
Abstract
The Deepwater Horizon (DWH) oil spill contaminated coastlines from Louisiana to Florida, burying oil up to 70 cm depth in sandy beaches, posing a potential threat to environmental and human health. The dry and nutrient-poor beach sand presents a taxing environment for microbial growth, raising the question how the biodegradation of the buried oil would proceed. Here we report the results of an in-situ experiment that (i) characterized the dominant microbial communities contained in sediment oil agglomerates (SOAs) of DWH oil buried in a North Florida sandy beach, (ii) elucidated the long-term succession of the microbial populations that developed in the SOAs, and (iii) revealed the coupling of SOA degradation to nitrogen fixation. Orders of magnitude higher bacterial abundances in SOAs compared to surrounding sands distinguished SOAs as hotspots of microbial growth. Blooms of bacterial taxa with a demonstrated potential for hydrocarbon degradation (Gammaproteobacteria, Alphaproteobacteria, Actinobacteria) developed in the SOAs, initiating a succession of microbial populations that mirrored the evolution of the petroleum hydrocarbons. Growth of nitrogen-fixing prokaryotes or diazotrophs (Rhizobiales and Frankiales), reflected in increased abundances of nitrogenase genes (nifH), catalyzed biodegradation of the nitrogen-poor petroleum hydrocarbons, emphasizing nitrogen fixation as a central mechanism facilitating the recovery of sandy beaches after oil contamination.
Similar content being viewed by others
Introduction
In April 2010, the Deepwater Horizon oil rig exploded and sank, which led to a discharge of approximately 4.9 million barrels of crude oil into the Gulf of Mexico (GoM) at a depth of 1544 m over the course of 86 days1,2. Released MC252 BP oil that reached the ocean surface was transported to coastal environments, impacting approximately 965 km of beaches from east Texas to west Florida3,4,5,6,7. When weathered oil reaches the shoreline, it is generally in the form of a highly viscous, buoyant emulsion (also known as mousse), and a large portion then mixes with solids to form oil-sediment residues that have been termed tar balls, sand mats or patties, and surface residual balls8,9,10,11. In order to avoid confusion, we will henceforth refer to these macroscopic oil-sediment residues as sediment oil agglomerates (SOAs), according to a recent review of nomenclature12. SOAs are oval-shaped residues, ranging from a few millimeters to centimeters in diameter, predominantly composed of sand (75–96% by mass) with a moisture content of less than 0.5%13,14,15,16.
After the DWH oil spill, oil-sediment residues were trapped and buried up to meters in depth in beaches from Louisiana to Florida13,16,17. SOAs in highly contaminated beaches of Louisiana were found to contain elevated concentrations of recalcitrant and toxic polycyclic aromatic hydrocarbons (PAHs) including C1- and C2-phenanthrenes, C2- and C3- dibenzothiophenes, along with other high molecular weight oil components16. After monitoring SOAs for four years in Alabama’s beaches, Yin et al. (2015) showed that high molecular weight PAHs-such as chrysene and alkylated chrysenes persisted with time. Studies have also demonstrated the toxicity of SOAs due to persistence of PAHs, oxygenated hydrocarbons, environmentally persistent free radicals (EPFRs), and human pathogens such as Vibrio vulnificus10,18,19,20,21. Although British Petroleum (BP) conducted Operation Deep Clean (ODC) to mechanically remove larger SOAs from the beach surface, SOAs remained buried to 50 cm depth4,17. Moreover, once SOAs are buried in the sediments, degradation of SOAs cannot occur by photooxidation, a critical weathering process for hydrocarbons in surficial environments22,23. Therefore, even after cleanup efforts, SOAs containing toxic PAHs can persist in the beach system for years and represent a potential long-term risk to ecosystem and human health14.
The biodegradation of larger, macroscopic oil-sediment residues such as SOAs is likely to be distinct from that of smaller oil droplets or particles in coastal zones13,16,24. The smaller surface area to volume ratio of SOAs may limit the access of hydrocarbons for biodegradation25. Depending on the porosity of the aggregate, biodegradation is also likely to be limited by the delivery of substrates, oxygen and nutrients, to sites where microorganisms mediate enzymatic breakdown of hydrocarbons. For example, Elango et al. (2014) observed that the C/N molar ratio, often used as a diagnostic variable for hydrocarbon biodegradability, ranged from 111 to 474 in SOAs, which is well above optimal C/N ratios (approximately 60) for aerobic hydrocarbon degradation26. These findings suggest that bioavailable nutrients are often a limiting factor for microbial SOA degradation. Pure culture studies have shown that some hydrocarbon-degrading bacteria have the potential to fix nitrogen27,28,29,30. However, a direct linkage between SOA degradation and nitrogen fixation is lacking.
Although the impacts of oil contamination on marine microbial communities are well documented, few studies have addressed the microbial community dynamics associated with SOAs that are often trapped in coastal ecosystems. Especially in the supratidal zone of beach sand environments, low moisture content, nutrient availability, and a low surface to volume ratio of larger residues may limit bacterial hydrocarbon degradation31. Thus, the main objectives of this study were to (i) identify the dominant microbial organisms that colonize SOAs buried in dry northeastern Gulf of Mexico beach sand (ii) elucidate long-term succession of microbial communities that recruit onto these SOAs, and (iii) to explore the potential coupling of SOA degradation and nitrogen fixation buried in dry beach sand. This study used SOAs that were collected at Pensacola Beach after the DWH oil spill. Standardized aliquots of this material were buried in Pensacola Beach sand and monitored for the succession of microbial communities, and nitrogen fixation potential for over 3 years.
Results and Discussion
In this study, an in situ experiment was conducted whereby standardized sediment-oil-agglomerates (sSOA) in stainless steel meshballs (3.8 cm diameter) were attached to PVC pipe and buried in the Pensacola Beach supratidal zone from 10 cm to 50 cm sediment depth (Fig. 1). Ten of these arrays were buried on 22 October, 2010 and retrieved 41, 89, 131, 181, 235, 279, 327, 445, 735, and 1152 days after burial. SOAs along with sandy sediment surrounding each of the sSOAs were collected to characterize the impacts to indigenous microbial communities. Control sands that showed no oil contamination were also collected from the surface of a nearby sand dune after 41, 89, 279, 445, 735, 1152 since the onset of the experiment. sSOAs, sSOA-surrounding sands, and control sands were then collected over a 3 year time series to investigate in situ biodegradation of SOAs and microbial community dynamics. Total enviromental DNA was extracted from the source material (sSOA at day 0), incubated SOAs, SOA-surrounding sands, and control sands and used for downstream analysis.

Experimental design employed to investigate the in situ biodegradation of sediment oil agglomerates (SOAs), showing replicate SOAs attached to PVC pipe at Pensacola Beach.
Diversity and composition of microbial communities in SOAs
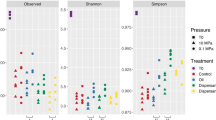
No significant trends in alpha diversity were observed over the time course of the experiment for microbial communities associated with SOAs and SOA-surrounding sands. Alpha diversity as determined by Shannon indices showed substantial variation (Fig. 2a) in SOAs and SOA-surrounding sands from both sediment depths, while diversity in control sands was more consistent with time. Whereas a previous study of buried small oil particles and oil films in the supratidal zone of Pensacola Beach (PB) showed a >50% reduction of Shannon indices during the initial 6 months after oil came ashore17, here we detected little to no reduction of Shannon indices in the SOAs. We attribute the lack of diversity change to the fact that the source material used in this study was already colonized by an established community of hydrocarbon-degrading bacteria. However, statistical analysis showed that Shannon indices in SOAs were distinct from those of control sands. The mean number of observed OTUs declined by 23% in SOAs and SOA-surrounding sands (827 ± 147 observed OTUs) in comparison to control sands (1069 ± 148 observed OTUs) (Fig. 2b).

Diversity of microbial communities in sediment oil agglomerates (SOAs), SOA-surrounding sands, and control sands over the 3 year time course. Incubation time is represented as days after SOA deployment. (a) Alpha-diversity is calculated based on Shannon indices and (b) the number of observed OTUs (Student’s t test: p-value = 0.015* and 0.001***).
In this study, the source material used for the experiment was produced from SOAs that had freshly formed from MC252-oil mousse washing onto the shore within a few months after the DWH oil spill (end of June 2010), and thus had a similar hydrocarbon composition as the weathered mousse32. SOAs formed as the mousse was stranded and mixed with beach sand. Microbial communities in the SOAs were distinct from those present in SOA-surrounding sands or in control sands, as determined by beta diversity of SSU rRNA genes according to the Bray Curtis distance metric (Fig. 3a). SOA community composition more closely resembled the source material in the beginning of the experiment and then strongly diverged from the source material and surrounding control sands across the time series, showing no evidence of recovery (Fig. 3a). To further examine the relationships between beta diversity and oil contamination, a canonical analysis of principal coordinates (CAP) analysis was performed exclusively on SOAs over time. For the first 6 months of the time series, community structure was strongly linked to total petroleum hydrocarbon concentrations, indicating that the input of labile hydrocarbon compounds was driving microbial community dynamics more than time or depth. Across intermediate time scales, after labile hydrocarbons were depleted, beta diversity appeared to vary with the concentration of polycyclic aromatic hydrocarbons. Subsequently, the influence of petroleum hydrocarbons diminished from the end of year 1 to year 3, and communities were structured by depth and time (Fig. 3b).

Effects of SOAs on Pensacola Beach microbial community. (a) The principal coordinates analysis PCoA and (b) constrained analysis of the principal coordinates (CAP) of bacterial communities. The ordination of the microbial community was constrained by the experimental variables to show how these factors affect the microbial community. The arrow’s length and direction indicate factors that have a significant effect on the microbial community organization.
The impact of oil contamination on microbial community composition was further examined in buried SOAs by investigating the dynamics of specific taxa over the 3-year time series. The mean of relative abundances from the two different depths were pooled for further analysis. A clear succession of microbial populations was observed in SOAs with time, whereas these same taxa were much lower in relative abundance and showed no noticeable pattern in SOA-surrounding sands and control sands (Fig. 4). At the phylum and class level, microbial communities in SOAs were dominated by Gammaproteobacteria (42 to 58% relative abundance), Alphaproteobacteria (38 to 58%), and Actinobacteria (up to 10%) (Fig. 4). Few studies are available from microbial communities of oil emulsions or SOAs/ tar balls and taxonomic characterization in previous work was often limited to the phylum or class level9,24,33. In an investigation of 3 mousse samples collected off the coast of Louisiana, Liu and Liu (2013) observed a high relative abundance, up to 75%, of either Alphaproteobacteria or Gammaproteobacteria, similar to the SOAs studied here33. In previous work on Louisiana beaches impacted by the DWH spill, Urbano et al. (2013) observed enrichment of Gammaproteobacteria and Alphaprotebacteria in tar balls collected in the drier supratidal zone, while Deltaproteobacteria were detected in tar balls from the wetter intertidal zone9. Bacosa et al. (2016) observed an enrichment of hydrocarbon-degrading bacteria, mainly Gammaproteobacteria, in tar balls collected within 13 months of the Texas city “Y” spill, although a phylum to class level classification of microbial communities was not provided in their study24. SOA communities appear to more closely resemble those of mousse rather than pristine sands or sands containing more diffuse oiled particles17,34,35. In particular, SOAs and mousse foster much higher levels of Alphaproteobacteria and Actinobacteria.

Relative abundance of Alphaproteobacteria (red), Gammaproteobacteria (green), and Actinobacteria (light blue) in SOAs (solid) and uncontaminated control sands (dotted) over the 3 year time course. Abundance is determined based on the total SSU rRNA gene sequences retrieved.
Similar to previous work in environments impacted by the DWH spill, the Gammaproteobacteria showed a maximum relative abundance of 58% early in the time course at approximately 100 days, whereas the Alphaproteobacteria peaked at 58% later at ~ 300 days, followed by a bloom of up to 10% Actinobacteria after 400 days34,35,36 (Fig. 4). Both Gammaproteobacteria (Alcanivorax, Marinobacter) and Alphaproteobacteria (Rhodobacteraceae) were enriched in Pensacola Beach sands after the Deepwater Horizon oil spill and were identified as key players in oil degradation in previous studies that monitored microbial community shifts after weathered oil contamination over a relatively shorter time period34,35. As in these studies of more diffuse oil contamination on shorelines34,37, groups of known hydrocarbon-degrading bacteria within the Gammaproteobacteria (e.g. Alcanivorax) responded first to oil in SOAs followed by members of the Alphaproteobacteria at later stages when recalcitrant oil hydrocarbons predominated. Major differences between the SOA time series presented here and time series of more diffuse oil contamination in buried sands17,33,34 are that microbial succession occurred over much longer time scales (>3 years) and no recovery occurred in the SOAs. In previous work at Pensacola Beach17,34,35, PHCs returned to background levels one year after oil came ashore, and a typical beach sand microbial community had reestablished that showed little to no evidence of oil hydrocarbon degradation potential.
At the order to genus level, three distinct maxima in microbial populations were observed with time in the SOAs, whereas these same taxa showed a much lower relative abundance in control samples (Fig. 5). Maxima in relative abundance are interpreted as microbial populations that respond to petroleum hydrocarbons available during the early (0–131 days of incubation), mid (131–235 days of incubation), and late stages (after 235 days of incubation) of the time series (Fig. 5; Supplemental Fig. 2). Early responders included the Caulobacterales within the class Alphaproteobacteria and Oceanospirillales within the class Gammaproteobacteria that increased in relative abundance during the first 100 days, from 0.5% to 12% and from 7% to 13%, respectively (Supplemental Fig. 2). At the genus level, the relative abundance of known alkane degraders such as Alcanivorax, Hyphomonas, Phenylobacterium, and Mycoplana increased early from 0.13–3% to 3–10% in SOAs and not in control sands (Fig. 5). Alcanivorax is known to degrade relatively short-chain alkanes and not being capable of degrading aromatic hydrocarbons38,39. In previous studies of oiled sands and tar balls, Alcanivorax spp. were the most abundant OTU in the oil-contaminated samples24,34. A high relative abundance of Alcanivorax (up to 20%) was also observed in a gravel beach after the Xingang oil spill in China40. The genus Hyphomonas, known to be able to utilize aromatic hydrocarbons, was enriched in oiled sand and microcosms with crude oil35,41,42. A DNA-based and stable isotope probing (SIP) study with [U-13C]anthraquinone from PAH-contaminated soil showed that the genus Phenylobacterium is responsible for anthraquinone degradation43. The genus Mycoplana is also known to degrade aromatic hydrocarbons44,45. Our observations are corroborated by petroleum hydrocarbon analysis, which revealed that short-chain alkanes (C15) and relatively low-molecular weight PAHs such as naphthalene were rapidly depleted over the first ~100 days when populations of early responders were enriched32.

Relative abundance of taxa detected at the genus level in SOAs (solid) and uncontaminated control sands (dotted) over the 3-year time course. Abundance is determined based on the total SSU rRNA gene sequences retrieved.
Distinct microbial groups were enriched during the mid phase, between 131 and 235 days, of the time series. The genus Pseudomonas within the order Pseudomonadales was enriched from 5 to 15.2% after 131 days in SOAs, whereas Pseudomonas remained generally below 1% abundance in control sands. Pseudomonas has been reported to produce biosurfactant during PAH degradation and was also enriched in previous studies of oil-contaminated sands34,35,46,47. The relative abundance of Rhizobiales, Actinomycetales, and Rhodospirillales increased in the later stages of the time series after 235 days. Parvibaculum within the order Rhizobiales is known to degrade both aliphatic and aromatic hydrocarbons, and this genus was highly enriched in SOAs and not in control sands late in our time series, in agreement with previous work in oiled sands35,48,49. Relative abundance of Mycobacterium, a known PAH degrader, increased from <0.01% to 9% during the time series (Fig. 5). Evidence of Mycobacterium was also detected using a DNA fingerprinting approach from SOAs collected from supratidal zone of coastal headland beach in Louisiana9. Mycobacterium is the only genus shown to degrade 4-ring PAHs such as the chrysene observed in SOAs9,50. Mycobacterium is also known to be adapted to low moisture content and periods of desiccation36, which resembles the characteristics of dry SOAs16. Another known PAH-degrader Stenotrophomonas was a major microbial group associated with tar balls in the intertidal zone of a headland beach9, but its relative abundance in this study was very low at 0–0.22% throughout our time course. Members of the genus Marinobacter, a generalist group known to degrade both alkane and PAHs, were enriched in many oil-impacted environments34,36,40,51,52 but their abundances in SOAs decreased during the experiment from 6% to near 0% (Fig. 5). Petroleum hydrocarbon analysis showed that mid-chain alkanes (C16–C30), phenanthrene, and dibenzothiophene were degraded from ~100 days to ~300 days after the SOA burial. This implies that secondary responders e.g. Pseudomonas and some of early the responders such as Hyphomonas and Mycoplana were capable of degrading these hydrocarbon compounds. After approximately 300 days of incubation, primarily long-chain alkanes (C30–C40) remained to be degraded32, which coincided with the rapid increase of the relative abundance of Parvibaculum. A few isolates of the genus Parvibaculum such as P. lavamentivorans and P. hydrocarboniclasticum are known to be capable of utilizing n-alkanes or linear alkylbenzenesulfonates49,53, which implies degradation of long-chain alkanes by Parvibaculum at a later stage of incubation.
The abundance of overall bacteria and diazotrophic communities in SOAs
The abundance of SSU rRNA genes on average was three orders of magnitude higher in SOAs in comparison to the surrounding sands or control sands. This bacterial bloom indicates that SOAs are hotspots of microbial growth. In the SOAs, overall bacterial abundance increased from 3.4 × 107 to 4.4 × 108 copies g−1 during the first 89 days, whereas overall bacterial abundance in SOA-surrounding sands and control sands remained at 2.0 × 105 to 2.4 × 107 copies g−1. Characterization of the microbial communities associated with the SOAs by SSU rRNA gene sequencing, as discussed above, revealed a number of abundant groups that contain members known to be capable of nitrogen fixation (diazotrophy) including the Rhizobiales, Frankiales, Rhodobacterales, and Rhodospirillales, and we hypothesized that microbial nitrogen fixation would be enhanced with time. In order to estimate nitrogen fixation potential during in situ SOA incubation in Pensacola Beach sand, the abundance of genes encoding nitrogenase enzyme (nifH), the best studied molecular marker for nitrogen fixation54, was quantified. Results revealed that both SSU rRNA gene and nifH abundance in SOAs differed from those of SOA-surrounding sands and control sands (Fig. 6). Initially, the ratio of nifH to SSU rRNA gene abundance remained 0–0.02 for the first 300 days and then showed a large increase to 0.44 towards the latter stages of the time course, suggesting that diazotrophs bloomed after 1 year. The abundance of nifH genes in SOAs then was one to three orders of magnitude higher in comparison to the nifH abundance in the surrounding sands and control sands (7.6 × 105 to 8.4 × 107 copies g−1 in SOAs; 2.3 × 104 to 7 × 104 in surrounding; 2.6 × 104 to 5.1 × 104 in control). Previous studies have shown that SOAs contain extremely high C/N ratios indicative of nitrogen limitation16. In addition, the results from this SOA experiment are corroborated by previous studies of more diffuse oil in sands at Pensacola Beach, which also showed elevated nitrogenase abundance in oil-contaminated sand layers35. Results indicate that a paucity of nitrogen as evidenced by a high C/N ratio triggered microbial nitrogen fixation to produce bio-available nitrogen from atmospheric nitrogen.

(a) SSU rRNA gene, (b) nifH gene abundance per gram sediment and (c) nifH gene abundance normalized to the abundance of SSU rRNA genes in 10 cm and 50 cm sediment depth intervals from SOAs and SOA-surrounding sands as well as control sands.
Sequencing of nifH amplicons was conducted to further elucidate the microbial groups responsible for diazotrophy in the oiled dry beach environment. Diazotroph microbial diversity as determined by Shannon indices decreased over the time series (Fig. 7). At the phylum to class level, results showed that the Alphaproteobacteria and Actinobacteria were the most dominant diazotroph groups in SOAs (relative abundances of 64–71% and 15–21%, respectively) across the time series (Supplemental Fig. 3a). At the order level, members of the Rhizobiales (36–44% relative abundance) of the Alphaproteobacteria and the Frankiales of the Actinobacteria (15–21%) were the most abundant throughout time series. The late bloom of diazotroph abundance is concurrent with the maximum relative abundance of Rhizobiales and Frankiales in the times series, determined by sequencing of SSU rRNA as well as nifH amplicons, which peaked after 400 days. At the genus level, Methylobacterium within the order Rhizobiales was the most abundant diazotroph group, which constituted from 25 to 33% of nifH gene relative abundance (Supplemental Fig. 3b). Methylobacterium increased rapidly from 25% relative abundance after 445 days of incubation to 31% abundance at the end of the time course. The second most abundant diazotroph group was Frankiales which comprised up to 15–21% relative abundance.

Alpha-diversity of nitrogenase gene sequences over the three-year time course of SOA samples incubated in Pensacola Beach sand.
Members of the Rhizobiales as well as Frankiales are well known as nitrogen-fixing symbionts associated with plant roots55, and free-living members of the Rhizobales are also thought to catalyze nitrogen fixation in a variety of ecosystems56. Methylobacterium was shown to grow on PAHs as well as produce biosurfactants in oil-contaminated systems57,58. Recently, it was shown that Frankia grows with PAHs as the sole carbon source and contains genes for alkane degradation59. Methylobacterium was also abundant in oil mousse collected from the sea surface and in salt marshes in the northern Gulf impacted by the DWH oil spill33,60. Therefore, multiple lines of evidence indicate a close coupling of petroleum hydrocarbon degradation to nitrogen fixation in SOAs. In our time series, nitrogen appears to become limiting after the first year, resulting in selection for microbial populations capable of coupling nitrogen fixation to hydrocarbon degradation.
Inferred metagenomic analysis
Based on microbial community composition in SOAs as determined by SSU rRNA gene amplicon sequencing, inferred metagenomic analysis was performed to assess the metabolic potential of the communities across the time series. Given the abundance of hydrocarbon-degraders and diazotrophs in the time course, our analysis focused on functional genes for hydrocarbon degradation and nitrogen fixation (Supplemental Fig. 4). The predicted relative abundance of alkane-1-monooxygenase (alkB) genes peaked within the first approximately 90 days post burial and then decreased rapidly, suggesting that relatively simple hydrocarbon substrates such as alkanes were utilized by bacteria at this early stage of the time series. Microbial groups that were predicted to contribute to alkane degradation include members of the Rhodobacteraceae, Pseudomonadaceae, Alcanivoraceae, and Alteromonadaceae. The predicted relative abundance of naphthalene 1,2-dioxygenase genes (nahAc, ndoB, nbzAc, dntAc) and other PAH dioxygenase genes (nidA, nidB) reached a maximum later in the time series, at approximately 200 and 300 days post initiation, respectively. Pseudomonas was predicted to contribute to the degradation of more recalcitrant, aromatic hydrocarbons later in the time series. Lastly, predicted relative abundance of nitrogenase genes increased rapidly after 400 days and peaked at approximately 750 days, indicating enhanced bacterial nitrogen fixation at a later stage of SOA incubation. Microbial groups that are predicted to contribute to nitrogenase gene abundance include Rhizobiales, Rhodobacterales, and Rhodospirillales (Supplemental Fig. 5).
Predictions from PICRUSt were corroborated by the chemical evolution of petroleum hydrocarbons, as determined in our companion study32, along with temporal trends in the abundance of overall bacteria and diazotrophs. The predicted abundance of hydrocarbon degradation genes peaked during the first 400 days in parallel with overall bacterial abundance as well as the degradation of alkanes and aromatic compounds. Predictions of alkane monooxygenase (alkB) abundance showed good agreement with the consumption of short chain (C15) alkanes, with both showing maximum changes during the first 100 days. Between 100 and 400 days, the consumption of longer chain alkanes (C18-C22) was not concurrent with the predicted alkB abundance, indicating that other enzyme pathways are responsible for the degradation of these compounds. Finally, the predicted abundance of nitrogenase shows good agreement with the observed nitrogenase abundance, with both showing the largest increases between 400 and 750 days. Thus, multiple evidence support the coupling of hydrocarbon degradation to diazotrophy in SOAs.
Conclusions
Macroscopic sediment-oil agglomerates (SOAs) were formed when MC252 oil from the Deepwater Horizon disaster reached the shores of the northern Gulf of Mexico and interacted with the sediment. Hydrocarbon-degrading bacteria were enriched and a succession of microbial populations was observed that paralleled the chemical evolution of the petroleum hydrocarbons over longer time scales (>3 years) in comparison to previous work on more diffuse oil contamination in beach sands. We provide evidence of bacterial blooms in SOAs, underlining that these large aggregates are hotspots of microbial growth. Our quantification of diazotrophs in large aggregates shows that nitrogen-fixing taxa predominate in oil-degrading microbial communities during the late stages of the time course when nutrients likely become depleted. The coupling of nitrogen fixation to hydrocarbon degradation thus represents a key process for the microbial decomposition of macroscopic oil aggregates.
Materials and Methods
Sample collection and experimental design
Sediment oil agglomerates (SOAs) were collected at Pensacola Beach, FL, USA (30.3261 N, 87.1744 W) on 30 June, 2010. SOAs were homogenized, and then filled into 3.8 cm diameter-stainless steel meshballs producing standardized SOAs. After determining the initial masses of each standardized SOA, 10 filled meshballs were attached in pairs to a PVC pipe (1.3 cm diameter) at 10 cm intervals (Fig. 1). Ten of these meshball arrays were buried in the supratidal zone at Pensacola Beach on 22 October, 2010 such that the meshballs were located at 10, 20, 30, 40 and 50 cm sediment depth (Fig. 1 and Supplemental Fig. 6). The arrays were retrieved 41, 89, 131, 181, 235, 279, 327, 445, 735, and 1152 days after burial, and the mass of each SOA was determined again before freezing at −20 °C in clean glass jars for further microbial community analysis. Together with the arrays, sandy sediments from the region surrounding each deployed SOA were also collected in order to identify possible impacts to indigenous microbial communities. This study analyzed SOAs and associated sand buried at 10 and 50 cm sediment depth. Control sand without oil contamination was collected from the surface of a nearby pristine sand dune at the same study site after 41, 89, 279, 445, 735, 1152 days of incubation.
Nucleic acid extraction and microbial community analysis
Total genomic DNA was extracted from SOAs using a MoBio PowerSoil DNA isolation kit (MoBio Laboratories, Carlsbad, CA) with slight modifications from the manufacturer’s protocol. Briefly, 0.25 g of thawed SOA sample was placed into a 2 ml bead tube and homogenized for 1 min using a Talboys High Throughput Homogenizer (Troemner, Thorofare, NJ). Overall microbial communities and nitrogen-fixing prokaryotes were characterized by targeting SSU rRNA and nitrogenase (nifH) genes, respectively. PCR amplification of SSU rRNA genes was performed using 515 F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′) primers as described by the Earth Microbiome Project (http://www.earthmicrobiome.org/emp- standard-protocols/dna-extraction-protocol/) for Illumina sequencing61. For nifH, the primer set IGK3 (5′-GCIWTHTAYGGIAARGGIGGIATHGGIAA-3′) and DVV (5′-ATIGCRAAICCICCRCAIACIACRTC-3′) was used for PCR amplification as described previously62. PCR products were barcoded using an Access Array Barcode Library (Fluidigm, South San Francisco, CA), purified using the E.Z.N.A Cycle Pure Kit (Omega Bio-tek, Norcross, GA), and pooled based on DNA concentration. Purified and pooled PCR amplicons were sequenced on an Illumina MiSeq platform at the DNA services facility at the University of Illinois at Chicago (https://rrc.uic.edu/). Sequence analysis was accomplished using the software QIIME ver. 1.9.163 and Mothur ver. 1.38.064. Sequences with quality score below 20 were removed using Mothur ver. 1.38.0 and clustered into operational taxonomic units (OTUs) by 97% and 92% sequence identity for SSU rRNA and nifH genes62, respectively using UCLUST65 implemented in QIIME ver. 1.9.1. Representative sequences were aligned against the SILVA ver. 123 database (https://www.arb-silva.de/) or nifH reference alignments database62, and chimeric sequences were removed using UCHIME65 implemented in Mothur ver. 1.38.0. These high-quality sequences were taxonomy assignment to the SILVA SSU rRNA or nfH reference alignments database with the RDP classification algorithm with a minimum confidence threshold of 50%. (https://www.arb-silva.de/, Gaby et al., 2018). The resultant OTU table was scaled using the CSS algorithm implemented in QIIME ver. 1.9.166. The microbial diversity calculations and statistical analyses were performed with default R functions or with “phyloseq” and “vegan” R packages67,68,69. For alpha diversity analysis, Shannon indices were calculated with QIIME ver. 1.9.1. To assess shifts in the diversity and community composition over time, a Bray-Curtis distance matrix was calculated from the rarefied OTU table and used for a principal coordinate analysis (PCoA) and canonical analysis of principal coordinates (CAP) analyses. The oil effect on community similarity and dispersion was estimated with a PERMANOVA and BETADISP statistical tests with 1,000 permutations. Additionally, CAP analysis was performed to assess the correlation between microbial community structure and the following variables: Incubation.Time + Depth + Total.Oil + Total.PAH + Total.Alkens using “capscale” function in R package vegan. The significance of the CAP models was tested using the “permutest” function in a vegan package with 999 permutations. Finally, the Mantel correlation test with 10,000 permutations was applied to determine the similarity between the patterns of the chemical components and microbial communities.
Based on microbial community composition in SOAs as determined by SSU rRNA gene amplicon sequencing, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was employed in order to predict metagenome functional content of differentially abundant OTUs70 and predict Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog functional profiles. The OTU table was normalized using the software QIIME ver. 1.9.163, and each OTU was divided by SSU rRNA gene abundance. The resultant OTU table was used to create the final metagenome functional predictions. All raw sequences have been uploaded to NCBI under Bioproject PRJNA450618.
Quantitative molecular analyses
To evaluate quantitative changes in the abundance of overall bacterial communities and nitrogen-fixing prokaryotes (or diazotrophs), quantitative PCR was performed with PowerUp SYBR Green Mastermix (Applied Biosystems, Foster City, CA) and 331 F (5′-TCCTACGGGAGGCAGCAGT-3′)/515 R (5′-ATTACCGCGGCTGCTGG-3′) primers targeting bacterial SSU rRNA genes or PolF (5′‐TGCGAYCCSAARGCBGACTC‐3′)/PolR (5′‐ATSGCCATCATYTCRCCGGA‐3′) primers targeting the nifH marker gene for nitrogen-fixing Bacteria and Archaea as previously described71,72,73. All reactions were performed in triplicate and analyzed using StepOne Software v. 2.3. Serially diluted pGEM-T Easy Vector plasmids (Promega, Madison, WI) containing either a full-length E. coli SSU rRNA gene or nifH gene were used to generate standard calibration curves for quantification of gene abundances. The efficiencies of the quantitative PCR assay ranged from 95.3 to 101%.
References
Reddy, C. M. et al. Composition and fate of gas and oil released to the water column during the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. USA 109, 20229–34 (2012).
Zukunft, P. F. Summary report for sub-sea and sub-surface oil and dispersant detection: sampling and monitoring. Oper. Sci. Advis. Team (2010).
Hayworth, J. S., Clement, T. P. & Valentine, J. F. Deepwater Horizon oil spill impacts on Alabama beaches. Hydrol. Earth Syst. Sci. 15, 3639–3649 (2011).
Wang, P. & Roberts, T. M. Distribution of Surficial and Buried Oil Contaminants across Sandy Beaches along NW Florida and Alabama Coasts Following the Deepwater Horizon Oil Spill in 2010. J. Coast. Res. 29, 144–155 (2013).
Barron, M., Awkerman, J. & Raimondo, S. Oil Characterization and Distribution in Florida Estuary Sediments Following the Deepwater Horizon Spill. J. Mar. Sci. Eng. 3, 1136–1148 (2015).
Michel, J. et al. Extent and Degree of Shoreline Oiling: Deepwater Horizon Oil Spill, Gulf of Mexico, USA. PLoS One 8, 1–9 (2013).
Nixon, Z. et al. Shoreline oiling from the Deepwater Horizon oil spill. Mar. Pollut. Bull. 107, 170–178 (2016).
Fingas, M. & Fieldhouse, B. Studies on crude oil and petroleum product emulsions: Water resolution and rheology. Colloids Surfaces A Physicochem. Eng. Asp. 333, 67–81 (2009).
Urbano, M., Elango, V. & Pardue, J. H. Biogeochemical characterization of MC252 oil: Sand aggregates on a coastal headland beach. Mar. Pollut. Bull. 77, 183–191 (2013).
Lemelle, K. R., Elango, V. & Pardue, J. H. Distribution, characterization, and exposure of MC252 oil in the supratidal beach environment. Environ. Toxicol. Chem. 33, 1544–1551 (2014).
Aeppli, C. et al. Oil weathering after the Deepwater Horizon disaster led to the formation of oxygenated residues. Environ. Sci. Technol. 46, 8799–8807 (2012).
Gustitus, S. A. & Clement, T. P. Formation, Fate, and Impacts of Microscopic and Macroscopic Oil-Sediment Residues in Nearshore Marine Environments: A Critical Review. Rev. Geophys. 55, 1130–1157 (2018).
Hayworth, J. S., Prabakhar Clement, T., John, G. F. & Yin, F. Fate of Deepwater Horizon oil in Alabama’s beach system: Understanding physical evolution processes based on observational data. Mar. Pollut. Bull. 90, 95–105 (2015).
Yin, F., John, G. F., Hayworth, J. S. & Clement, T. P. Long-term monitoring data to describe the fate of polycyclic aromatic hydrocarbons in Deepwater Horizon oil submerged off Alabama’s beaches. Sci. Total Environ. 508, 46–56 (2015).
Operational Science Advisory Team (OSAT-2). Summary Report for fate and effects of remnant oil in the beach environment. Gulf Coast Incid. Manag. Team 1–36 (2011).
Elango, V., Urbano, M., Lemelle, K. R. & Pardue, J. H. Biodegradation of MC252 oil in oil:sand aggregates in a coastal headland beach environment. Front. Microbiol. 5, 161 (2014).
Huettel, M. et al. Degradation of Deepwater Horizon oil buried in a Florida beach influenced by tidal pumping. Mar. Pollut. Bull. 126, 488–500 (2018).
John, G. F., Han, Y. & Clement, T. P. Weathering patterns of polycyclic aromatic hydrocarbons contained in submerged Deepwater Horizon oil spill residues when re-exposed to sunlight. Sci. Total Environ. 573, 189–202 (2016).
Kiruri, L. W., Dellinger, B. & Lomnicki, S. Tar balls from deep water horizon oil spill: Environmentally persistent free radicals (EPFR) formation during crude weathering. Environ. Sci. Technol. 47, 4220–4226 (2013).
Tao, Z., Bullard, S. & Arias, C. High numbers of Vibrio vulnificus in tar balls collected from oiled areas of the north-central Gulf of Mexico following the 2010 BP Deepwater Horizon oil spill. Ecohealth 8, 507–511 (2011).
White, H. K. et al. Long-term weathering and continued oxidation of oil residues from the Deepwater Horizon spill. Mar. Pollut. Bull. 113, 380–386 (2016).
Radović, J. R. et al. Assessment of photochemical processes in marine oil spill fingerprinting. Mar. Pollut. Bull. 79, 268–277 (2014).
Prince, R. C. et al. The roles of photooxidation and biodegradation in long-term weathering of crude and heavy fuel oils. Spill Sci. Technol. Bull. 8, 145–156 (2003).
Bacosa, H. P., Thyng, K. M., Plunkett, S., Erdner, D. L. & Liu, Z. The tarballs on Texas beaches following the 2014 Texas City “Y” Spill: Modeling, chemical, and microbiological studies. Mar. Pollut. Bull. 109, 236–244 (2016).
Salleh, A. B., Ghazali, F. M., Abd Rahman, R. N. Z. & Basri, M. Bioremediation of Petroleum Hydrocarbon Pollution. Indian J. Biotechnol. 2, 411–425 (2003).
Dibble, J. T. & Bartha, R. Effect of Environmental Parameters on the Biodegradation of Oil Sludge. Appl. Environ. Microbiol. 37, 729–739 (1979).
Coty, V. F. Atmospheric nitrogen fixation by hydrocarbon-oxidizing bacteria. Biotechnol. Bioeng. 9, 25–32 (1967).
Toccalino, P. L., Johnson, R. L. & Boone, D. R. Nitrogen limitation and nitrogen fixation during alkane biodegradation in a sandy soil. Appl. Environ. Microbiol. 59, 2977–2983 (1993).
Chen, Y. P., Lopez-de-Victoria, G. & Lovell, C. R. Utilization of aromatic compounds as carbon and energy sources during growth and N2-fixation by free-living nitrogen fixing bacteria. Arch. Microbiol. 159, 207–212 (1993).
Prantera, M. T., Drozdowicz, A., Leite, S. G. & Soares, A. Degradation of gasoline aromatic hydrocarbons by two N2 -fixing soil bacteria. Biotechnol. Lett. 24, 85–89 (2002).
Medina-Bellver, J. I. et al. Evidence for in situ crude oil biodegradation after the Prestige oil spill. Environ. Microbiol. 7, 773–779 (2005).
Bociu, I. et al. Decomposition of sediment-oil-agglomerates in a Gulf of Mexico sandy beach. Sci Rep 9, 10071 https://doi.org/10.1038/s41598-019-46301-w (2019).
Liu, Z. & Liu, J. Evaluating bacterial community structures in oil collected from the sea surface and sediment in the northern Gulf of Mexico after the Deepwater Horizon oil spill. MicrobiologyOpen 2, 492–504 (2013).
Kostka, J. E. et al. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl. Environ. Microbiol. 77, 7962–7974 https://doi.org/10.1128/AEM.05402-11 (2011).
Rodriguez-R, L. et al. Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9, 1928–1940 https://doi.org/10.1038/ismej.2015.5 (2015).
Lamendella, R. et al. Assessment of the deepwater horizon oil spill impact on gulf coast microbial communities. Front. Microbiol. 5, 1–13 (2014).
Alonso-Gutierrez, J. et al. Bacterial communities from shoreline environments (Costa da Morte, northwestern Spain) affected by the Prestige oil spill. Appl. Environ. Microbiol. 75, 3407–3418 (2009).
Liu, C. & Shao, Z. Alcanivorax dieselolei sp. nov., a novel alkane-degrading bacterium isolated from sea water and deep-sea sediment. Int. J. Syst. Evol. Microbiol. 55, 1181–1186 (2005).
Schneiker, S. et al. Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 24, 997–1004 (2006).
Zhang, K. et al. Periodically spilled-oil input as a trigger to stimulate the development of hydrocarbon-degrading consortia in a beach ecosystem. Sci. Rep. 7, 1–9 (2017).
Coulon, F., McKew, B. A., Osborn, A. M., McGenity, T. J. & Timmis, K. N. Effects of temperature and biostimulation on oil-degrading microbial communities in temperate estuarine waters. Environ. Microbiol. 9, 177–186 (2007).
Kappell, A. D. et al. The polycyclic aromatic hydrocarbon degradation potential of Gulf of Mexico native coastal microbial communities after the Deepwater Horizon oil spill. Front. Microbiol. 5, 1–13 (2014).
Rodgers-Vieira, E. A., Zhang, Z., Adrion, A. C., Gold, A. & Aitken, M. D. Identification of anthraquinone-degrading bacteria in soil contaminated with polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 81, 3775–3781 (2015).
Brinda Lakshmi, M., Muthukumar, K. & Velan, M. Immobilization of Mycoplana sp. MVMB2 Isolated from Petroleum Contaminated Soil onto Papaya Stem (Carica papaya L.) and Its Application on Degradation of Phenanthrene. Clean - Soil, Air, Water 40, 870–877 (2012).
Brinda Lakshmi, M., Anandaraj, V. P. & Velan, M. Bioremediation of Phenanthrene by Mycoplana sp. MVMB2 Isolated from Contaminated Soil. Clean - Soil, Air, Water 41, 86–93 (2013).
Deziel, E. et al. Biosurfactant production by a soil pseudomonas strain growing on polycyclic aromatic hydrocarbons. These include: Biosurfactant Production by a Soil Pseudomonas Strain Growing on Polycyclic Aromatic Hydrocarbons. Appl. Environ. Microbiol. 62, 1908–1912 (1996).
Prabhu, Y. & Phale, P. S. Biodegradation of phenanthrene by Pseudomonas sp. strain PP2: novel metabolic pathway, role of biosurfactant and cell surface hydrophobicity in hydrocarbon assimilation. Appl. Microbiol. Biotechnol. 61, 342–351 (2003).
Lai, Q. et al. Parvibaculum indicum sp. nov., isolated from deep-sea water. Int. J. Syst. Evol. Microbiol. 61, 271–274 (2011).
Rosario-Passapera, R. et al. Parvibaculum hydrocarboniclasticum sp. nov., a mesophilic, alkane-oxidizing alphaproteobacterium isolated from a deep-sea hydrothermal vent on the East Pacific Rise. Int. J. Syst. Evol. Microbiol. 62, 2921–2926 (2012).
Khan, A. A., Kim, S. J., Paine, D. D. & Cerniglia, C. E. Classification of a polycyclic aromatic hydrocarbon-metabolizing bacterium, Mycobacterium sp. strain PYR-1, as Mycobacterium vanbaalenii sp. nov. Int. J. Syst. Evol. Microbiol. 52, 1997–2002 (2002).
Atlas, R. M. et al. Oil Biodegradation and Oil-Degrading Microbial Populations in Marsh Sediments Impacted by Oil from the Deepwater Horizon Well Blowout. Environ. Sci. Technol. 49, 8356–8366 (2015).
Wang, J. et al. Biodegradation of dispersed Macondo crude oil by indigenous Gulf of Mexico microbial communities. Sci. Total Environ. 557–558, 453–468 (2016).
Schleheck, D., Tindall, B. J., Rosselló-Mora, R. & Cook, A. M. Parvibaculum lavamentivorans gen. nov., sp. nov., a novel heterotroph that initiates catabolism of linear alkylbenzenesulfonate. Int. J. Syst. Evol. Microbiol. 54, 1489–1497 (2004).
Gaby, J. C. & Buckley, D. H. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7 (2012).
Erlacher, A. et al. Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front. Microbiol. 6, 1–9 (2015).
Sellstedt, A. & Richau, K. H. Aspects of nitrogen-fixing actinobacteria, in particular free-living and symbiotic frankia. FEMS Microbiol. Lett. 342, 179–186 (2013).
Salam, L. B., Obayori, O. S. & Raji, S. A. Biodegradation of Used Engine Oil by a Methylotrophic Bacterium, Methylobacterium Mesophilicum Isolated from Tropical Hydrocarbon- contaminated Soil Biodegradation of Used Engine Oil by a Methylotrophic Bacterium, Methylobacterium Mesophilicum Isolated. Pet. Sci. Technol. 33, 186–195 (2015).
Nzila, A., Thukair, A., Sankara, S., Chanbasha, B. & Musa, M. M. Isolation and characterization of naphthalene biodegrading Methylobacterium radiotolerans bacterium from the eastern coastline of the Kingdom of Saudi Arabia. Arch. Environ. Prot. 42, 25–32 (2016).
Rehan, M. & Swanson, E. Frankia as a Biodegrading Agent Frankia as a Biodegrading Agent, 10.5772/61825 (2016).
Engel, A. S. et al. Salt marsh bacterial communities before and after the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 83 (2017).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
Gaby, J. C. et al. Diazotroph community characterization via a high-throughput nifH amplicon sequencing and analysis pipeline. Appl Environ Microbiol 84, e01512-17. https://doi.org/10.1128/AEM.01512-17 (2018).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–6 (2010).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202 (2013).
R Core Team, R Foundation for Statistical Computing, Vienna, A. R: A language and environment for statistical computing. Available online at, https://www.r-project.org/ (2018).
Dixon, P. Computer program review VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Mcmurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8, e61217 (2013).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Warren, M. J. et al. Molybdenum-based diazotrophy in a Sphagnum peatland in northern Minnesota. Appl Environ Microbiol 83, e01174-17. https://doi.org/10.1128/AEM.01174-17 (2017).
Carrell, A. A. et al. Experimental warming alters the community composition, diversity, and N 2 fixation activity of peat moss (Sphagnum fallax) microbiomes. Glob. Chang. Biol. 25, 2993–3004 https://doi.org/10.1111/gcb.14715 (2019).
Kolton, M., Marks, A., Wilson, R. M., Chanton, J. P. & Kostka, J. E. Impact of Warming on Greenhouse Gas Production and Microbial Diversity in Anoxic Peat From a Sphagnum -Dominated Bog (Grand Rapids, Minnesota, United States). Front. Microbiol. 10, 1–13 https://doi.org/10.3389/fmicb.2019.00870 (2019).
Acknowledgements
This research was made possible by grants from The Gulf of Mexico Research Initiative to the C-IMAGE II, C-IMAGE III, and Deep-C consortia. All data are publicly available through the Gulf of Mexico Research Initiative information & Data Cooperative (GRIIDC) at https:/data.gulfresearchinitiative.org (DOI: 10.7266/N7WS8RSV).
Author information
Authors and Affiliations
Contributions
B.S. and M.H. performed the experiments. B.S. and M.K. analyzed the data. B.S., I.B., M.K., M.H. and J.E.K. discussed the data and contributed to manuscript preparation. The manuscript was written by B.S., M.H., M.K., and J.E.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, B., Bociu, I., Kolton, M. et al. Succession of microbial populations and nitrogen-fixation associated with the biodegradation of sediment-oil-agglomerates buried in a Florida sandy beach. Sci Rep 9, 19401 (2019). https://doi.org/10.1038/s41598-019-55625-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55625-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
