarcoglanidinae), and the two species of Listrura (Glanapteryginae). Potamoglanis is sister to the Northwestern Clade composed of Sarcoglanis simplex, Stauroglanis gouldingi, Typhlobelus guacamaya and the two species of Pygidianops. That clade is the sister to the so-called Vandelliinae-group, a node with strong support that includes the Tridentinae (sensu stricto), Stegophilinae, and Vandelliinae. Tridentinae (minus Potamoglanis) is at the base of the whole group with Stegophilinae and Vandelliinae appearing as sister taxa. Our results do not support the recent allocation of Potamoglanis hasemani in the Tridentinae (contra; Henschel et al. 2017, see Discussion) and resolve Tridens sp. 2 (Madeira) as the sister group to the clade formed by Tridensimilis brevis and Tridentopsis pearsoni. All SH tests using the 75% complete UCE matrix showed that topologies supporting the monophyly of the Glanapteryginae and the Sarcoglanidinae, respectively, their mutual sister-group relationships, as well as the alternative hypotheses of relationships of Potamoglanis32,35,36 were significantly worse (P < 0.001) than the recovered phylogeny (Table 1).
With nine of 11 genera of the Stegophilinae represented in our analysis (only Apomatoceros and Schultzichthys are missing), the internal relationships of the subfamily were well resolved and mostly in agreement with a recent morphological revision36. Our hypothesis divides the Stegophilinae into two major subgroups. The largest clade (Pareiodon Clade) contains Homodiaetus (Ho. anisitsi and Ho. passarellii) at the base and two subclades: one composed of the monotypic Megalocentor echthrus and Henonemus (He. intermedius, He. punctatus, and Henonemus sp.) and the second by the monotypic Pareiodon sister to monotypic Acanthopoma sister group to Pseudostegophilus. A few analyses do not support the monophyly of Pseudostegophilus (Supplementary Figs. 6–8). The second major stegophiline group (Stegophilus Clade) clusters Ochmacanthus (O. alternus, O. reinhardti, and two unnamed species of Ochmacanthus from the Itaya and Itatá Rivers) as sister group to the clade composed of Stegophilus panzeri and the monotypic Haemomaster venezuelae. In the Bayesian and ML analyses with 90% complete matrices the monophyly of the Stegophilinae is not recovered (Supplementary Figs. 2 and 5) and the resulting lineage, Stegophilus Clade + Vandelliinae shows extremely low support (<0.5 posterior probability and 50% bootstrap).
Two of the four vandelliine genera were included in our analysis, Paracanthopoma and Vandellia. The monophyly of each genus and the whole subfamily is strongly supported, but several species-level interrelationships showed low support (Fig. 5). Some minor differences were observed in the interspecific relationships of Vandellia across the different analyses (Supplementary Figs. 1, 3 and 8).
Discussion
The present phylogenomic analysis is the largest molecular dataset ever assembled for Trichomycteridae. Interfamilial and intergeneric relationships are mostly congruent with previous hypotheses based on morphology20,29,34,37 and multilocus datasets21,30,32. The species tree was less resolved with low bootstrap values and switched the positions of some taxa compared to topologies supported by analyses of the concatenated dataset. Inconsistencies between trees38,39 can be caused by different factors such as gene duplication40 horizontal transfer41, and incomplete sorting of ancestral polymorphism42,43,44,45. Discrepancies in bootstrap values are associated with the traditional concatenation approach, insufficient data46,47,48 and issues inherent to multilocus bootstrapping methods49,50. However, the most significant reason why the species have different relationships amongst species trees should be attributable to deep coalescence processes, where multiples lineages tend to persist into the deeper portion of the species tree51. This pattern is common in groups with rapid and/or recent diversification, where the species tree are characterized by short branches52. In this case, gene lineages persist through the species trees and coalesce with gene lineages that are not from the most closely related species due to the short evolutionary time that is not sufficient for the fixation of gene lineages by genetic drift49.
Notwithstanding those limitations, our phylogenomic hypothesis supports the monophyly of Trichomycteridae. This result is congruent with all morphological studies, which provide a high number of unequivocal synapomorphies for the family20,29,34,37. Additionally, our topology obtained supports the monophyly and recognition of most clades previously recognized within the Trichomycteridae. For instance, Copionodontinae and Trichogeninae have been considered basal lineages since the first phylogenetic studies incorporating those taxa20. Members of these subfamilies show several plesiomorphic character-states not present in the remaining trichomycterids and the sister group relationship between Copionodontinae and Trichogeninae has been evidenced in other studies20,29,37.
Our results support the monophyly of the Clade C taxa that corresponds to the classic definition of the family, that is, the Trichomycteridae prior to the discoveries of trichogenines and copionodontines in the late 20th century37,53. In the present analysis, Clade C is basally divided into two lineages: TSVSG clade and Trichomycterinae sensu Datovo & Bockmann29. Monophyly of TSVSG clade composed of the Tridentinae, Stegophilinae, Vandelliinae, Sarcoglanidinae, and Glanapteryginae was first proposed by Costa & Bockmann54 and corroborated by all subsequent morphological20,29,36,55 and molecular studies30,32. The major taxonomic change in the TSVSG clade was the formal incorporation of Potamoglanis, formerly referred to as the Trichomycterus hasemani-group. This group was long proposed to be related to the TSVSG clade, but its closer affinities are contentious. De Pinna56 was the first to draw attention to the presence of some highly derived features of Potamoglanis hasemani, such as its small body size and the presence of a single enormous cranial fontanel. These characters, although not unique, are shared with tridentines and de Pinna35 suggested the inclusion of Potamoglanis in that subfamily. The cladistic analysis of DoNascimiento36, with 49 terminal taxa and 520 morphological characters, refuted de Pinna’s35 hypothesis and supported Potamoglanis as the sister group of the clade comprising tridentines, stegophilines, and vandelliines (Fig. 6d). In the molecular study of Ochoa et al.30, Potamoglanis was the sister group to Sarcoglanis (Fig. 6e). More recently, Henschel et al.32 performed a multilocus analysis proposing Potamoglanis as sister group to tridentines (Fig. 6f). Based on that finding and ignoring prior hypotheses30,36, Henschel et al.32 formally erected the genus Potamoglanis as a member of an expanded Tridentinae. Nevertheless, the present analysis obtained a result similar to that of Ochoa et al.30 by grouping Potamoglanis with the sarcoglanidines and glanapterygines from the Amazon and Orinoco basins. It is worth mentioning that Potamoglanis does share some anatomical features traditionally considered to be synapomorphic at different levels of the Glanapteryginae-group, such as the possession of a toothless lateral process on the premaxilla and a long anterior process on the hyomandibula32,57. Moreover, all three synapomorphies proposed by Henschel et al.32 to support Potamoglanis as a tridentine are blatantly problematic as they are shared by some sarcoglanidines and glanapterygines. The first synapomorphy, expanded single cranial fontanel, is present in most members of the basal-most sarcoglanidine genus Ammoglanis (pers. obs.58,59). The second synapormophy, origin of the dorsal fin just above or posterior to the anal-fin origin, is absent in P. anhanga57 and present in Listrura picinguabae60 and some L. costai61 and not comparable in most glanapterygines, which lack a dorsal fin. The third proposed synapomorphy for Potamoglanis is the short ventral process of the opercle (p. 732), however Potamoglanis has a process comparable in size or larger32,57 than that observed in several sarcoglanidines and glanapterygines (pers. obs.; see also Stauroglanis gouldingi62, Glanapteryx anguilla35; Microcambeva barbata55; Ammoglanis pulex59; Microcambeva draco63; Pygidianops amphioxus64 and several species of Typhlobelus65). In light of these issues and the conflicting hypotheses of relationships for Potamoglanis with or within the TSVSG clade, we consider the assignment of Potamoglanis to any trichomycterid subfamily impetuous. Therefore, for the sake of nomenclatural stability, the present paper follows the traditional concept of the Tridentinae, i.e. not including Potamoglanis20,34.

Phylogenetic hypothesis for Trichomycteridae.
There has been some debate on the relationships of the Sarcoglanidinae and Glanapteryginae. The two subfamilies are traditionally considered sister taxa by most anatomical studies29,34,55,66 (Fig. 6a–c). However, the most recent morphological analysis36 and all molecular phylogenies of the Trichomycteridae30,32 refuted the monophyly of the Glanapteryginae-group (Fig. 6d–g). Based on morphology, DoNascimiento36 resolved the Sarcoglanidinae and Glanapteryginae as successive sister taxa to the Vandelliinae-group. Based on molecules Ochoa et al.30 placed Glanapteryginae at the base of the TSVSG clade; however, Sarcoglanidinae was non-monophyletic, with Stauroglanis closer to vandelliines and Sarcoglanis closer to Potamoglanis. In the topology of Henschel et al.32, tridentines plus Potamoglanis are intercalated between Glanapteryginae (at the base of the TSVSG clade) and the clade (Sarcoglanidinae + (Stegophilinae + Vandelliinae)). Our analysis shows even more striking result in which neither Glanapteryginae nor Sarcoglanidinae are monophyletic. Members of both subfamilies are clustered into two clades that are successive sister taxa to the Vandelliinae-group. Interestingly, our analysis groups the glanapterygines and sarcoglanidines from the Atlantic coastal drainages (Listrura, Microcambeva, and Trichomycteridae n. gen.) into one clade (Southeastern Clade), and members of both subfamilies from the Amazon and Orinoco basins (Pygidianops, Sarcoglanis, Stauroglanis and Typhlobelus) along with Potamoglanis (Amazon and Paraguay basins) into a second clade (Northwestern Clade). The glanapterygines and sarcoglanidines grouped in the latter clade curiously share several reductive features, such as extreme reductions in the pigmentation and numbers of opercular odontodes, interopercular odontodes, and premaxillary teeth (pers. obs.58,62).
Dismantling the Glanapteryginae-group is a drastic change that obviously demands further investigation, but this result is not so unexpected. A critical appraisal of the osteological characters listed to support various nodes of the Glanapteryginae-group demonstrates that several putative synapomorphies are highly homoplastic or vaguely delimited. For instance, the quadrate with a “posteriorly directed process” (p. 725)55 is considered a synapomorphy for the clade Glanapteryginae + Sarcoglanidinae54,55, but a large number of its members obviously lack this feature (pers. obs.; Glanapteryx anguilla62; Ammoglanis pulex59; Microcambeva ribeirae67; M. draco63; Pygidianops amphioxus68; and several species of Typhlobelus65). A second putative synapomorphy for the Glanapteryginae-group, hyomandibula with a long anterior process, is also problematic. The feature is also present in Potamoglanis and vandelliines59. Moreover, diagnoses and interrelationships among the putative basal-most genera of the Glanapteryginae and Sarcoglanidinae are particularly unstable and the limits of each subfamily are increasingly blurry69. For instance, new data suggest that Ammoglanis pulex is actually a glanapterygine, rather than a sarcoglanidine as originally described70. Allocation of newly discovered taxa (e.g., trichomycterid n. gen.) into one or another subfamily is often difficult and possibly arbitrary (pers. obs.70). These examples indicate the critical need for a taxonomically comprehensive revisionary study of the Glanapteryginae and Sarcoglanidinae, ideally combining morphological and molecular data.
Within the TSVSG clade, all morphological studies have postulated the existence of a monophyletic lineage consisting of Tridentinae, Stegophilinae, and Vandelliinae20,29,34,36 (Fig. 6a–d). This lineage, the Vandelliinae-group, is strongly supported by a profusion of anatomical specializations. Previous molecular analyses failed to recover the monophyly of the Vandelliinae-group and presented conflicting topologies within the TSVSG clade30,32 (Fig. 6e,f). The present study resolves a monophyletic Vandelliinae-group (Fig. 6g). Our tree also supports the Tridentinae as sister group to the clade Stegophilinae + Vandelliinae, thus agreeing with the topologies obtained by most previous analyses20,32,34,36.
Only Baskin34 tested the interrelationships among all four genera traditionally assigned to Tridentinae. In that morphological analysis, Miuroglanis and Tridentopsis are placed as the successive sister taxa to the node Tridens + Tridensimilis. As subsequent analyses never sampled all tridentine genera, Baskin’s34 scheme prevailed as the only cladistic study among tridentines for over 45 years. The recent analyses of DoNascimiento36 and Henschel et al.32, did not include Miuroglanis and Tridentopsis, respectively. Nevertheless, both analyses obtained a topology compatible with that of Baskin34. Unfortunately, we could not sample Miuroglanis and our resolution of tridentines differs from the past analyses by obtaining Tridensimilis as sister group to the clade Tridens + Tridentopsis. Our topology should be seen as provisional inasmuch the identification of “Tridens sp. Madeira” is inconclusive. That species does not perfectly fit the traditional definition of the genus and seems to exhibit morphological conditions apparently intermediate between Tridens and Tridensimilis.
Our hypothesis of interrelationships among stegophilines is almost identical to the comprehensive revision of the subfamily published by DoNascimiento36. In both analyses, Stegophilinae has a basal dichotomy into a clade clustering Haemomaster, Ochmacanthus, and Stegophilus and another grouping all remaining genera. The only difference between the two topologies is the placement of the monotypic Acanthopoma as sister group to Pseudostegophilus in our analysis. DoNascimiento’s36 tree placed Acanthopoma in a basal polytomy with Pareiodon and Pseudostegophilus. In the pioneering molecular analysis of the Vandelliinae-group, Fernandez & Schaefer21 obtained a sister -group relationship between Acanthopoma and Pareiodon in a rather distal position within Stegophilinae, as sister group to the clade (Pseudostegophilus (Apomatoceros + Henonemus)).
Different studies arrived at conflicting hypotheses of relationships among the vandelliine genera29,32,34,36. Our analysis includes representatives of Paracanthopoma and Vandellia and support the monophyly of each genus. However, proper resolution of the Vandelliinae will depends of a broad taxonomic revision of the group, since only a small fraction of its true diversity has been carefully studied and several species and genera await formal descriptions.
Among trichomycterids, controversy has surrounded the phyletic status and composition of the Trichomycterinae. Most traditional studies considered the subfamily to be a non-monophyletic assemblage that includes some taxa more closely related to the TSVSG clade than to other trichomycterines (Fig. 6a,b,d). Three trichomycterine subgroups were explicitly proposed as aligned with the TSVSG clade: Scleronema, Ituglanis, and Potamoglanis (previously referred to as Trichomycterus hasemani group). The inclusion of the lattermost genus within the TSVSG clade is unequivocal on both morphological20,29,36,56 and molecular grounds30,32. On the other hand, a Scleronema-Ituglanis-TSVSG relationship was rejected by Datovo & Bockmann29, who additionally provided morphological evidence for the grouping of these genera with the remaining trichomycterines (excluding Potamoglanis) into a monophyletic Trichomycterinae (Fig. 6c). This hypothesis was subsequently corroborated by all molecular analyses30,32, including the present one (Clade D; Fig. 6e,g).
Interrelationships within Trichomycterinae have never been extensively surveyed using morphological data, notwithstanding some proposals of small putative subgroups with restricted geographic distributions20,71,72,73,74,75,76. The multilocus analysis of Ochoa et al.30 was the first study to employ a substantial taxonomic sampling of the subfamily. That topology divided the Trichomycterinae into two major lineages and six main subclades (D1, D2, D3, D4, D5, and E). The present UCE analysis expands the previous sampling of trichomycterines by roughly 15% and the resulting subfamily tree exhibits only three most significant divergences from Ochoa et al.30 the placement of an undescribed trichomycterine from the Caribbean coast of Venezuela (previously unsampled) at the base of the whole subfamily and the non-monophyly of Clades D1 and D2. As in Ochoa et al.30, most trichomycterines are divided into two major clades: the herein termed Eremophilus lineage and the Trichomycterus lineage.
Within the Eremophilus lineage, a novel sister group relationship between Ituglanis and the clade comprising Bullockia plus the Chilean species of Trichomycterus is consistently supported, either by multilocus analyses30 and the present genomic study. Monophyly of the clade consisting of Bullockia and the Chilean species of Trichomycterus is also congruent between both molecular analyses and was likewise recovered in the morphological study by DoNascimiento36.
In the Trichomycterus lineage, the sister group relationship between Scleronema and a clade of species traditionally assigned to Trichomycterus and transferred to the recently erected genus Cambeva was originally proposed by Ochoa et al.30 and is also recovered herein. Recognition of a monophyletic subset of species of Cambeva (originally comprising C. crassicaudatus, C. igobi, and C. stawiarski), was first advanced by Wosiacki & de Pinna74,77, based on shared derived characters of the caudal skeleton. Subsequently, Datovo & Bockmann29 provided evidence from the head musculature supporting a clade consisting of C. davisi and C. stawiarski. Datovo et al.78 included C. perkos in that group based on the shared presence of a high number of branchiostegal rays and, more recently, Terán et al.79 found morphological support to include in that group also their newly described C. ytororo. Katz et al.31 diagnosed Cambeva based on a combination of plesiomorphic or general character states and some derived characters uniquely shared by Cambeva and Scleronema.
A notable finding of the present genomic analysis is consistent support for several subclades within Trichomycterus previously identified in multilocus analyses30. These subclades coincide with circumscribed geographic regions that had a denser taxonomic sampling in both multilocus and UCE analyses. One of these main subclades includes species from South and Southeastern Brazil and is subdivided into multiple lineages that are partially congruent with species groups proposed by morphology. Thus, the clade including T. pauciradiatus, T. piratymbara, T. reinhardti, and T. septemradiatus, was originally proposed as containing T. reinhardti and T. pauciradiatus and defined by the shared color pattern consisting of a broad dark brown stripe along the lateral midline, bordered above by a light yellow longitudinal zone80. Composition of this clade was later expanded to include T. piratymbara and T. septemradiatus31. However, some emblematic species groups within Trichomycterus have not been recovered herein or in previous molecular analyses, such as the T. brasiliensis species-complex that has transited through multiple definitions and main changes in its taxonomic composition73,81,82. A major effort is needed to increase the taxonomic sampling of Trichomycterus from the Andes and Guiana shield regions, whose few representative species analyzed in Ochoa et al.30 were found to have shifting positions in the present analyses, even jumping to different main clades (e.g. T. cf. kneri and T. punctulatus). Ideally, increased taxonomic representation in the molecular analyses should be accompanied by a comprehensive morphological survey in order to provide phylogenetic diagnoses for each of these remnant groups of Trichomycterus.
As mentioned earlier, Trichomycterinae concentrates most of the species-level diversity and most of the taxonomic problems inherent to the family. The identification of trichomycterine subgroups is necessary to advance in the systematics of the Trichomycteridae, but should avoid the premature and unreasonable proposition of new taxa. The recent erection of the genus Cambeva31 is a prime example. Ochoa et al.30 obtained a fully resolved trichomycterine phylogeny, but prudently concluded that taxonomic changes at moment were premature. Among the original results of that study, was a clade composed of nine species of Trichomycterus from southeastern Brazil that appeared as the sister group of Scleronema. One year later, Katz et al.31 transferred these species of Trichomycterus to a new genus, Cambeva, based on a molecular matrix, including only nine sequences of the 25 species allocated in Cambeva. Most, if not all, osteological characters proposed by Katz et al.31 to diagnose Cambeva and the clade Cambeva + Scleronema are incorrect as revealed by a test survey of the literature (e.g.)73,83,84,85,86 and examination of a few clear and stained specimens (AD, pers. obs.). The study of Katz et al.31 does not address any of the crucial issues that should be taken into account before proposing any changes in the subfamilial classification, such as the elucidation of the controversial identity of the type species of Trichomycterus (T. nigricans), a reasonable sampling of the traditional trichomycterine genera (Hatcheria, Rhizosomichthys, and Silvinichthys are lacking), and the taxonomic allocation of the large number of species that remained in a polyphyletic Trichomycterus. The study also ignores most morphological characters proposed in previous studies for delimiting subgroups of their Cambeva74,77,78. Katz et al.31 further included in the new genus ten species neither examined for osteology nor sampled for DNA based on “general external appearance and occurrence in the same basins that Cambeva is distributed”. The arbitrary creation of new taxon based on an equivocal anatomical character possibly incur in a setback instead of a significant advance in trichomycterid taxonomy.
Finally, the phylogenetic hypothesis proposed here provides a strong framework for future analyses of biogeography, niche modeling, and evolutionary history of multiple traits. At the same time, it also highlights the major challenges to understanding of the systematics of this remarkable family of catfishes.
Material and Methods
Taxon sampling
The procedures used for the sampling, maintenance and analysis of the tissue fishes samples are in agreement with Brazilian law regulated by the National Council for the Control of Animal Experimentation (CONCEA) approved by the protocol 1058/2017 and ethical principles in animal research formulated by the Brazilian Society of Science in Laboratory Animals and authorized by the Bioscience Institute/UNESP Ethics Committee on the Use of Animals (CEUA). Tissue samples and voucher specimens used in this study are deposited in the collections of Laboratório de Biologia e Genética de Peixes, Universidade Estadual Paulista, Botucatu, Brazil (LBP), Instituto Nacional de Pesquisa da Amazônia, Manaus, Brazil (INPA), The Academy of Natural Sciences of Drexel University, Philadelphia, USA (ANSP), Colección de Zoologia de la Universidad del Tolima, Ibagué, Colombia (CZUT-IC) and Colección Zoologica de Referencia del Museo de Ciencias Naturales Federico Carlos Lehmann Valencia del INCIVA (IMCN), Cali, Colombia. Supplementary Table 1 synthesizes pertinent data from all samples belonging to the ingroup and outgroups. Our analysis included representatives of the all eight subfamilies, and from 30 genera and 139 species of Trichomycteridae with a total of 153 individuals. Representatives of all remaining families of Loricarioidei were included as outgroups and the resulting trees were rooted in the characiform Leporinus striatus.
Library preparation, target enrichment and sequencing of UCEs
DNA extractions were carried out from approximately 25 mg of tissue using Qiagen DNeasy Tissue kits following the manufacturer’s protocols. After a visual analysis of the quality of DNA on the agarose gel, we quantified 2 µl of each sample using fluorometry (Qubit, Life Technologies) to obtain an ideal concentration between 10–40 ng/µl were used for the analysis. Library preparation and sequencing were performed at Arbor Biosciences (AB; arborbiosci.com; Ann Arbor, MI, USA). AB staff sheared 1–2 µg of DNA to 400–600 bps in length using a Diagenode Bioruptor Standard (UCD 200) with 6–8 cycles of sonication (depending on DNA quality) to prepare the libraries. The DNA libraries from 153 individuals were prepared using the Nextera (Epicentre Biotechnologies, Inc.) library preparation protocol for solution-based target enrichment following Faircloth et al.2 and increasing the number of PCR cycles following the tagmentation reaction to 20 as recommended by Faircloth et al.5. AB staff used the Nextera library preparation protocol of in vitro transposition followed by PCR to prune the DNA and attach sequencing adapters87. The Epicentre Nextera kit was used to prepare transposase-mediated libraries with insert sizes averaging 100 bp (95% CI: 45 bp) following Adey et al.87. The libraries were enriched using a probe set developed for application to ostariophysan fishes to generate sequences data for approximately 2500 UCE loci16. DNA was converted to Illumina sequencing libraries with a slightly modified version of the NEBNext(R) Ultra (TM) DNA Library Prep Kit for Illumina(R). After ligation of sequencing primers, libraries were amplified using KAPA HiFi HotStart ReadyMix (Kapa Biosystems) for six cycles using the manufacturer’s recommended thermal profile and dual P5 and P7 indexed primers see Meyer et al.88 for primer configuration89. After purification with SPRI beads, libraries were quantified with the Quant-iT(TM) Picogreen(R) dsDNA Assay kit (ThermoFisher). AB staff enriched pools comprising 100 ng each of eight libraries (800 ng total) using the MYbaits(R) Target Enrichment system (MYcroarray) followed manual version 3.0. After capture cleanup, the bead-bound library was resuspended in the recommended solution and amplified for 10 cycles, using a universal P5/P7 primer pair and KAPA HiFi reagents. After purification, each captured library pool was quantified with PicoGreen and combined with all other pools in projected equimolar ratios prior to sequencing. Sequencing was performed across two Illumina HiSeq paired-end 100 bp lanes using v4 chemistry.
Sequence data processing
The standard PHYLUCE pipeline was used for processing target-enriched UCE data90. The matrices used in this study were deposited at figshare (doi: 10.6084/m9.figshare.7857485) and sequences are available at NCBI Sequence Read Archive (SRA) submissions: (PRJNA530617). After sequencing, reads were trimmed for adapter contamination, low-quality bases and sequences containing ambiguous bases, using the Illumiprocessor software pipeline, included in the PHYLUCE. We assembled reads and generated consensus contigs for each species using ABySS using a kmer value of 55 (version 2.0.2)91.
Following assembly, we screened the resulting assemblies for those contigs matching enriched UCE loci using the “match_contigs_to_probes” program and discarded putative paralogs with the standard PHYLUCE algorithm. We created a fasta file containing all data for all taxa. The monolithic fasta files were used to generate the alignments with MUSCLE92 and the resulting alignments were trimmed, using the algorithm implemented by the seqcap_align_2.py script within PHYLUCE. Every alignment was cleaned from the locus name using “phyluce_align_remove_locus_name_from_nexus_lines”. From the trimmed alignments, we created three matrices with 50, 75, and 90% of completeness in order to evaluate the role of missing data in our matrices, tree topology and clade support values. For each matrix we prepared a concatenated alignment in phylip format and every matrix was analyzed using maximum likelihood (ML) algorithm in RAxML v8.2.X93 to compare the topologies with different levels of completeness.
Phylogenetic analyses
The best-fitting partitioning scheme was obtained using the Sliding-Window Site Characteristics (SWSC) approach94 to divide each UCE into three data blocks, based on the pattern of variation of entropy, multinomial likelihood and GC content to generate a partition that accounts for heterogeneity in rates and patterns of evolution within each UCE. Posteriorly the data blocks were analyzed using standard algorithms in PartitionFinder v295. The best partitioning scheme grouped loci with having the same substitution model to be used in subsequent analyses. We performed maximum likelihood (ML) inferences on the three concatenated matrices using RAxML ver. 8.1.393, assuming a general time reversible model of rate substitution and gamma-distributed rates among sites (GTRGAMMA). The number of alternative runs and a posteriori bootstrapping analysis were conducted using the autoMRE function of bootstopping criteria96. Bayesian inference was performed in ExaBayes version 1.597, with two independent runs, each with four chains (one cold and three heated) with 1 million iterations with priors for parameters by default. Tree space was sampled every 100 generations to yield a total of 10,001 trees. We assessed convergence of the posterior distribution examining the ESS > 200 (effective sample size) and evaluating posterior trace distribution in Tracer v1.6.098. We obtained the 50% most credible set of trees from the posterior distribution of possible topologies using the consensus algorithm of ExaBayes with 25% burn-in.
A coalescent analysis of species tree was inferred from individual gene trees to account for heterogeneous gene histories that may influence accurate resolution of phylogenetic relationships51. Optimal gene trees for each UCEs under maximum likelihood in RAxML were generated and the unrooted gene trees were used to estimate the true species tree in ASTRAL v 5.6.1.99, under the multi-species coalescent model and bootstrap option.
To compare the topologies obtained for each completeness matrix and for each inference method we used the software ViPhy33 which incorporate information on similarity scores and allow highlighting of similar structures in multiples trees. We also explored different alternative hypotheses of relationships in Trichomycteridae according with previous morphological and molecular studies. To test the monophyly of Glanapteryginae and Sarcoglanidinae, as well as the different hypotheses of relationships of Potamoglanis, we estimated first the best constrained tree for every hypotheses using the concatenated matrix of 75% afterward we compared the best constrained tree with unconstrained tree using Shimodaira-Hasegawa (SH) test100 through the -f option in RAxML, and subsequently, p-values were extracted to do Bonferroni correction for the different comparisons using “padjust” in R.
Ethical approval and informed consent
The procedures used for the sampling, maintenance and analysis of the tissue fishes samples are in agreement with Brazilian law regulated by the National Council for the Control of Animal Experimentation (CONCEA) approved by the protocol 1058/2017 and ethical principles in animal research formulated by the Brazilian Society of Science in Laboratory Animals and authorized by the Bioscience Institute/UNESP Ethics Committee on the Use of Animals (CEUA).
Data availability
The matrices used in this study were deposited at figshare (doi: 10.6084/m9.figshare.7857485) and sequences are available at NCBI Sequence Read Archive (SRA) submissions: (PRJNA530617).
References
Crawford, N. G. et al. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol. Lett. 8, 783–786 (2012).
Faircloth, B. C. et al. Ultraconserved Elements Anchor Thousands of Genetic Markers Spanning Multiple Evolutionary Timescales. Syst. Biol. 61, 717–726 (2012).
Mccormack, J. E. et al. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 22, 746–754 (2012).
Smith, B. T. et al. The drivers of tropical speciation. Nature 515, 406–409 (2014).
Faircloth, B. C., Sorenson, L., Santini, F. & Alfaro, M. E. A Phylogenomic Perspective on the Radiation of Ray- Finned Fishes Based upon Targeted Sequencing of Ultraconserved Elements (UCEs). Plos One 8 (2013).
Harrington, R. C. et al. Phylogenomic analysis of carangimorph fishes reveals flatfish asymmetry arose in a blink of the evolutionary eye. BMC Evol. Biol. 1–14, https://doi.org/10.1186/s12862-016-0786-x (2016).
McGee, M. D. et al. Replicated divergence in cichlid radiations mirrors a major vertebrate innovation. Proc. R. Soc. B Biol. Sci. 283, 20151413 (2016).
Chakrabarty, P. et al. Phylogenomic Systematics of Ostariophysan fishes: Ultraconserved Elements Support the Surprising Non-monophyly of Characiformes. Syst. Biol. 66, 881–895 (2017).
Alfaro, M. E. et al. Explosive diversification of marine fishes at the Cretaceous-Palaeogene boundary. Nat. Ecol. Evol. 2, 688–696 (2018).
Roxo, F. F. et al. Phylogenomic reapprasial of the Neotropical catfish family Loricariidae (Teleostei:Siluriformes) using ultraconserved elements. Mol. Phylogenet. Evol., https://doi.org/10.1016/j.ympev.2017.07.007 (2019).
Alda, F. et al. Resolving Deep Nodes in an Ancient Radiation of Neotropical Fishes in the Presence of Conflicting Signals from Incomplete Lineage Sorting. Syst. Biol. 68, 573–593 (2019).
Crawford, N. G. et al. A phylogenomic analysis of turtles. Mol. Phylogenet. Evol. 83, 250–257 (2015).
Siepel, A. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005).
Derti, A., Roth, F. P., Church, G. M. & Wu, C. Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants. Nat. Genet. 38, 1216–1220 (2006).
Gilbert, P. S. et al. Genome-wide ultraconserved elements exhibit higher phylogenetic informativeness than traditional gene markers in percomorph fishes. Mol. Phylogenet. Evol. 92, 140–146 (2015).
Faircloth, B. C. et al. A target enrichment bait set for studying relationships among ostariophysan fishes. Copeia, https://doi.org/10.1101/432583 (2018).
Fricke, R., Eschemeyer, W. N. & van der Laan, R. Eschemeyer’s Catalog of Fishes:Genera, Species, References. Available at, http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp, (Accessed: 20th November 2017) (2019).
Zuanon, J. & Sazima, I. Vampire catfishes seek the aorta not the jugular: candirus of the genus Vandellia (Trichomycteridae) feed on major gill arteries o fhost fishes Aqua. J. Ichthyol. Aquat. Biol. 8, 31–36 (2004).
Winemiller, K. O. & Yan, H. Y. Obligate mucusfeeding in aSouth American trichomycterid catfish (Pisces:Ostariophysi). Copeia 522–514 (1989).
de Pinna, M. C. C. Phylogenetic Relationships of Neotropical Siluriformes (Teleostei:Ostariophysi):Historical Overview and Synthesis of Hypotheses. In Phylogeny and Classification of Neotropical Fishes (eds. Malabarba, L. R., Reis, R. E., Vari, R. P., Lucena, Z. M. S. & Lucena, C. A. S.) 279–330 (EDIPUCRS, 1998).
Fernández, L. & Schaefer, S. A. Molecular Phylogenetics and Evolution Relationships among the Neotropical Candirus (Trichomycteridae, Siluriformes) and the evolution of parasitism based on analysis of mitochondrial and nuclear gene sequences. 52, 416–423 (2009).
Goulding, M. Ecologia da pesca do rio Madeira (1979).
Goulding, M. The fishes and the forest: explorations in Amazoninan Natural History. (University of California Press, 1980).
Kelley, W. A. & Atz, J. W. A pygiidid catfish taht can suck blood from goldfish. Copeia 702–704 (1964).
Machado, F. A. & Sazima, I. Comportamento alimentar do peixe hematófago Branchioica bertonii (Siluriformes, Trichomycteridae). Cienc. Cult. 35, 344–348 (1983).
Wosiacki, W. B. New species of the catfish genus Trichomycterus (Siluriformes, Tri- chomycteridae) from the headwaters of the rio São Francisco basin, Brazil. Zootaxa 592, 1–12 (2004).
de Pinna, M. C. C. & Wosiacki, W. B. Family Trichomycteridae. In Checklist of the Freshwater Fishes of South and Central America (ed. Reis, R.E., Kullander, S.O., Ferraris, C.J., J.) 270–290 (EDIPUCRS, 2003).
Fernández, L. & Schaefer, S. A. New Trichomycterus (Siluriformes: Trichomycteridae) from an Offshore Island of Colombia Author. Cope 2005, 68–76 (2005).
Datovo, A. & Bockmann, F. A. Dorsolateral head muscles of the catfish families Nematogenyidae and Trichomycteridae (Siluriformes: Loricarioidei): comparative anatomy and phylogenetic analysis. Neotrop. Ichthyol. 8, 193–246 (2010).
Ochoa, L. E. et al. Multilocus analysis of the catfish family Trichomycteridae (Teleostei: Ostariophysi: Siluriformes) supporting a monophyletic Trichomycterinae. Mol. Phylogenet. Evol. 115, 71–81 (2017).
Katz, A. M., Barbosa, M. A., de Oliveira Mattos, J. L. & da Costa, W. J. E. M. Multigene analysis of the catfish genus Trichomycterus and description of a new South American trichomycterine genus (Siluriformes, Trichomycteridae). Zoosystematics Evol. 94, 557–566 (2018).
Henschel, E., Mattos, J. L. O., Katz, A. M. & Costa, W. J. E. M. Position of enigmatic miniature trichomycterid catfishes inferred from molecular data (Siluriformes). Zool. Scr. 1–10, https://doi.org/10.1111/zsc.12260 (2017).
Bremm, S. et al. Interactive visual comparison of multiple trees. In 2011 IEEE Conference on Visual Analytics Science and Technology (VAST) 31–40, https://doi.org/10.1109/VAST.2011.6102439 (2011).
Baskin, J. N. Structure and relationships of the Trichomcyteridae. (City University of New York, 1973).
De Pinna, M. C. C. Redescription of Glanapteryx anguilla, with Notes on the Phylogeny of Glanapteryginae (Siluriformes, Trichomycteridae). Proc. Acad. Nat. Sci. Philadelphia 141, 361–374 (1989).
DoNascimiento, C. Morphological Evidence for the Monophyly of the Subfamily of Parasitic Catfishes Stegophilinae (Siluriformes, Trichomycteridae) and Phylogenetic Diagnoses of Its Genera. Copeia 103, 933–960 (2015).
de Pinna, M. C. C. A new subfamily of Trichomycteridae (Teleostei, Siluriformes), lower loricarioid relationships and a discussion on the impact of additional taxa for phylogenetic analysis. Zool. J. Linn. Soc. 106, 175–229 (1992).
Maddison, W. P. & Wiens, J. J. Gene Trees in Species Trees. Syst. Biol. 46, 523–536 (1997).
Slowinski, J. B. & Page, R. D. How should species phylogenies be inferred from sequence data? Syst. Biol. 48, 814–825 (1999).
Fitch, W. M. Distinguishing homologous from analogous proteins. Syst. Zool. 19, 99–113 (1970).
Cummings, M. P. Transmission patterns of eukaryotic transposable elements: arguments for and against horizontal transfer. Trends Ecol. Evol. 9, 141–145 (1994).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–95 (1989).
Takahata, N. & Nei, M. Gene genealogy and variance of interpopulational nucleotide differences. Genetics 110, 325–344 (1985).
Hudson, R. R. & Coyne, J. A. Mathematical consequences of the genealogical species concept. Evolution 56, 1557–1565 (2002).
Carstens, B. C. & Knowles, L. L. Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: An example from melanoplus grasshoppers. Syst. Biol. 56, 400–411 (2007).
McCormack, J. E. et al. A Phylogeny of Birds Based on Over 1,500 Loci Collected by Target Enrichment and High-Throughput Sequencing. Plos One 8, 1–11 (2013).
Kubatko, L. S. & Degnan, J. H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56, 17–24 (2007).
Roch, S. & Steel, M. Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theor. Popul. Biol. 100, 56–62 (2015).
Sayyari, E. & Mirarab, S. Fast Coalescent-Based Computation of Local Branch Support from Quartet Frequencies. Mol. Biol. Evol. 33, 1654–1668 (2016).
Huang, H., He, Q., Kubatko, L. S. & Knowles, L. L. Sources of error inherent in species-tree estimation: Impact of mutational and coalescent effects on accuracy and implications for choosing among different methods. Syst. Biol. 59, 573–583 (2010).
Degnan, J. H. & Rosenberg, N. A. Discordance of Species Trees with Their Most Likely Gene Trees. Plos Genet. 2, e68 (2006).
Degnan, J. H. & Rosenberg, N. A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340 (2009).
Britski, H. A. & Ortega, H. Trichogenes longipinnis, novo gênero de Trichomycterinae do sudeste do Brasil (Pisces, Siluriformes). Rev. Bras. Zool. 1, 211–216 (1982).
Costa, W. J. E. M. & Bockmann, F. A. Un nouveau genre néotropical de la famille des Trichomycteridae (Siluriformes: Loricarioidei). Rev. Française d’Aquariologie Herpétologie 20, 43–46 (1993).
Costa, W. J. E. M. & Bockmann, F. A. A new genus and species of Sarcoglanidinae (Siluriformes: Trichomycteridae) from southeastern Brazil, with a re-examination of subfamilial phylogeny. J. Nat. Hist. 28, 715–730 (1994).
de Pinna, M. C. C. A New Sarcoglanidine Catfish, Phylogeny of Its Subfamily, and an Apprasail of the Phyetic Status of the Trichomycterinae (Teleostei, Trichomycteridae). Am. Museum Novit. (1989).
Dutra, G. M., Wosiacki, W. B. & Pinna, M. C. Cde Trichomycterus anhanga, a new species of miniature catfish related to T. hasemani and T. johnsoni (Siluriformes: Trichomycteridae) from the Amazon basin, Brazil. Neotrop. Ichthyol. 10, 225–231 (2012).
Costa, W. J. E. M. A new genus and species of Sarcoglanidinae (Siluriformes: Trichomycteridae) from the Araguaia basin, central Brazil, with notes on subfamilial phylogeny. Ichthyol. Explor. Freshwaters 5, 207–216 (1994).
de Pinna, M. C. C. & Winemiller, K. O. A new species of Ammoglanis (Siluriformes: Trichomycteridae) from Venezuela. Ichthyol. Explor. Freshwaters 11, 255–264 (2000).
Villa-verde, L. & Costa, W. J. E. M. A new glanapterygine catfish of the genus Listrura (Siluriformes: Trichomycteridae) from the southeastern Brazilian coastal plains. Zootaxa 50, 43–50 (2006).
Villa-Verde, L., Lazzarotto, H. & Lima, S. M. Q. A new glanapterygine catfish of the genus Listrura (Siluriformes: Trichomycteridae) from southeastern Brazil, corroborated by morphological and molecular data. Neotrop. Ichthyol. 10, 527–538 (2012).
de Pinna, M. C. C. A new Sarcoglanidine catfish, phylogeny of Its Subfamily, and an Appraisal of the Phyletic Status of the Trichomycterinae (Teleostei, Trichomycteridae). Am. Museum Novit. 2950, 1–39 (1989).
Mattos, J. L. O. & Lima, S. M. Q. Microcambeva draco, a new species from northeastern Brazil (Siluriformes: Trichomycteridae). Ichthyol. Explor. Freshwaters 21, 233–238 (2010).
de Pinna, M. C. C. & Kirovsky, A. L. A new species of sand-dwelling catfish, with a phylogenetic diagnosis of Pygidianops Myers (Siluriformes: Trichomycteridae: Glanapteryginae). Neotrop. Ichthyol. 9, 493–504 (2011).
de Pinna, M. C. C. & Zuanon, J. The Genus Typhlobelus: Monophyly and Taxonomy, with Description of a New Species with a Unique Pseudotympanic Structure (Teleostei: Trichomycteridae). Copeia 2013, 441–453 (2013).
de Pinna, M. C. C., Helmer, J. L., Britski, H. A. & Nunes, L. R. A new species of Trichogenes from the rio Itapemirim drainage, southeastern Brazil, with comments on the monophyly of the genus (Siluriformes: Trichomycteridae). Neotrop. Ichthyol. 8, 707–717 (2010).
Costa, W. J. E. M. & Lima, S. M. Q. & carlos roberto S. F. Bizerril. Microcambeva ribeirae sp. n. (Teleostei: Siluriformes: Trichomyc- teridae): a new sarcoglanidine catfish from the Rio Ribeira do Iguape basin, southeastern Brazil. Zootaxa 563, 1–10 (2004).
Pinna, M. C. C. D & Kirovsky, A. L. A new species of sand-dwelling catfish, with a phylogenetic diagnosis of Pygidianops Myers (Siluriformes: Trichomycteridae: Glanapteryginae). 9, 493–504 (2011).
de Pinna, M. C. C. & Winemiller, K. O. A new species of Ammoglanis (Siluriformes: Trichomycteridae) from Venezuela. Ichthyol. Explor. Freshwaters 11, 225–264 (2000).
de Pinna, M. C. C. The dawn of phylogenetic research on Neotropical fishes: A commentary and introduction to Baskin (1973), With an overview… The dawn of phylogenetic research on Neotropical fishes: a commentary. Neotrop. Ichthyol. 14, e150127 (2016).
Costa, W. J. E. M. Description de huit nouvelles espèces du genre Trichomycterus (Siluriformes:Trichomycteridae), du Brésil oriental. Rev. Française d’ Aquariol. Herpetol. 18, 101–110 (1992).
Wosiacki, W. B. Estudo das relações Filogenéticas de Trichomycterinae (Telesotei: Siluriformes:Trichomycteridae) com uma proposta de classificação. (Universidade de São Paulo, 2002).
Barbosa, M. A. & Costa, W. J. E. M. Trichomycterus potschi (Siluriformes: Loricarioidei) a new trichomycterid catfish from coastal streams of southeastern Brazil. Ichthyol. Explor. Freshwaters 14, 281–287 (2003).
Wosiacki, W. B. & de Pinna, M. A New Species of the Neotropical Catfish Genus Trichomycterus (Siluriformes: Trichomycteridae) Representing a New Body Shape for the Family. Copeia 273–278, https://doi.org/10.1643/CI-06-237 (2008).
Barbosa, M. A. & Costa, W. J. E. M. Validade, relações filogenéticas e redescrição de Eremophilus candidus Ribeiro 1949 (Teleostei, Siluriformes, Trichomycteridae). Arq. do Mus. Nac. Rio Janeiro 61, 179–188 (2003).
Fernández, L. & Osinaga, K. A new Trichomycterus (Siluriformes: Trichomycteridae) from Aguarague National Park of the Bolivian preandean region, with comments on relationships within of the genus. Environ. Biol. Fishes 75, 385–393 (2006).
Wosiacki, W. B. & De Pinna, M. Trichomycterus igobi, a new catfish species from the rio Iguaçu drainage: The largest head in Trichomycteridae (Siluriformes: Trichomycteridae). Neotrop. Ichthyol. 6, 17–23 (2008).
Datovo, A., Carvalho, M. & Ferrer, J. A new species of the catfish genus. Zootaxa 44, 33–44 (2012).
Terán, G. E. et al. Living in the waterfalls: A new species of Trichomycterus (Siluriformes: Trichomycteridae) from Tabay stream, Misiones, Argentina. PLoS One 12, 1–13 (2017).
Alencar, A. R. & Costa, W. J. E. M. Trichomycterus pauciradiatus, a new catfish species from the upper rio Parana basin, southeastern Brazil (Siluriformes: Trichomycteridae). Zootaxa 49, 43–49 (2006).
Barbosa, M. A. & Costa, W. J. E. M. Description of a new species of the catfish genus Trichomycterus (Teleostei: Si lu rifor mes: Trichomycteridae) from the rio Paraíba do Sul basin, southeastern. Vertebr. Zool. 60, 193–197 (2010).
Bockmann, F. A. & Sazima, I. Trichomycterus maracaya, a new catfish from the upper rio Paraná southern Brazil (Siluriformes: Trichomycteridae), with notes on the T. brasiliensis species-complex. Neotrop. Ichthyol. 2, 61–74 (2004).
Barbosa, M. A. & Costa, W. J. E. M. Description of a new species of catfish from the upper rio Paraíba do Sul basin, south-eastern Brazil (Teleostei: Siluriformes: Trichomycteridae) and re-description of Trichomycterus itatiayae. Aqua, Int. J. Ichthyol. 14 (2008).
Ardila-Rodriguez, C. A. Trichomycterus maldonadoi (Siluriformes: Trichomycteridae), especie nueva de la cuenca alta del río Sinú, Colombia. Dahlia 11, 13–22 (2011).
Ardila-Rodriguez, C. A. Trichomycterus nietoi sp. nov. (Siluriformes: Trichomycteridae) Una nueva especie de pez del río Guachaca Sierra Nevada de Santa Marta. Departamento del Magdalena-Colombia. Univ. Metrop. Barranquilla 2–20 (2014).
Ferrer, J. & Malabarba, L. R. Taxonomic review of the genus Trichomycterus Valenciennes (siluriformes: Trichomycteridae) from the laguna dos Patos system, Southern Brazil. Neotrop. Ichthyol. 11, 217–246 (2013).
Adey, A. et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 11, R119 (2010).
Meyer, M. & Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. (2010).
Esposti, M. D. et al. Mitochondrial cytochrome b: evolution and structure of the protein. Biochim. Biophys. Acta 1143, 243–71 (1993).
Faircloth, B. C. Phyluce is a software package for the analysis of conserved genomic loci. Bioinformatics 32, 786–788 (2016).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Edgar, R. C. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Tagliacollo, V. A. & Lanfear, R. Estimating improved partitioning schemes for UltraConserved Elements (UCEs). Mol. Biol. Evol. 35, 1798–1811 (2018).
Lanfear, R., Frandsen, P. B., Wright, A. M., Sendfel, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetics analyses. Mol. Biol. Evol. 34, 772–773 (2016).
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R., Moret, B. M. & Stamatakis, A. How Many Bootstrap Replicates Are Necessary? In Proceedings of the 13th Annual International Conference on Research in Computational Molecular Biology 184–200 (Springer-Verlag), https://doi.org/10.1007/978-3-642-02008-7_13(2009).
Aberer, A. J., Kobert, K. & Stamatakis, A. ExaBayes: Massively Parallel Bayesian Tree Inference for the Whole-Genome Era. Molecular Biology and Evolution 31, 2553–2556 (2014).
Rambaut, A., Suchard, M. A., Xie, D., Baele, G. & Suchard, M. A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. (2018).
Mirarab, S. et al. Astral: Genome-scale coalescent-based species tree estimation. Bioinformatics 30, 541–548 (2014).
Shimodaira, H. & Hasegawa, M. Multiple Comparison of Log-Likelihood with Applications to Phylogenetic Inference. Mol. Biol. Evol. 16, 1114–1116 (1999).
Acknowledgements
We are grateful to M. Mehanna and R. Devidé for their help collecting specimens, J. Garcia-Melo and F. Villa-Navarro for help us with samples from Colombia. Most analyses were performed on zungaro and Brycon severs at IBB/UNESP funded by São Paulo Research Foundation (FAPESP proc. 2014/26508-3), and portions of analyses were supported by the Center for Scientific Computing (NCC/GridUNESP) at São Paulo State University. This research was supported by FAPESP (proc. 2015/13382-4 and 2014/06853-8 to LEO; 2016/19075-9 to AD; 2014/05051-5 and 2015/00691-9 to FFR; 2016/11313-8 to BFM; and 2014/26508-3 to CO). Samples from ANSP/INPA collected by MHS during fieldwork supported by NSF DEB 0107751 (PI Jon Armbruster), All Catfish Species Inventory, NSF DEB-0315963 (PI Lawrence Page) and iXingu Project, NSF DEB-1257813 (PI MHS) with help from numerous students and colleagues, especially Nathan Lujan, Lúcia Rapp Py-Daniel and Leandro Sousa. We would like to thank staff at the biological collection of: Auburn University Museum of Natural History, MZUSP, ANSP, CZUT-IC and CAS and the follow institutions: UCLA-Los Angeles and Instituto de Biociências, Universidade Estadual Paulista-UNESP.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.O. and L.E.O.; data curation: L.E.O. and F.F.R.; formal analysis: L.E.O., A.D. and J.C.; funding acquisition: C.O., F.F. and M.A.; investigation: L.E.O., B.M., F.F.R., G.S.C. and M.H.S.; methodology: M.A., J.C.; project administration: L.E.O., A.D. and C.D.; resources: C.O., F.F., M.A. and M.H.S.; supervision: C.O., M.A.; validation: L.E.O.; visualization: A.D., writing-original draft: A.D., C.D. and L.E.O., writing-review and editing: L.E.O., A.D., C.D. and M.H.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Third party rights
The author(s) declare no use additional material that are not own intellectual property.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ochoa, L.E., Datovo, A., DoNascimiento, C. et al. Phylogenomic analysis of trichomycterid catfishes (Teleostei: Siluriformes) inferred from ultraconserved elements. Sci Rep 10, 2697 (2020). https://doi.org/10.1038/s41598-020-59519-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59519-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
