
Abstract
Accumulation of bile acids (BAs) may mediate development of necrotizing enterocolitis (NEC). Serial fecal samples were collected from premature infants with birth weight (BW) ≤ 1800 g, estimated gestational age (EGA) ≤ 32 weeks, and <30 days old prior to initiation of enteral feeding. Nine infants that developed Bell’s Stage ≥ II NEC were matched with control infants based on BW, EGA, day of life (DOL) enteral feeding was initiated and DOL of the first sample. From each subject, five samples matched by DOL collected were analyzed for BA levels and composition. Fifteen individual BA species were measured via LC-MS/MS and total BA levels were measured using the Diazyme Total Bile Acid Assay kit. No statistically significant differences in composition were observed between control and NEC at the level of individual species (p = 0.1133) or grouped BAs (p = 0.0742). However, there was a statistically significant difference (p = 0.000012) in the mean coefficient of variation (CV) between the two groups with infants developing NEC having more than four-fold higher mean CV than controls. Importantly, these variations occurred prior to NEC diagnosis. These data suggest fluctuations in total fecal BA levels could provide the basis for the first predictive clinical test for NEC.
Similar content being viewed by others

Introduction
Necrotizing enterocolitis (NEC), a hemorrhagic inflammatory necrosis that mainly affects the distal ileum and colon1, is the most commonly diagnosed gastrointestinal emergency of premature infants2,3. Mortality rates range from 20–40 percent2,4,5,6 and survivors encounter hospital stays on average of 11 days longer than infants born at similar estimated gestational age that did not develop NEC7. Disease-associate costs of surgical NEC can extend beyond initial diagnosis, including development of short bowel syndrome and increased risk of neurodevelopmental morbidity8,9. While prematurity10,11, enteral feeding12 and intestinal bacterial colonization13,14 are recognized as major risk factors, definitive mechanisms of NEC pathophysiology have yet to be established and treatments remain primarily supportive. Methods that reliably predict which preterm infants are likely to develop NEC would greatly enhance clinical practice.
Bile acids (BAs) emulsify, absorb and transport fats and sterols in the intestine and liver. The primary BAs—cholic acid (CA) and chenodeoxycholic acid (CDCA)—are synthesized in hepatocytes, conjugated with either glycine or taurine, and then transported from the liver to the gall bladder for storage. After secretion into the intestine, BAs undergo bacterial dehydroxylation and conversion to their secondary forms—deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA). Bacterial deconjugation of BAs occurs in the colon, and most are recirculated to the liver via the portal vein15. Alterations of these highly regulated processes may result in accumulation of cytotoxic, hydrophobic BAs (e.g., LCA, DCA, and CDCA) within enterocytes, with subsequent damage to the intestinal epithelium16,17.
We were the first to show accumulation of ileal BAs is critical to experimental NEC pathophysiology18, the mechanisms by which BAs accumulate in ileal enterocytes19, active transport of ileal BAs is required to decrease protective responses20, and the concomitant dysregulation of hepatic BA transporters21 during development of NEC. This novel paradigm encompasses major disease-associated findings and risk factors: 1) the more premature an infant, the more likely they will develop NEC10,11. Many of the key processes of BA homeostasis are not fully developed in neonates, but reach maturity at weaning22,23,24,25,26,27. 2) Formula-fed preemies are 6–10 times more likely to develop NEC12 and have higher fecal BA levels than breast-fed preemies28. Further, formula feeding is required to develop experimental NEC29,30. 3) The majority of BA reclamation occurs in the ileum and colon, the primary sites of NEC injury31, and ileal BA levels increase 24-hours before inflammation or histological damage in experimental NEC32. 4) Bacterial colonization is required for both development of NEC and formation of secondary BAs. While no specific pathogen has been conclusively associated with NEC33,34,35,36,37,38,39,40,41,42,43, disease cannot be developed in germ free conditions13,14 and pneumatosis intestinalis, the most pathognomonic radiological finding of NEC, is thought to be caused by bacterial overgrowth44. Bacteria are also required for formation of more toxic, hydrophobic BAs45,46,47, which are higher in both experimental18 and human NEC48.
To further characterize how BAs are dysregulated in human NEC, we prospectively collected fecal samples from premature infants and determined BA levels and composition from matched-pairs diagnosed with NEC versus those without NEC. Contrary to a previous study48, we found no statistically significant differences in BA composition between control and NEC. However, there was a statistically significant difference (p = 0.000012) in the mean coefficient of variation (CV) in the two groups with infants developing NEC having more than four-fold higher mean CV than controls. Importantly, these differences occurred prior to NEC diagnosis. These data strongly suggest the potential for utilization of total BA variability as the first predictive test for this devastating disorder.
Results
Matched pairs used for analysis
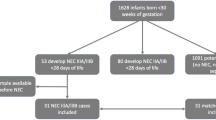
One hundred-sixty-nine infants were enrolled in the study and 12 developed NEC. Our final dataset was composed of nine NEC subject pairs matched to control subjects based on estimated gestational age (EGA), birth weight (BW), day of life (DOL) of initiation of enteral feeding and the DOL the first sample was collected. Five samples from each subject pair were matched based on DOL. All NEC subject samples measured were collected prior to NEC diagnosis and all subjects received antibiotics in the first week of life. Table 1 shows the characteristics of the matched pairs used for data analyses.
BA composition
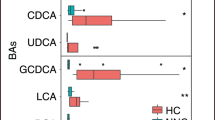
UDCA, CA, GCA, TCA, CDCA, GCDCA, TCDCA and TDCA comprised the main components of fecal BAs in the study population. Mean composition of the 15 individual bile acid species and of the five grouped species (all UDCA, CA, CDCA, DCA, and LCA) for NEC and Control subjects are shown in Fig. 1A, B, respectively. No statistically significant difference in overall composition was detected with either the 15 individual bile acids (p-value = 0.1133) or with the five bile acid groups (p-value = 0.0742).

Mean (center) composition of individual (A) and grouped BAs (B) in control versus NEC. Grouped BAs include the unconjugated and conjugated species of UDCA, CA, CDCA, DCA and LCA. BAs are shown from top to bottom from least to most hydrophobic.
BA quantity
The mean difference between NEC and control subjects in quantity of total BA, as well as of each individual BA species and of the five BA groups, is shown in Fig. 2. After False Discovery Rate adjustment, no statistically significant difference was detected in any of the 21 comparisons (p-values all > 0.20).

Mean difference in quantity of each bile acid between control and NEC pairs. Error bars represent ± 1 standard deviation as measured over the nine paired differences.
BA variation
Within pairs, the NEC subjects consistently had higher CV of total BA than the paired control, with the NEC subject having at least a 3-fold higher CV of total BA. The mean CV of total BA for NEC subjects was more than 4-times that of the Controls (Table 2). Notably, this variation is apparent well before NEC diagnosis (Fig. 3) and there was there was no overlap between the two groups: the smallest observed CV in the NEC subjects (0.5252) was larger than the largest CV of the control Subjects (0.3399, Table 2). The mean CV for each of the 15 individual BAs and for the five BA groups are shown in Tables 3 and 4, respectively. After adjusting for False Discovery Rate with these 21 comparisons, we detected a statistically significant difference in the mean CV of total BAs (p-value = 0.000012, Table 2), but not in any of the 15 individual BA species (Table 3) nor in any of the five BA groups (Table 4).

Total BA levels for each sample in each subject pair. Red arrows indicate day of life of NEC diagnosis.
Discussion
Despite advances in neonatal practice, NEC remains the most common GI emergency of premature infants. Immunologic49,50,51,52,53 and microbial initiators37,41,54,55,56,57 have been examined, but the pathophysiology of NEC has yet to be clearly elucidated and there are no predictive tests available. Studies in animal models of NEC strongly suggest accumulation of BAs play a significant role in disease development18,19,20,21. We found no statistically significant differences between control and NEC for any mean individual BA species, nor did we find statistically significant differences in mean total BAs. However, the mean CV for total BAs was statistically significantly higher in premature infants that develop NEC compared to matched controls. Importantly, variability occurred throughout the NEC subject’s measured samples and prior to NEC diagnosis. There was also no overlap in the CVs of total BAs, enhancing its potential as a prognostic measure.
These data show profound differences between magnitude of fluctuations of total fecal BAs between premature infants that develop NEC and those that do not. This suggests neonatal ileum may be especially sensitive to pulsatile exposure to BAs. In addition, fluctuation of total BA levels appears to be a better predictor of NEC development than mean overall BA levels. While exposure to varying levels of hydrophobic BAs has been shown to increase proliferation of cells in esophageal reflux and Barrett’s esophagus58,59,60, cytotoxic effects of intermittent exposure to BAs in neonatal intestine has not been published. While previous research reported a statistically significant increase in total unconjugated BAs in NEC patients compared to controls48, we did not observe a statistically significant increase in mean BA quantity for any individual BA, BA group, or total BAs. However, our study differed from theirs in that we used matched pairs, analyzed more samples per subject, looked at slightly different outcomes, and had lower power due to a smaller sample size and correcting for multiple comparisons (that is, we required a lower p-value for “statistical significance”). While we observed a trend toward higher BAs in NEC, extreme variation between an individual NEC patient’s samples prevented reaching statistical significance in this relatively modest sample size. However, this variability is in itself, a potential predictor of NEC development.
Although no single pathogen has been definitively associated with NEC55,56,57, the intestinal microbiome has a profound effect on the composition of intestinal BAs. Conversion of BAs from primary to secondary forms requires bacterial dehydroxylation by species of Clostridium and Eubacterium in the intestine61,62,63,64,65,66 and Clostridia are abundant in the feces of infants with NEC14,67,68. Further, deconjugation of BAs depends on separate bacterial conversions69. While postnatal antibiotic use has been suggested as an additional risk factor for NEC70,71, antibiotic use in all subjects from both groups were similar (treatment within the first seven days of life). Further investigation of changes in the microbiome during development of NEC, specifically with regard to bacteria capable of BA transformations, is ongoing but exceeds the scope of this work.
Whether fluctuations in total BAs leads to NEC or is a consequence of other pathophysiologic changes that occur prior to NEC has yet to be determined. The intestinal epithelial barrier is inherently leaky in any neonate but tight junctions (TJs), an integral component of intestinal barrier integrity, are altered in NEC72,73,74,75,76,77 and disrupted after continuous exposure to hydrophobic BAs78,79,80. Pulsatile exposure to BAs may produce more profound changes in TJs and barrier function that allows aberrant movement of proinflammatory mediators out of the intestinal epithelium as well as bacterial products into cells or systemic circulation if epithelial integrity is compromised.
The apical sodium-dependent bile acid transporter (ASBT) is the primary transporter of BAs across the apical membrane of enterocytes81,82,83. To prevent toxicity from intracellular accumulation of BAs, BAs repress ASBT via interaction with the farnesoid X receptor (FXR)84,85. However, we determined that surface expression of ileal ASBT is increased in NEC, leads to accumulation of intra-enterocyte BAs, and increases the incidence and severity of experimental NEC18,19. Further, ASBT is also increased on the surface of ileal surgical samples from infants diagnosed with NEC19. Consequently, changes in ASBT regulation after exposure to fluctuating BA levels must be examined as a potential mechanism to explain how pulsatile exposure to BAs may be more cytotoxic than continuous exposure.
The statistical power of our analyses was limited by two key factors. First, there were large standard deviations in the quantities of several BAs, some of which were attributed to individual subjects (e.g., elevated levels UDCA for a single subject in the NEC group and TCDCA for a single subject in the control group). This affected both the analysis of mean BA quantity as well as of the overall composition. Second, the sample size was limited to only nine pairs of subjects. This is, in part, due to the limited number of subjects that develop NEC, but also to the issue of finding appropriate controls (both subject pairs as well as sample pairs) for comparison. The variability in stooling frequency coupled with inability to control stooling frequency further contributed to our limited sample size. It is also possible that variation in stooling frequency affected the measured response (the amount, either absolute or proportional, of various BAs). Sample collection has begun at two additional NICUs which will increase the number of NEC patients, allowing for analysis of matched pairs in a ratio of 1:2 (NEC:Controls) and compaction of tolerance for EGA in future studies. Parameters used to establish the matched pairs did not include gender or type of enteral feeding. Gender is not considered a risk factor for NEC86, BAs do not differ significantly by gender87,88 and the type of enteral feeding was quite similar between matched pairs (Table 1). The only pair with significant differences in diet—Pair 7—the control subject was the one given formula (and only for four feedings), which is considered a risk factor for NEC12. Thus, we considered the differences in gender and feed type unlikely to have influenced our results.
In conclusion, variation in total BAs occurred well in advance of NEC diagnosis with no overlap in CV between the two groups (all nine NEC subjects had CV of total BA greater than that of any of the control subjects). While larger clinical studies must be performed, these data suggest the CV of total BAs is a biomarker of NEC and has the potential to be developed as the first predictive test for this devastating disorder.
Methods
Study participants
Following approval by the University of Arizona Institutional Review Board, premature infants were enrolled prospectively via informed, written parental consent at Banner University Medical Center Tucson. All research was performed in accordance with relevant regulations. The inclusion criteria—birth weight (BW) ≤ 1800 g, estimated gestational age (EGA) ≤ 32 weeks, and < 30 days old prior to initiation of enteral feeding—were chosen because NEC occurs almost exclusively in premature infants, the most premature infants are more likely to develop disease, and most cases occur after the initiation of enteral feeding89,90,91. Exclusion criteria included conditions not related to prematurity including blood-culture positive sepsis or genetic syndromes and were based on eliminating subjects that could develop NEC-like syndromes due to other confounding problems not related to the most common risk factors for NEC. Definitions of NEC diagnosis and time of diagnosis were defined as any subject with Bell’s Stage ≥ II (modified Bell’s staging criteria) and radiographic evidence of NEC, respectively.
Sample collection and analysis
Post-meconium fecal samples were collected from the diaper for up to four weeks after initiation of enteral feeding. Samples were placed in sterile microtubes, frozen in the NICU at −20 °C and transported to the laboratory weekly where they were then stored at −80 °C until processing. Analysis of various freezing protocols prior to actual sample collection determined this freezing protocol did not alter BA levels or composition (data not shown). For analysis, samples were thawed, weighed, and mixed with an equal volume of nanopure water. After homogenization, samples were centrifuged to separate fecal water from the solids and the fecal water was frozen at −80 until BAs were assayed28,92. BA composition was determined using the ThermoFinnigan TSQ Quantum Ultra Mass Spectrometer with High Performance Liquid Chromatography System (Arizona Cancer Center Analytical Core Shared Service). Fifteen separate BA species—UDCA, CA, DCA, CDCA, LCA, along with their glycol and tauro conjugates—were evaluated93. The LC-MS/MS method utilized to determine individual BA species does not measure hyocholic acid (HCA) or hyodeoxycholic (HDCA), which can contribute significantly to total BAs in infants92. Therefore, to determine total BA levels, we utilized a commercially available kit that measures all BAs via an enzymatic cycling method with spectrophotometric readout18,19. Total BAs were calculated using the Diazyme Total Bile Acids Assay Kit (Diazyme Laboratories, Poway, CA, USA).
Statistical analyses
Due to the nested-nature of samples, pairing was accomplished via a two-stage process: 1) appropriate control subjects were first paired to each NEC subject, then 2) paired samples within those subjects were selected. A control subject was considered an appropriate match for a NEC subject if the following four tolerances were met: EGA within 2.5 weeks, BW within 150 g, initiation of enteral feeding within 3.5 days, and the day of life of the first sample was within 3.5 days. Once a set of potential control pairs was identified using these tolerances for each NEC subject, the single control pair was selected for each NEC subject as follows. Because a single control subject could be a potential match for more than one NEC subject, we first ensured that all NEC subjects with potential controls were paired with a control (if a control was a potential pair with a NEC subject that had several potential control-subjects and also was a potential pair with a NEC subject with only one potential control, it would be paired with the NEC subject with only one potential control). Of the remaining controls that were potential matches to a NEC subject, we looked for pairs that had at least five samples on the same day (days from birth) as the control. Our final dataset was composed of nine NEC subject pairs, each with five paired samples. All NEC subject samples measured were collected prior to NEC diagnosis and all subjects received antibiotics in the first week of life.
To assess whether there was a difference in BA composition between NEC and control subjects, we conducted two separate analyses, one using each of the fifteen BAs analyzed and another where the BAs were combined into groups: UDCA group (UDCA + GUDCA + TUDCA), CA group (CA + GCA + TCA), CDCA group (CDCA + GCDCA + TCDCA), DCA group (DCA + GDCA + TDCA), and LCA group (LCA + GLCA + TLCA). Prior to analysis, composition data was additive log-ratio (ALR) transformed94. The ALR-transformation takes the natural-log of the proportion for each of the D−1 components divided by the proportion of the Dth component. For the ALR-transformation, the Dth (denominator) component was CA (for the analysis of the 15 BAs) and the CA group (for the analysis of the five BA groups). Due to the complex, hierarchically paired structure of the data, a standard MANOVA was not appropriate. Instead, a permutation test was performed. In order to preserve the nested and paired structure of the data, for each permutation we only randomly permuted which subject within a pair was given the label “NEC” vs. “control”. Subject and sample pairs were maintained. As there were only nine pairs, there were only 29 = 512 permutations. The test statistic used in the permutation test was the Pillai’s Trace for diagnosis (NEC vs. control) from a MANOVA with fixed effects for Sample Pair95.
To visualize a representative composition of BAs and BA groups for NEC vs. control subjects, the compositional mean was calculated for each diagnosis group by determining the geometric means of the proportion of each BA over all 45 samples for a diagnosis group and standardizing to 1 by dividing by the sum of those geometric means96.
In addition to examining overall composition, we also examined whether there was a difference between control and NEC subjects in the mean quantity and in the mean coefficient of variation (CV) of the quantity of total BA as well as in each of the 15 BAs and each of the five BA groups. For these analyses we, again, used the same nine pairs of subjects and the same five pairs of samples within each pair of subjects. For each BA, the difference in the quantity of that BA between sample pairs was calculated. We then took the average of these five differences within each Subject Pair. A 1-sample t-test was used to test whether the mean difference in quantity was equal to zero or not. The CV of each BA over the five samples was calculated for each subject and a paired t-test was conducted on the nine pairs of CVs. Because testing for differences in mean quantity and differences in mean CV each involved a total of 21 tests, we adjusted p-values for 21 multiple comparisons using the false discovery rate (FDR) method of Benjamini and Hochberg97.
Data availability
Data are available from the authors upon reasonable request.
References
Israel, E. J. Neonatal necrotizing enterocolitis, a disease of the immature intestinal mucosal barrier. Acta Paediatr. Suppl. 396, 27–32 (1994).
Lemons, J. A. et al. Very low birth weight outcomes of the National Institute of Child health and human development neonatal research network, January 1995 through December 1996. NICHD Neonatal Research Network. Pediatrics 107, E1 (2001).
Kafetzis, D. A., Skevaki, C. & Costalos, C. Neonatal necrotizing enterocolitis: an overview. Curr. Opin. Infect. Dis. 16, 349–355 (2003).
de Souza, J. C., da Motta, U. I. & Ketzer, C. R. Prognostic factors of mortality in newborns with necrotizing enterocolitis submitted to exploratory laparotomy. J Pediatr Surg 36, 482-486, doi:S0022-3468(01)90000-4 (2001).
Fitzgibbons, S. C. et al. Mortality of necrotizing enterocolitis expressed by birth weight categories. J Pediatr Surg 44, 1072–1075; discussion 1075-1076, doi:S0022-3468(09)00160-2 (2009).
Thyoka, M. et al. Advanced necrotizing enterocolitis part 1: mortality. Eur. J. Pediatr. Surg. 22, 8–12, https://doi.org/10.1055/s-0032-1306263 (2012).
Ganapathy, V., Hay, J. W. & Kim, J. H. Costs of Necrotizing Enterocolitis and Cost-Effectiveness of Exclusively Human Milk-Based Products in Feeding Extremely Premature Infants. Breastfeed Med, https://doi.org/10.1089/bfm.2011.0002 (2011).
Vohr, B. R. et al. Neurodevelopmental and functional outcomes of extremely low birth weight infants in the National Institute of Child Health and Human Development Neonatal Research Network, 1993-1994. Pediatrics 105, 1216–1226 (2000).
Allendorf, A. et al. Necrotizing enterocolitis as a prognostic factor for the neurodevelopmental outcome of preterm infants - match control study after 2years. J. Pediatr. Surg. 53, 1573–1577, https://doi.org/10.1016/j.jpedsurg.2018.01.006 (2018).
Gephart, S. M., McGrath, J. M., Effken, J. A. & Halpern, M. D. Necrotizing enterocolitis risk: state of the science. Adv. Neonatal Care 12, 77–87, https://doi.org/10.1097/ANC.0b013e31824cee9400149525-201204000-00004 (2012).
Schnabl, K. L., Van Aerde, J. E., Thomson, A. B. & Clandinin, M. T. Necrotizing enterocolitis: a multifactorial disease with no cure. World J. Gastroenterol. 14, 2142–2161 (2008).
Schanler, R. J., Hurst, N. M. & Lau, C. The use of human milk and breastfeeding in premature infants. Clin. Perinatol. 26, 379–398 (1999). vii.
Bousseboua, H. et al. Experimental cecitis in gnotobiotic quails monoassociated with Clostridium butyricum strains isolated from patients with neonatal necrotizing enterocolitis and from healthy newborns. Infect. Immun. 57, 932–936 (1989).
Sangild, P. T. et al. Diet- and colonization-dependent intestinal dysfunction predisposes to necrotizing enterocolitis in preterm pigs. Gastroenterology 130, 1776–1792, https://doi.org/10.1053/j.gastro.2006.02.026 (2006).
Hofmann, A. F. Bile Acids: The Good, the Bad, and the Ugly. N. Physiol. Sci. 14, 24–29 (1999).
Craven, P. A., Pfanstiel, J., Saito, R. & DeRubertis, F. R. Relationship between loss of rat colonic surface epithelium induced by deoxycholate and initiation of the subsequent proliferative response. Cancer Res. 46, 5754–5759 (1986).
Milovic, V., Teller, I. C., Faust, D., Caspary, W. F. & Stein, J. Effects of deoxycholate on human colon cancer cells: apoptosis or proliferation. Eur. J. Clin. Invest. 32, 29–34 (2002).
Halpern, M. D. et al. Bile acids induce ileal damage during experimental necrotizing enterocolitis. Gastroenterology 130, 359–372, doi:S0016-5085(05)02179-7 (2006).
Halpern, M. D. et al. Apical sodium-dependent bile acid transporter upregulation is associated with necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 299, G623-631, doi:ajpgi.00242.2010 (2010).
Martin, N. A. et al. Active transport of bile acids decreases mucin 2 in neonatal ileum: implications for development of necrotizing enterocolitis. PLoS One 6, e27191, https://doi.org/10.1371/journal.pone.0027191PONE-D-11-13529 (2011).
Cherrington, N. J. et al. The hepatic bile acid transporters Ntcp and Mrp2 are downregulated in experimental necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 304, G48–56, doi:ajpgi.00317.2012 (2012).
Shneider, B. L., Setchell, K. D. & Crossman, M. W. Fetal and neonatal expression of the apical sodium-dependent bile acid transporter in the rat ileum and kidney. Pediatr. Res. 42, 189–194 (1997).
Hwang, S. T. & Henning, S. J. Ontogenic regulation of components of ileal bile acid absorption. Exp. Biol. Med. (Maywood) 226, 674–680 (2001).
Suchy, F. J. & Balistreri, W. F. Uptake of taurocholate by hepatocytes isolated from developing rats. Pediatr. Res. 16, 282–285 (1982).
Suchy, F. J., Courchene, S. M. & Balistreri, W. F. Ontogeny of hepatic bile acid conjugation in the rat. Pediatr. Res. 19, 97–101 (1985).
Balistreri, W. F., Heubi, J. E. & Suchy, F. J. Immaturity of the enterohepatic circulation in early life: factors predisposing to “physiologic” maldigestion and cholestasis. J. Pediatr. Gastroenterol. Nutr. 2, 346–354 (1983).
Heubi, J. E., Balistreri, W. F. & Suchy, F. J. Bile salt metabolism in the first year of life. J. Lab. Clin. Med. 100, 127–136 (1982).
Hammons, J. L., Jordan, W. E., Stewart, R. L., Taulbee, J. D. & Berg, R. W. Age and diet effects on fecal bile acids in infants. J. Pediatr. Gastroenterol. Nutr. 7, 30–38 (1988).
Caplan, M. S., Amer, M. & Jilling, T. The role of human milk in necrotizing enterocolitis. Adv. Exp. Med. Biol. 503, 83–90 (2002).
Dvorak, B. et al. Maternal milk reduces severity of necrotizing enterocolitis and increases intestinal IL-10 in a neonatal rat model. Pediatr. Res. 53, 426–433 (2003).
Kosters, A. & Karpen, S. J. Bile acid transporters in health and disease. Xenobiotica 38, 1043–1071, https://doi.org/10.1080/00498250802040584 (2008).
Halpern, M. D. & Dvorak, B. Does Abnormal Bile Acid Metabolism Contribute to NEC? Semin Perinatol 32, 114–121, doi:S0146-0005(08)00012-8 (2008).
Mai, V. et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One 6, e20647, https://doi.org/10.1371/journal.pone.0020647 (2011).
Normann, E., Fahlen, A., Engstrand, L. & Lilja, H. E. Intestinal microbial profiles in extremely preterm infants with and without necrotizing enterocolitis. Acta Paediatr. 102, 129–136, https://doi.org/10.1111/apa.12059 (2013).
Torrazza, R. M. et al. Intestinal microbial ecology and environmental factors affecting necrotizing enterocolitis. PLoS One 8, e83304, https://doi.org/10.1371/journal.pone.0083304 (2013).
Wang, Y. et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 3, 944–954, https://doi.org/10.1038/ismej.2009.37 (2009).
Morrow, A. L. et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 1, 13, https://doi.org/10.1186/2049-2618-1-13 (2013).
Stewart, C. J. et al. The preterm gut microbiota: changes associated with necrotizing enterocolitis and infection. Acta Paediatr. 101, 1121–1127, https://doi.org/10.1111/j.1651-2227.2012.02801.x (2012).
Claud, E. C. et al. Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants. Microbiome 1, 20, https://doi.org/10.1186/2049-2618-1-20 (2013).
Li, M., Wang, M. & Donovan, S. M. Early development of the gut microbiome and immune-mediated childhood disorders. Semin Reprod Med 32, 74-86, https://doi.org/10.1055/s-0033-1361825.
McMurtry, V. E. et al. Bacterial diversity and Clostridia abundance decrease with increasing severity of necrotizing enterocolitis. Microbiome 3, 11, https://doi.org/10.1186/s40168-015-0075-8 (2015).
Zhou, Y. et al. Longitudinal analysis of the premature infant intestinal microbiome prior to necrotizing enterocolitis: a case-control study. PLoS One 10, e0118632, https://doi.org/10.1371/journal.pone.0118632PONE-D-14-36153 (2015).
Ward, D. V. et al. Metagenomic Sequencing with Strain-Level Resolution Implicates Uropathogenic E. coli in Necrotizing Enterocolitis and Mortality in Preterm Infants. Cell Rep. 14, 2912–2924, https://doi.org/10.1016/j.celrep.2016.03.015 (2016).
Kliegman, R. M. & Fanaroff, A. A. Necrotizing enterocolitis. N. Engl. J. Med. 310, 1093–1103 (1984).
Velardi, A. L. et al. Cell type-dependent effect of phospholipid and cholesterol on bile salt cytotoxicity. Gastroenterology 101, 457–464 (1991).
Shekels, L. L., Beste, J. E. & Ho, S. B. Tauroursodeoxycholic acid protects in vitro models of human colonic cancer cells from cytotoxic effects of hydrophobic bile acids. J. Lab. Clin. Med. 127, 57–66 (1996).
Powell, A. A., LaRue, J. M., Batta, A. K. & Martinez, J. D. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem. J. 356, 481–486 (2001).
Hulzebos, C. V. et al. Fecal Bile Salts and the Development of Necrotizing Enterocolitis in Preterm Infants. PLoS One 12, e0168633, https://doi.org/10.1371/journal.pone.0168633 (2017).
McElroy, S. J. et al. Tumor Necrosis Factor Receptor 1-Dependent Depletion of Mucus in Immature Small Intestine: A Potential Role in Neonatal Necrotizing Enterocolitis. Am J Physiol Gastrointest Liver Physiol 301, G656–G666, doi:ajpgi.00550.2010 (2011).
Halpern, M. D. et al. Reduction of Experimental Necrotizing Enterocolitis with Anti-TNF-{alpha}. Am. J. Physiol. Gastrointest. Liver Physiol 290, G757–764 (2006).
Halpern, M. D. et al. Up-regulation of IL-18 and IL-12 in the ileum of neonatal rats with necrotizing enterocolitis. Pediatr. Res. 51, 733–739 (2002).
Halpern, M. D. et al. Decreased development of necrotizing enterocolitis in IL-18-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol 294, G20–26 (2008). doi:00168.2007.
Hackam, D. J., Afrazi, A., Good, M. & Sodhi, C. P. Innate immune signaling in the pathogenesis of necrotizing enterocolitis. Clin. Dev. Immunol. 2013, 475415, https://doi.org/10.1155/2013/475415 (2013).
Lu, P., Sodhi, C. P. & Hackam, D. J. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiology 21, 81–93, https://doi.org/10.1016/j.pathophys.2013.11.007 (2014).
Carlisle, E. M. & Morowitz, M. J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 25, 382–387, https://doi.org/10.1097/MOpipiP.0b013e3283600e91 (2013).
Elgin, T. G., Kern, S. L. & McElroy, S. J. Development of the Neonatal Intestinal Microbiome and Its Association With Necrotizing Enterocolitis. Clin. Ther. 38, 706–715, https://doi.org/10.1016/j.clinthera.2016.01.005 (2016).
Torrazza, R. M. & Neu, J. The altered gut microbiome and necrotizing enterocolitis. Clin. Perinatol. 40, 93–108, https://doi.org/10.1016/j.clp.2012.12.009 (2013).
Green, N. H. et al. Pulsatile exposure to simulated reflux leads to changes in gene expression in a 3D model of oesophageal mucosa. Int. J. Exp. Pathol. 95, 216–228, https://doi.org/10.1111/iep.12083 (2014).
Hao, Y. et al. Gene expression changes associated with Barrett’s esophagus and Barrett’s-associated adenocarcinoma cell lines after acid or bile salt exposure. BMC Gastroenterol. 7, 24, https://doi.org/10.1186/1471-230X-7-24 (2007).
Triadafilopoulos, G. Acid and bile reflux in Barrett’s esophagus: a tale of two evils. Gastroenterology 121, 1502–1506 (2001).
Batta, A. K. et al. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J. Biol. Chem. 265, 10925–10928 (1990).
Stellwag, E. J. & Hylemon, P. B. 7alpha-Dehydroxylation of cholic acid and chenodeoxycholic acid by Clostridium leptum. J. Lipid Res. 20, 325–333 (1979).
Wells, J. E., Williams, K. B., Whitehead, T. R., Heuman, D. M. & Hylemon, P. B. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7alpha-dehydroxylating bacteria in human feces. Clin. Chim. Acta 331, 127–134 (2003).
White, B. A., Lipsky, R. L., Fricke, R. J. & Hylemon, P. B. Bile acid induction specificity of 7 alpha-dehydroxylase activity in an intestinal Eubacterium species. Steroids 35, 103–109 (1980).
Kitahara, M., Takamine, F., Imamura, T. & Benno, Y. Assignment of Eubacterium sp. VPI 12708 and related strains with high bile acid 7alpha-dehydroxylating activity to Clostridium scindens and proposal of Clostridium hylemonae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 50(Pt 3), 971–978, https://doi.org/10.1099/00207713-50-3-971 (2000).
Hirano, S., Nakama, R., Tamaki, M., Masuda, N. & Oda, H. Isolation and characterization of thirteen intestinal microorganisms capable of 7 alpha-dehydroxylating bile acids. Appl. Env. Microbiol. 41, 737–745 (1981).
Brower-Sinning, R. et al. Mucosa-associated bacterial diversity in necrotizing enterocolitis. PLoS One 9, e105046, https://doi.org/10.1371/journal.pone.0105046 (2014).
Dai, D. & Walker, W. A. Role of bacterial colonization in neonatal necrotizing enterocolitis and its prevention. Zhonghua Min. Guo Xiao Er Ke Yi Xue Hui Za Zhi 39, 357–365 (1998).
Jones, B. V., Begley, M., Hill, C., Gahan, C. G. & Marchesi, J. R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl Acad. Sci. USA 105, 13580–13585, https://doi.org/10.1073/pnas.0804437105 (2008).
Cotten, C. M. et al. Prolonged duration of initial empirical antibiotic treatment is associated with increased rates of necrotizing enterocolitis and death for extremely low birth weight infants. Pediatrics 123, 58–66, https://doi.org/10.1542/peds.2007-3423 (2009).
Kuppala, V. S., Meinzen-Derr, J., Morrow, A. L. & Schibler, K. R. Prolonged initial empirical antibiotic treatment is associated with adverse outcomes in premature infants. J. Pediatr. 159, 720–725, https://doi.org/10.1016/j.jpeds.2011.05.033 (2011).
Bein, A., Eventov-Friedman, S., Arbell, D. & Schwartz, B. Intestinal tight junctions are severely altered in NEC preterm neonates. Pediatr. Neonatol. 59, 464–473, https://doi.org/10.1016/j.pedneo.2017.11.018 (2018).
Moore, S. A. et al. Intestinal barrier dysfunction in human necrotizing enterocolitis. J. Pediatr. Surg. 51, 1907–1913, https://doi.org/10.1016/j.jpedsurg.2016.09.011 (2016).
Bergmann, K. R. et al. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am. J. Pathol. 182, 1595–1606, doi:S0002-9440(13)00097-7 (2013).
Khailova, L. et al. Bifidobacterium bifidum improves intestinal integrity in a rat model of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol 297, G940–949 (2009).
Hackam, D. J., Upperman, J. S., Grishin, A. & Ford, H. R. Disordered enterocyte signaling and intestinal barrier dysfunction in the pathogenesis of necrotizing enterocolitis. Semin. Pediatr. Surg. 14, 49–57 (2005).
Clark, J. A. et al. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am. J. Physiol. Gastrointest. Liver Physiol 291, G938-949, doi:00090.2006 (2006).
Bernstein, H. et al. Deoxycholate-induced colitis is markedly attenuated in Nos2 knockout mice in association with modulation of gene expression profiles. Dig. Dis. Sci. 52, 628–642, https://doi.org/10.1007/s10620-006-9608-0 (2007).
Fasano, A. et al. Bile acids reversible effects on small intestinal permeability. An in vitro study in the rabbit. Dig. Dis. Sci. 35, 801–808 (1990).
Raimondi, F. et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am. J. Physiol. Gastrointest. Liver Physiol 294, G906-913, doi:00043.2007 (2008).
Dawson, P. A. et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem. 278, 33920–33927 (2003).
Lazaridis, K. N. et al. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc. Natl Acad. Sci. U S A 97, 11092–11097 (2000).
Wong, M. H., Oelkers, P., Craddock, A. L. & Dawson, P. A. Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. J. Biol. Chem. 269, 1340–1347 (1994).
Makishima, M. et al. Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 (1999).
Neimark, E., Chen, F., Li, X. & Shneider, B. L. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 40, 149–156 (2004).
Carter, B. M. & Holditch-Davis, D. Risk factors for necrotizing enterocolitis in preterm infants: how race, gender, and health status contribute. Adv. Neonatal Care 8, 285–290, https://doi.org/10.1097/01.ANC.0000338019.56405.29 (2008).
Polkowska, G., Polkowski, W., Kudlicka, A., Wallner, G. & Chrzastek-Spruch, H. Range of serum bile acid concentrations in neonates, infants, older children, and in adults. Med. Sci. Monit. 7(Suppl 1), 268–270 (2001).
Kawasaki, H., Yamanishi, Y., Miyake, M., Mura, T. & Ikawa, S. Age- and sex-related profiles of serum primary and total bile acids in infants, children and adults. Tohoku J. Exp. Med. 150, 353–357 (1986).
Caplan, M. S. & Jilling, T. New concepts in necrotizing enterocolitis. Curr. Opin. Pediatr. 13, 111–115 (2001).
Neu, J. Necrotizing enterocolitis: the search for a unifying pathogenic theory leading to prevention. Pediatr. Clin. North. Am. 43, 409–432 (1996).
Uauy, R. D. et al. Necrotizing enterocolitis in very low birth weight infants: biodemographic and clinical correlates. National Institute of Child Health and Human Development Neonatal Research Network. J. Pediatr. 119, 630–638 (1991).
Kimura, A. et al. Fetal bile acid metabolism during infancy: analysis of 1 beta-hydroxylated bile acids in urine, meconium and feces. Hepatology 20, 819–824 (1994).
Torchia, E. C., Labonte, E. D. & Agellon, L. B. Separation and quantitation of bile acids using an isocratic solvent system for high performance liquid chromatography coupled to an evaporative light scattering detector. Anal. Biochem. 298, 293–298 (2001).
Aitchinson, J. in 2nd Compositional Data Analysis Workshop.
Pillai, K. C. S. Some New Test Criteria in Multivariate Analysis. Ann. Math. Statist. 26, 117–121 (1955).
van den Boogaart, K. G., Tolosana-Delgado, R. In Analyzing Compositional Data with R Ch. 3, 73–94 (Springer-Verlag, 2013).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B (Methodol.) 57, 289–300 (1995).
Acknowledgements
The National Institute of Diabetes and Digestive and Kidney Diseases (R21DK102058 and RO1DK117652 to M.D.H.) and the National Cancer Institute (P30 CA023074) of the National Institutes of Health supported this research.
Author information
Authors and Affiliations
Contributions
M.D.H. designed the study and wrote the manuscript, tables and figures. S.K. performed all statistical analyses and wrote the manuscript, tables and figures. S.M.G. performed data acquisition. A.K., J.S., C.M.C., S.B., K.E.B., G.I.D., H.J., R.R. and M.S. performed experimental work. A.D.B. provided clinical expertise. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Knapp, S., Kehring, A., Stepp, J. et al. Elevated Coefficient of Variation in Total Fecal Bile Acids Precedes Diagnosis of Necrotizing Enterocolitis. Sci Rep 10, 249 (2020). https://doi.org/10.1038/s41598-019-57178-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-57178-0
This article is cited by
-
Targeted LC-MS/MS profiling of bile acids reveals primary/secondary bile acid ratio as a novel biomarker for necrotizing enterocolitis
Analytical and Bioanalytical Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
