
Abstract
The mucosal surfaces of fish harbour microbial communities that can act as the first-line of defense against pathogens. Infectious diseases are one of the main constraints to aquaculture growth leading to huge economic losses. Despite their negative impacts on microbial diversity and overall fish health, antibiotics are still the method of choice to treat many such diseases. Here, we use 16 rRNA V4 metataxonomics to study over a 6 week period the dynamics of the gill and skin microbiomes of farmed seabass before, during and after a natural disease outbreak and subsequent antibiotic treatment with oxytetracycline. Photobacterium damselae was identified as the most probable causative agent of disease. Both infection and antibiotic treatment caused significant, although asymmetrical, changes in the microbiome composition of the gills and skin. The most dramatic changes in microbial taxonomic abundance occurred between healthy and diseased fish. Disease led to a decrease in the bacterial core diversity in the skin, whereas in the gills there was both an increase and a shift in core diversity. Oxytetracycline caused a decrease in core diversity in the gill and an increase in the skin. Severe loss of core diversity in fish mucosae demonstrates the disruptive impact of disease and antibiotic treatment on the microbial communities of healthy fish.
Similar content being viewed by others
Introduction
Mucosal surfaces of animals harbour microbial communities (i.e., microbiomes), which can act as the first-line of defense against pathogens, either through competition or production of antibiotic compounds1,2,3. Furthermore, microbiomes are thought to have evolved to optimize the immune response of each organ and promote homeostasis3,4. Usually a diverse microbiome is associated with healthy phenotypes, but disruptions to this equilibrium can lead to an increase in abundance of opportunistic pathogens and disease susceptibility5,6.
Many factors can shape the composition of the fish microbiomes, including host species7, stress8,9, diet10, water quality11, host physiology12,13 and infection6,14,15. Importantly, healthy mucosal surfaces, such as the skin and gills, are naturally colonized by pathogens16,17,18 from the surrounding waters that can integrate into the host’s microbial community19,20. A shift in the abundance of such pathogens on the fish mucosae can lead to microbial imbalance (i.e. dysbiosis) and disease21, which is usually accompanied by a reduction in bacterial diversity6,14,22.
Stress imposed by fish farming conditions can also result in changes in microbiome composition that may lead to an increase in disease susceptibility8. As infectious disease is one of the main constraints to aquaculture growth and profitability, it is crucial to have a better understanding of the host-symbiont-pathogen nexus. The European seabass Dicentrarchus labrax is one of the main farmed fish species in southern Europe, totaling 103.476 tons in landings (10% of global aquaculture production) between 2002 and 201123. This important food resource is susceptible to several bacterial pathogens: Photobacterium damselae, which causes photobacteriosis, Vibrio spp. causing vibriosis, and Tenacibaculum maritimum causing tenacibaculosis, just to name a few24. All of these pathogens can induce bacterial septicemia resulting in high mortalities in fish farms25,26,27. Photobacterium damselae in particular, is increasingly being reported as the main etiologic agent affecting fish farms worldwide and has been also described in molluscs, crustaceans and mammals28,29,30,31,32,33,34. Control of photobacteriosis in fish farms is challenging, and mortality can reach 60–80% rates in farmed European seabass35,36. Although vaccination is available, immunization is still not fully effective36,37, and in many cases antibiotic treatment remains the preferred option to control such pathogens (e.g. Oxytetracycline32,38).
Most commonly, the impact of antibiotic use on fish health is assessed through toxicological studies39,40. The few studies that have investigated the effects of antibiotics on the microbiome of fish have focused on gut dysbiosis41,42,43,44,45,46. Not surprisingly, a decrease in microbial diversity was detected42,46, along with an increased susceptibility to secondary infection, reduced host growth41,44 and higher mortality43. Importantly, these studies also reported bacterial pathogens acquiring resistance after antibiotic treatment, suggesting that farmed fish microbiomes could become reservoirs for antibiotic resistant genes42,43,44. In fact, several studies showed an increase in resistance to tetracycline and streptomycin antibiotics in strains of P. damselae sampled from both wild and farmed fish hosts35,38,47,48.
In the present study, we characterized the dynamics of the gill and skin microbiomes of the seabass Dicentrarchus labrax before, during and after a disease outbreak potentially caused by Photobacterium damselae, and subsequent antibiotic treatment with oxytetracycline49. We describe the dysbiosis caused by disease and antibiotic treatment in microbial diversity of both mucosae over 3 weeks. Towards this aim, we used 16S rRNA high-throughput sequencing (metataxonomics) and amplicon sequence variance analysis to examine changes in both alpha- and beta-diversity, as well as differences in taxa proportion over time.
Results
Approximately, a total of 3.6 million raw reads were generated, while the number of sequences per sample ranged from 2,354 to 50,564 (Supplementary Table S1). A total of 6,485 unique ASVs were detected, but after normalization and depletion of Archaea and Algae ASVs, a total of 3,827 ASVs (1,560,279 sequences) and 3,741 ASVs (1,904,115 sequences) were analyzed for the gill and skin microbiomes, respectively (Supplementary Table S1). Taxa showing a mean proportion ≥4% in any state were considered the most abundant taxa. Analyses of alpha- and beta-diversity showed no significant differences (P > 0.05; data not shown) between samples from the two healthy time points as well as between the samples from the three recovery time points (Fig. 1) for both gill and skin microbiomes. Therefore, in all our subsequent analyses, samples from Aug 21 (Healthy 1) and Aug 29 (Healthy 2) were combined into the “healthy” state; and samples from Sep 19 (Recovery 1), Sep 26 (Recovery 2) and Oct 3 (Recovery 3) were combined into the “recovery” state in order to increase sample size (Fig. 1).

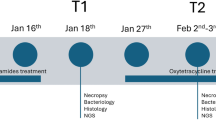
Schematic illustration of the experimental design and health status of each sampling point. Ten fish were sampled for gill and skin microbial communities at each sampling point, totaling 70 fish sampled in this experiment.
Gill bacterial composition and diversity
No significant differences were detected in alpha-diversity across all states (RRPP, P > 0.5), with the exception of the Shannon index (RRPP, P = 0.04) (Table 1, Fig. 2A). There were, however, significant differences between healthy and recovery states for all alpha-diversity indices (RRPP, P ≤ 0.03; Table 1). Beta-diversity also varied greatly between states (PCoA, Fig. 3A), with significant differences both in overall and pairwise comparisons in almost all the tests (Adonis, P < 0.05; Table 1).

Mean values and standard deviations of Shannon, Faith’s phylogenetic (PD), ACE and Fisher alpha-diversity estimates plotted for the gill (A) and skin (B) microbiomes of Dicentrarchus labrax (seabass) during the four different states. H1 – Healthy 1; H2 – Healthy 2; D – Diseased; T – Treatment; R1 – Recovery 1; R2 – Recovery 2; R3 – Recovery 3.

PCoA plot computed with weighted Unifrac distance for gill (A) and skin (B). Each dot represents a microbiome sample and is coloured by sampling point.
Bacteroidetes, Proteobacteria and Verrucomicrobia were the most abundant phyla retrieved from the seabass gill microbiome across states, accounting for 83% to 93% of the sequences altogether (Table 2). The most abundant genera were NS3a marine group, Polaribacter 4, Pseudomonas and Rubritalea, which were present in all four states (Table 2, Supplementary Fig. S1A). Other relatively abundant genera were: (a) Polynucleobacter, which accounted for 4–7% of the sequences in the diseased, treatment and recovery states, but only 0.2% in the healthy state; (b) Stenotrophomonas, represented by 5% of the sequences in the healthy state, and 2–3% in the remainder states; and (c) Photobacterium, which accounted for 5% of the sequences in the diseased state but ≤1 in all other states (Table 2, Supplementary Fig. S1A).
Taxa mean proportions varied between states: (i) in healthy versus diseased states 7 taxa increased and 3 decreased; (ii) in diseased versus treatment states 3 taxa increased, 2 decreased and 6 remained almost constant; and (iii) finally, in treatment versus recovery states 3 taxa decreased, 6 increased and 1 remained constant (Table 2, Fig. 4A). The 3 most abundant bacterial phyla and the 8 most abundant genera all varied significantly (P ≤ 0.04) in their mean proportions across the four studied states (Fig. 4A, Table 1). In addition, 9 of these taxa varied significantly (P ≤ 0.04) between healthy and disease states, whereas 5 varied significantly (P ≤ 0.04) between treatment and recovery states (Table 1). Only 2 genera varied significantly (P ≤ 0.03) between disease and treatment states (Table 1).

Alluvial plots of relative frequency of most abundant (>4%) taxa recovered from the gill (A) and skin (B) of the seabass for healthy, diseased, treatment and recovery states.
Of the 55 ASVs recovered from the gill core microbiome across the four states, 21 were present in the healthy state, 26 in the diseased state, 5 in the treatment state and 10 in the recovery state (Fig. 5A). Four of these ASVs were unique to the healthy state, 11 unique to the diseased state and one to the recovery state (Fig. 5A). Of the 11 unique ASVs recovered from the gill core microbiome of diseased fish, one was identified as Photobacterium damselae. There were 8 other ASVs belonging to the Photobacterium genus, of which 7 were unique to the diseased state and one was found in all four states (Supplementary Table S1). This suggests that P. damselae is the most likely causative agent of the disease in the diseased fishes.

Core microbiota of seabass gill (A) and skin (B) at the ASV level. Distinctive bars represent relative abundance of each ASV for healthy, diseased, treatment and recovery states, labeled to the lowest taxonomic level possible.
Skin bacterial composition and diversity
Alpha-diversity estimates varied significantly across all states (RRPP, P ≤ 0.002; Table 1, Fig. 2B) and between states in the skin microbiome. They decreased significantly between healthy and diseased fish and increased significantly between diseased and treatment states (RRPP, P ≤ 0.003; Table 1, Fig. 2B). Beta-diversity estimates show significant differences across all states and between states (Adonis, P < 0.05; Table 1).
As for the skin microbiome, Bacteroidetes, Proteobacteria and Verrucomicrobia were the most abundant phyla retrieved across states, accounting for 87% to 93% of the sequences altogether (Table 2). The genera NS3a marine group, Polaribacter 4, Pseudomonas and Stenotrophomonas were the most abundant in all four states (Table 2, Supplementary Fig. S1). Moreover, Pseudoalteromonas accounted for 5% of the sequences in the treatment state, but only 0.1–1% in the remaining states (Table 2, Supplementary Fig. S1).
Mean proportions of the bacterial taxa varied significantly between states: (i) in healthy versus diseased states 4 taxa increased and 4 decreased; (ii) in diseased versus treatment states 5 taxa increased and 3 decreased; and (iii) in treatment versus recovery states 5 taxa increased, 2 decreased and 1 remained constant (Table 2, Fig. 4B). The 3 most abundant phyla and 5 most abundant genera all varied significantly (P ≤ 0.03) across the four states (Fig. 4B, Table 1). All taxa varied significantly between healthy and diseased states (P ≤ 0.01); all except one varied significantly between diseased and treatment states (P ≤ 0.02); and 2 genera varied significantly (P ≤ 0.02) between treatment and recovery states (Table 1).
A total of 43 ASVs formed the core microbiome of all four states, 17 were present in the healthy state, 8 were present in the diseased state, 33 in the treatment state and 8 in the recovery state (Fig. 5B). It is worth noticing that 2 ASVs were unique to the healthy state and 16 ASVs were unique to the treatment state.
Discussion
In this study, we investigated the dynamics of the gill and skin microbiomes in 140 samples of the farmed seabass Dicentrarchus labrax during a natural disease outbreak and subsequent antibiotic treatment with oxytetracycline. We used high-throughput sequencing technology to generate 16S rRNA bacterial ASVs and examine changes in microbial composition and diversity over six weeks. We identified Photobacterium damselae as the most probable causative agent of disease.
The most abundant taxa found in the gill and skin microbiomes of healthy farmed seabass (Dicentrarchus labrax) belonged to the Proteobacteria, Bacteroidetes and Verrucomicrobia phyla. These phyla have been previously described as the most abundant in the gill and skin microbiomes of several teleosts14,50,51,52, including the seabass16,53,54. At the genus level, the most abundant taxa were the NS3a marine group, Polaribacter 4, Pseudomonas, and Stenotrophomonas in the gills and skin (Table 2, Supplementary Fig. S1, Fig. 4), and Rubritalea in the gills (Table 2, Supplementary Fig. S1, Fig. 4). These results are mainly in accordance with previously described microbiomes of healthy seabass16,54, including fish retrieved from the same farmed population during winter months16. However, one of the most abundant genera in the healthy seabass gill microbiome was Polynucleobacter16, which in the present study only accounted for 0.2% of the sequences in the healthy state, and 4–7% in the other three studied states (Table 2). Another compositional difference was the high abundance of Stenotrophomonas found in both tissues in apparently healthy individuals (Table 2) in this study, but not in Rosado et al.16. Several environmental factors known to impact microbiome composition, such as seasonality55,56 and water temperature57, could be driving these differences between our two studies.
The composition and diversity of the gill and skin seabass microbiomes varied differently during infection. Whereas in the skin there was a significant decrease in alpha-diversity between healthy and diseased fish, there were no significant differences in the gill microbiome. An overall decrease in microbial richness was also reported for the skin of Atlantic salmon as a result of infection with salmonid alphavirus6 and sea lice15; but interestingly, as in the present study, Legrand et al.14 reported significant differences in microbial richness between the skin of healthy and enteritis-infected yellowtail kingfish, but not in the gills.
Significant changes in beta-diversity occurred in both gills and skin, showing clear signs of dysbiosis in both tissues. In the skin microbiome of diseased fish, the abundance of taxa from the non-pathogenic NS3a marine group and Polaribacter 4 decreased, whereas the pathogenic Pseudomonas and Stenotrophomonas significantly increased. Pseudomonas spp. almost doubled their abundance and largely dominated the skin microbiome of diseased fish. While the genus Stenotrophomonas contains important globally emergent fish pathogens (e.g. Stenotrophomonas maltophilia58,59), Pseudomonas harbors both opportunist fish pathogens (e.g. P. baetica, P. chlororaphis60,61; amongst others8,62) and taxa with known antimicrobial activity against fish pathogens (e.g. Flavobacterium psychrophilum63). For example, P. fluorescens is an important pathogen of carp and salmon64,65, but is also known to inhibit the growth of Saprolegnia, an oomycete that causes huge losses in aquaculture66,67. Importantly, a ten-fold increase of Pseudoalteromonas, which was not amongst the most abundant taxa in healthy fish, occurred in the skin of diseased fish. Species from this genus can inhibit the growth of both Vibrio spp. and Photobacterium damselae6,68,69,70,71, hence an increase of Pseudoalteromonas could lead to a decrease of the other two genera, as we have seen in the skin microbiomes of seabass transitioning from healthy to diseased states (from 2% to 0.7% and from 0.3% to 0.2%, respectively). In the gills of diseased fish, the majority of the most abundant bacterial genera in the healthy state (NS3a marine group, Polaribacter 4, Pseudomonas, Rubritalea and Stenotrophomonas) decreased significantly in abundance during infection, with the exception of Polynucleobacter. Amongst the most abundant taxa in the gill, only Photobacterium spp. was exclusively associated with diseased fish, where it showed a 25-fold increase. Similarly, all studies addressing the effects of parasitic infection on fish microbiomes reported significant changes in microbial composition6,14,15. Importantly, all of these studies reported an increase of potentially pathogenic taxa, which highlights the opportunistic nature of such pathogens6,14,15. Although Photobacterium damselae was only highly abundant in the diseased gill microbiome, the tissue with more significant shifts in overall bacterial composition (alpha-diversity) between healthy and diseased states was the skin. This is not totally unexpected, since it has been shown that this pathogen can unequally affect the microbiome of distinctive mucosal surfaces such as the skin and gill14.
The effects of the disease in the core microbiomes were also significant and again different between tissues, with a shift of core species in the gill and a decrease of core diversity in the skin from healthy to diseased states. A shift of the microbial assemblages with enrichment of specific groups was also described for the gill microbiome of the yellowtail killifish as a result of enteritis14.
Antibiotics administration can negatively impact host physiology in different ways (e.g., inhibiting mitochondrial gene expression72; decreasing enzymatic activity46), leading to dysbiosis and the emergence of antibiotic resistant bacteria35,42,43,44,73. Specifically, the reported effects of oxytetracycline in the gut microbiome of the Atlantic salmon showed a clear reduction in taxonomic diversity, becoming almost exclusively composed of the oxytetracycline resistant Aeromonas spp., which include the salmon pathogens Aeromonas sobria and A. salmonicida74. Similarly, in zebrafish, long-term exposure (6 weeks) to environmental concentrations of oxytetracycline, prompted both a decrease in gut microbial diversity and higher mortality when fish were challenged with the pathogen A. hydrophila46. The impact of broad-spectrum antibiotics in the skin microbiome of Gambusia affinis have also been assessed41,42. In this case, the use of rifampicin led to a decrease of diversity in the skin microbiome after 2.6 days of antibiotic administration. Additionally, as reported for zebrafish and Atlantic salmon, fish subjected to rifampicin antibiotic administration were more susceptible to infection due to osmotic stress and exhibited less growth compared to the control group, an effect that lasted one month after treatment41,42. A key difference with the present study is that the fish used by Carlson et al.41,42 were healthy before antibiotic administration. Importantly, our results showed that skin core diversity was higher in healthy than in recovery individuals, indicating a negative effect of disease and antibiotic use.
In the present study, administration of oxytetracycline resulted in a dramatic reduction of Photobacterium abundance in the gill microbiome, with this genus no longer being one of the most abundant taxa in the treatment and recovery states. This was expected given the reported sensitivity of P. damselae to several antibiotics, including oxytetracycline38. Pseudoalteromonas, however, remained one of the most abundant taxa in the skin microbiome during treatment perhaps due to the host innate immune response mediated by the skin microbiome, given the ability of this genus to produce antimicrobial metabolites that are correlated with host homeostasis68.
Previous studies on the impact of antibiotics on fish skin microbiomes showed that, even though stabilization of bacterial communities during recovery occurs, neither diversity nor composition returns to healthy-like values in the short term (after 1 week)41,42. Here the relative frequency of the most abundant taxa found in the skin microbiome of the seabass during the recovery period, which corresponded to 3 weeks, was similar to that in healthy individuals (P ≥ 0.1 for all taxa except the NS3a marine group). In the gill microbiome, however, differences in taxa proportions between the healthy and recovery states were significant for almost all of the most abundant taxa. Hence, although dysbiosis due to infection was more noticeable in the skin than in the gill, the microbial communities present in the skin seem to be more resilient than those of the gill. Importantly, although the abundance of Photobacterium damselae in the gill seemed to have been controlled through antibiotic administration, it increased significantly in the recovery state, surpassing its initial proportion in the healthy state.
In summary, the mucosal surfaces of fish, such as the gill and the skin, are constantly exposed to several pathogens in the aquatic environment and are crucial to prevent and/or control disease1. It has been shown that both infectious diseases and antibiotic treatment lead to a decrease in microbial diversity, which translates into a decrease in host immunity14,42. Here we described microbial changes in the gill and skin of adult seabass in response to a natural disease outbreak followed by a succeeding treatment with oxytetracycline. We showed that the gill and skin microbiomes are highly disturbed by both infection and antibiotic treatment, ultimately decreasing their diversity.
Methods
Ethical statement
This study monitored a natural infection and subsequent antibiotic treatment as part of routine procedures in a commercial fish farm. All animals were handled by the fish farm employees, our sampling through swabbing was non-invasive and fish were released unharmed with no mortalities observed. According to the Portuguese legislation DL N° 113/2013, our work does not involve animal experimentation and therefore is exempted from the need of ethical approval.
Experimental design, sample collection and preparation
Ten individuals of seabass were collected once a week between August 21 and October 3, 2016, from the same rearing tank in a commercial fish farm located in the estuarine environment of the Ria Formosa (Portimão), southern Portugal. Fish were hatched at September 26, 2014 and entered the growth facility at March 6, 2015. Fish were kept in an open water circulation system in a semi-intensive farming facility, where water is supplied to each tank from the estuary. Fish were kept at a density of ca. 3 kg/m3 corresponding to roughly 100 fish/tank with fish weighting on average 281 g. Given that it was not possible to tag individual fish and the unlikelihood of re-sampling the same individuals every week, a subset of samples believed to be representative of the population was chosen, i.e. 10 individuals (~ 10%), and for statistical purposes individuals were considered as pseudo-replicates. All fish were fed with the same commercial feed and they shared the same clinical history. Individuals were randomly caught using a fishing pole and skin and gill swabs were collected immediately using tubed sterile dry swabs (Medical Wire & Equipment, UK). Skin samples were taken by swabbing several times along the right upper lateral part of the fish from head to tail, while gill swabs were taken from the right filaments between the first and second arch. Due to the non-invasive nature of our sampling procedure, it was not possible to ascertain the sex of the individuals sampled; however, we do not expect this to impact our conclusions since, to the best of our knowledge, no gender bias in microbiome composition has ever been reported for skin or gill of piscine hosts. Swabs were immediately stored at −20 °C until transported on dry ice to the CIBIO-InBIO laboratory by airmail where they were kept at −80 °C until further processing.
To assess gill and skin microbiome dynamics in the seabass during a disease outbreak and under oxytetracycline treatment after infection, fish were sampled in 4 different states: healthy, diseased, treatment and recovery (Fig. 1). During the healthy state (August 21 and 29), all fish specimens were considered healthy due to a lack of visible disease symptoms, such as external lesions or behavioural alterations. On September 8 fish began to die in the farming tanks, showing symptoms of disease, and treatment with oxytetracycline antibiotic (a broad-spectrum tetracycline) was initiated, being administrated at 35 g/Kg through commercial feed for at lasted 8 days. On the same day, smears from spleen and kidney were collected for culture using Bionor kits DE020, MONO-VA-50 for Vibrio anguillarum and DL020, MONO-Pp-50 for Photobacterium piscicida. Agglutination essays were not conclusive and, at that stage, the causative agent of the disease was unknown. We do not have samples from September 8, hence we used the samples from our closest time point, September 5, which we classified as potentially diseased (i.e., diseased state). Antibiotic treatment lasted until September 16 and fish were sampled on September 12; this sample point corresponded to the treatment state. Then, three additional time points were sampled (September 19 and 26, and October 3), when fish were no longer dying or presented signs of infection; these three time points corresponded to the recovery state.
Total DNA from 140 fish samples (70 skin and 70 gills) was extracted using the PowerSoil DNA Isolation Kit (QIAGEN, Netherlands), following the manufacturer’s protocol. DNA extractions were shipped in dry ice to the University of Michigan Medical School (USA) for amplification and sequencing on a single run of the Illumina MiSeq platform according to the protocol of Kozich et al.75 Each sample was amplified for the V4 (~250 bp) hypervariable region of the 16S rRNA gene, using the primers in Caporaso et al.76 This region has been widely used to characterize microbiomes from vertebrates (Earth Microbiome Project77), including fish42,78,79,80.
Data and statistical analysis
Raw FASTQ files were analyzed using the Quantitative Insights Into Microbial Ecology 2 (QIIME2; release 2018.4) platform. Clean sequences were aligned against the SILVA (132 release) reference database81 using the DADA2 pipeline82. A feature table containing amplicon sequence variants (ASVs) was constructed and normalized using the negative binomial distribution83. The core microbiome was assessed at the ASV level for the gill and skin of seabass for each state (healthy, diseased, treatment and recovery) separately. An ASV was considered as part of the core microbiome if present in 100% of the samples from each state. Core diversity is here defined as number of ASVs represented in a given group.
Microbial alpha-diversity (intra-sample) was calculated using Shannon, ACE, Fisher and Faith’s phylogenetic diversity (PD) indices as implemented in the R package phyloseq84. Microbial beta-diversity (inter-sample) was estimated using phylogenetic Unifrac (unweighted and weighted) and Bray-Curtis distances. Dissimilarity between samples was assessed by principal coordinates analysis (PCoA). Variation in microbial alpha-diversity and the mean proportions of the most abundant taxa (with more than 4% of all reads) were assessed using linear models with randomized residuals in a permutation procedure (RRPP). Differences in community composition (beta-diversity) were tested using permutational multivariate analysis of variance (PERMANOVA) with 1,000 permutations as implemented in the adonis function of the R vegan package85. All statistical analyses were carried out separately for the gills and skin. All statistical analyses were performed in R-studio v1.0.14386.
Data availability
The raw sequences are available at NCBI Sequence Read Archive (SRA) database within the BioProject ID PRJNA575053.
References
Trivedi, B. Microbiome: the surface brigade. Nature. 492, S60–S61 (2012).
Gómez, G. D. & Balcázar, J. L. A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol. Med. Microbiol. 52, 145–154 (2007).
Kelly, C. & Salinas, I. Under pressure: interactions between commensal microbiota and the teleost immune system. Front. Immunol. 8, 559 (2017).
Lee, Y. K. & Mazmanian, S. K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330, 768–1773 (2010).
Mohammed, H. H. & Arias, C. R. Potassium permanganate elicits a shift of the external fish microbiome and increases host susceptibility to columnaris disease. Vet. Res. 46, 82 (2015).
Reid, K. M. et al. Salmonid alphavirus infection causes skin dysbiosis in Atlantic salmon (Salmo salar L.) post-smolts. PloS One. 12, e0172856 (2017).
Larsen, A., Tao, Z., Bullard, S. A. & Arias, C. R. Diversity of the skin microbiota of fishes: evidence for host species specificity. FEMS Microbiol. Ecol. 85, 483–494 (2013).
Boutin, S., Bernatchez, L., Audet, C. & Derôme, N. Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS One. 8, e84772 (2013).
Zha, Y., Eiler, A., Johansson, F. & Svanbäck, R. Effects of predation stress and food ration on perch gut microbiota. Microbiome 6, 28 (2018).
Chiarello, M. et al. Skin microbiome of coral reef fish is highly variable and driven by host phylogeny and diet. Microbiome 6, 147 (2018).
Galbraith, H. et al. Exposure to synthetic hydraulic fracturing waste influences the mucosal bacterial community structure of the brook trout (Salvelinus fontinalis) epidermis. AIMS Microbiol. 4, 413–427 (2018).
Ye, L., Amberg, J., Chapman, D., Gaikowski, M. & Liu, W. T. Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J. 8, 541 (2014).
Pratte, Z. A., Besson, M., Hollman, R. D. & Stewart, F. J. The gills of reef fish support a distinct microbiome influenced by host-specific factors. Appl. Environ. Microbiol. 84, AEM-00063 (2018).
Legrand, T. P. et al. The inner workings of the outer surface: skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front. Microbiol. 8, 2664 (2018).
Llewellyn, M. S. et al. Parasitism perturbs the mucosal microbiome of Atlantic salmon. Sci. Rep. 7, 43465 (2017).
Rosado, D., Pérez-Losada, M., Severino, R., Cable, J. & Xavier, R. Characterization of the skin and gill microbiomes of the farmed seabass (Dicentrarchus labrax) and seabream (Sparus aurata). Aquaculture 500, 57–64 (2019).
Givens, C. E., Ransom, B., Bano, N. & Hollibaugh, J. T. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 518, 209–223 (2015).
Li, E. et al. Gut microbiota and its modulation for healthy farming of Pacific white shrimp Litopenaeus vannamei. Rev. Fish. Sci. Aquac. 26, 381–399 (2018).
Califano, G. et al. Molecular taxonomic profiling of bacterial communities in a gilthead seabream (Sparus aurata) hatchery. Front. Microbiol. 8, 204 (2017).
Rud, I. et al. Deep sequencing of the bacterial microbiota in commercial-scale recirculating and semiclosed aquaculture systems for Atlantic salmon post-smolt production. Aquac. Eng. 78, 50–62 (2017).
Hess, S., Wenger, A. S., Ainsworth, T. D. & Rummer, J. L. Exposure of clownfish larvae to suspended sediment levels found on the Great Barrier Reef: impacts on gill structure and microbiome. Sci. Rep. 5, 10561 (2015).
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H. & Derome, N. Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5, 117 (2014).
FAO Aquaculture Department: Statistics and Information Service FishStatJ: Universal software for fishery statistical time series (2011).
Toranzo, A. E., Magarinos, B. & Romalde, J. L. A review of the main bacterial fish diseases in mariculture systems. Aquaculture. 246, 37–61 (2005).
Faílde, L. D. et al. Immunohistochemical diagnosis of tenacibaculosis in paraffin‐embedded tissues of Senegalese sole Solea senegalensis Kaup, 1858. J. Fish Dis. 37, 959–968 (2014).
Zlotkin, A., Hershko, H. & Eldar, A. Possible transmission of Streptococcus iniae from wild fish to cultured marine fish. Appl. Environ. Microbiol. 64, 4065–4067 (1998).
Romalde, J. L. et al. Streptococcus phocae, an emerging pathogen for salmonid culture. Vet. Microbiol. 130, 198–207 (2008).
Rivas, A. J., Balado, M., Lemos, M. L. & Osorio, C. R. The Photobacterium damselae subsp. damselae hemolysins damselysin and HlyA are encoded within a new virulence plasmid. Infect. Immun. 79, 4617–4627 (2011).
Tao, Z. et al. An outbreak of Photobacterium damselae subsp. damselae infection in cultured silver pomfret Pampus argenteus in Eastern China. Aquaculture 492, 201–205 (2018).
Terceti, M. S., Ogut, H. & Osorio, C. R. Photobacterium damselae subsp. damselae, an emerging fish pathogen in the Black Sea: evidences of a multiclonal origin. Appl. Environ. Microbiol. 82, 3736–3745 (2016).
Terceti, M. S. et al. Molecular epidemiology of Photobacterium damselae subsp. damselae outbreaks in marine rainbow trout farms reveals extensive horizontal gene transfer and high genetic diversity. Front. Microbiol. 9, 2155 (2018).
Labella, A., Berbel, C., Manchado, M., Castro, D. & Borrego, J. J. Photobacterium damselae subsp. damselae, an emerging pathogen affecting new cultured marine fish species in southern Spain. In Recent advances in fish farms. InTech (2011).
Pedersen, K., Skall, H. F., Lassen‐Nielsen, A. M., Bjerrum, L. & Olesen, N. J. Photobacterium damselae subsp. damselae, an emerging pathogen in Danish rainbow trout, Oncorhynchus mykiss (Walbaum), mariculture. J. Fish Dis. 32, 465–472 (2009).
Rivas, A. J., Lemos, M. L. & Osorio, C. R. Photobacterium damselae subsp. damselae, a bacterium pathogenic for marine animals and humans. Front. Microbiol. 4, 283 (2013).
Essam, H. M., Abdellrazeq, G. S., Tayel, S. I., Torky, H. A. & Fadel, A. H. Pathogenesis of Photobacterium damselae subspecies infections in sea bass and sea bream. Micro. Pathogenesis. 99, 41–50 (2016).
Bakopoulos, V. et al. Vaccination trials of sea bass, Dicentrarchus labrax (L.), against Photobacterium damsela subsp. piscicida, using novel vaccine mixtures. J. Fish Dis. 26, 77–90 (2003).
Byadgi, O. et al. Immunogenicity of inactivated formalin-killed Photobacterium damselae subsp. piscicida combined with Toll-like receptor 9 agonist in Cobia rachycentron canadum. Aquaculture. 492, 369–378 (2018).
Abdel-Aziz, M., Eissa, A. E., Hanna, M. & Okada, M. A. Identifying some pathogenic Vibrio/Photobacterium species during mass mortalities of cultured Gilthead seabream (Sparus aurata) and European seabass (Dicentrarchus labrax) from some Egyptian coastal provinces. Int. J. Vet. Sci. Med. 1, 87–95 (2013).
Rodrigues, S., Antunes, S. C., Correia, A. T. & Nunes, B. Rainbow trout (Oncorhynchus mykiss) pro-oxidant and genotoxic responses following acute and chronic exposure to the antibiotic oxytetracycline. Ecotoxicology 26, 104–117 (2017).
Yonar, M. E., Yonar, S. M. & Silici, S. Protective effect of propolis against oxidative stress and immunosuppression induced by oxytetracycline in rainbow trout (Oncorhynchus mykiss, W.). Fish Shellfish Immunol. 31, 318–325 (2011).
Carlson, J. M., Hyde, E. R., Petrosino, J. F., Manage, A. B. & Primm, T. P. The host effects of Gambusia affinis with an antibiotic-disrupted microbiome. Comp. Biochem. Phys. C. 178, 163–168 (2015).
Carlson, J. M., Leonard, A. B., Hyde, E. R., Petrosino, J. F. & Primm, T. P. Microbiome disruption and recovery in the fish Gambusia affinis following exposure to broad-spectrum antibiotic. Infect. Drug Resist. 10, 143 (2017).
Pindling, S., Azulai, D., Zheng, B., Dahan, D. & Perron, G. G. Dysbiosis and early mortality in zebrafish larvae exposed to subclinical concentrations of streptomycin. FEMS Microbiol. Lett. 365, fny188 (2018).
Liu, Y. et al. Gibel carp Carassius auratus gut microbiota after oral administration of trimethoprim/sulfamethoxazole. Dis. Aquat. Organ. 99, 207–213 (2012).
Narrowe, A. B. et al. Perturbation and restoration of the fathead minnow gut microbiome after low-level triclosan exposure. Microbiome. 3, 6 (2015).
Zhou, L. et al. Environmental concentrations of antibiotics impair zebrafish gut health. Environ. Pollut. 235, 245–254 (2018).
Chiu, T. H., Kao, L. Y. & Chen, M. L. Antibiotic resistance and molecular typing of Photobacterium damselae subsp. damselae, isolated from seafood. J. Appl. Microbiol. 114, 1184–1192 (2013).
Nonaka, L. et al. Novel conjugative transferable multiple drug resistance plasmid pAQU1 from Photobacterium damselae subsp. damselae isolated from marine aquaculture environment. Microbes Environ. 27, 263–272 (2012).
Rigos, G. & Troisi, G. M. Antibacterial agents in Mediterranean finfish farming: a synopsis of drug pharmacokinetics in important euryhaline fish species and possible environmental implications. Revi. Fish Biol. Fisher. 15, 53–73 (2005).
Boutin, S., Sauvage, C., Bernatchez, L., Audet, C. & Derôme, N. Inter individual variations of the fish skin microbiota: host genetics basis of mutualism? PLoS One. 9, e102649 (2014).
Lowrey, L., Woodhams, D. C., Tacchi, L. & Salinas, I. Topographical mapping of the rainbow trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community in the skin with antifungal properties. Appl. Environ. Microb. AEM-01826 (2015).
Tapia-Paniagua, S. T., Ceballos-Francisco, D., Balebona, M. C., Esteban, M. Á. & Moriñigo, M. Á. Mucus glycosylation, immunity and bacterial microbiota associated to the skin of experimentally ulcered gilthead seabream (Sparus aurata). Fish Shellfish Immunol. 75, 381–390 (2018).
Chiarello, M., Villéger, S., Bouvier, C., Bettarel, Y. & Bouvier, T. High diversity of skin-associated bacterial communities of marine fishes is promoted by their high variability among body parts, individuals and species. FEMS Microbiol. Ecol. 91 (2015).
Pimentel, T., Marcelino, J., Ricardo, F., Soares, A. M. & Calado, R. Bacterial communities 16S rDNA fingerprinting as a potential tracing tool for cultured seabass Dicentrarchus labrax. Sci. Rep. 7, 11862 (2017).
Larsen, A. M., Bullard, S. A., Womble, M. & Arias, C. R. Community structure of skin microbiome of gulf killifish, Fundulus grandis, is driven by seasonality and not exposure to oiled sediments in a Louisiana salt marsh. Microb. Ecol. 70, 534–544 (2015).
de Bruijn, I., Liu, Y., Wiegertjes, G. F. & Raaijmakers, J. M. Exploring fish microbial communities to mitigate emerging diseases in aquaculture. FEMS Microbiol. Ecol. 94, 161 (2017).
Lokesh, J. & Kiron, V. Transition from freshwater to seawater reshapes the skin associated microbiota of Atlantic salmon. Sci. Rep. 6, 19707 (2016).
Brooke, J. S. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41 (2012).
Abraham, T. J. & Adikesavalu, H. Association of Stenotrophomonas maltophilia in African catfish, Clarias gariepinus (Burchell, 1822) fry mortalities with dropsy. Int. J. Aquac. 6 (2016).
López, J. R. et al. Pseudomonas baetica sp. nov., a fish pathogen isolated from wedge sole, Dicologlossa cuneata (Moreau). Int. J. Syst. Evol. Microbiol. 62, 874–882 (2012).
Hatai, K., Egusa, S., Nakajima, M. & Chikahata, H. Pseudomonas chlororaphis as a fish pathogen. B. Jpn. Soc. Sci. Fish. 41 (1975).
Pridgeon, J. W. & Klesius, P. H. Major bacterial diseases in aquaculture and their vaccine development. Anim. Sci. Rev. 7, 1–16 (2012).
Korkea‐Aho, T. L., Heikkinen, J., Thompson, K. D., Von Wright, A. & Austin, B. Pseudomonas sp. M174 inhibits the fish pathogen Flavobacterium psychrophilum. J. Appl. Microbiol. 111, 266–277 (2011).
Austin, B. & Austin, D. A. Characteristics of the diseases. In Bacterial Fish Pathogens: Diseases of Farmed and Wild Fish15–46 (Springer, 2012).
Loch, T. P., Scribner, K., Tempelman, R., Whelan, G. & Faisal, M. Bacterial infections of Chinook salmon, Oncorhynchus tshawytscha (Walbaum), returning to gamete collecting weirs in Michigan. J. Fish Dis. 35, 39–50 (2012).
Liu, Y. et al. Diversity of aquatic Pseudomonas species and their activity against the fish pathogenic oomycete Saprolegnia. PloS One. 10, e0136241 (2015).
van West, P. Saprolegnia parasitica, an oomycete pathogen with a fishy appetite: new challenges for an old problem. Mycologist 20, 99–104 (2006).
Offret, C. et al. Spotlight on antimicrobial metabolites from the marine bacteria Pseudoalteromonas: chemodiversity and ecological significance. Mar. Drugs. 14, 129 (2016).
Richards, G. P. et al. Mechanisms for Pseudoalteromonas piscicida-induced killing of vibrios and other bacterial pathogens. Appl. Environ. Microbiol. AEM-00175 (2017).
Lloyd, M. M. & Pespeni, M. H. Microbiome shifts with onset and progression of Sea Star Wasting Disease revealed through time course sampling. Sci. Rep. 8, 16476 (2018).
Papaleo, M. C. et al. Sponge-associated microbial Antarctic communities exhibiting antimicrobial activity against Burkholderia cepacia complex bacteria. Biotechnol. Adv. 30, 272–293 (2012).
Morgun, A. et al. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut 64, 1732–1743 (2015).
Gaulke, C. A., Martins, M. L., Watral, V., Kent, M. L. & Sharpton, T. J. Parasitic Infection by Pseudocapillaria tomentosa is associated with a longitudinal restructuring of the Zebrafish gut microbiome. bioRxiv, p.076596 (2016).
Navarrete, P., Mardones, P., Opazo, R., Espejo, R. & Romero, J. Oxytetracycline treatment reduces bacterial diversity of intestinal microbiota of Atlantic salmon. J. Aquat. Anim. Health. 20, 177–183 (2008).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. 108, 4516–4522 (2011).
Gilbert, J. A., Jansson, J. K. & Knight, R. The Earth Microbiome project: successes and aspirations. BMC Biol. 12, 69 (2014).
Llewellyn, M. S. et al. The biogeography of the Atlantic salmon (Salmo salar) gut microbiome. ISME J. 10, 1280 (2015).
Nielsen, S., Walburn, J. W., Vergés, A., Thomas, T. & Egan, S. Microbiome patterns across the gastrointestinal tract of the rabbitfish Siganus fuscescens. Peer J. 5, e3317 (2017).
Wang, J. et al. Effects of fish meal replacement by soybean meal with supplementation of functional compound additives on intestinal morphology and microbiome of Japanese seabass (Lateolabrax japonicus). Aquac. Res. 48, 2186–2197 (2017).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581 (2016).
McMurdie, P. J. & Holmes, S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10, e1003531 (2014).
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8, e61217 (2013).
Oksanen, J. et al. The vegan package. Community Ecology Package, http://r-forge.r-project.org/projects/vegan (2008).
Studio, R. RStudio: Integrated Development Environment for R. RStudio Inc, Boston, Massachusetts (2012).
Acknowledgements
This work was funded by the European Regional Development Fund (ERDF) through the COMPETE program and by National Funds through FCT - Foundation for Science and Technology (project PTDC/MAR-BIO/0902/2014 -POCI-01-0145-FEDER-016550; project PTDC/BIA-MIC/27995/2017 POCI-01-0145-FEDER-027995; and by a “Projecto de Investigação Exploratória”: IF/00764/2013); the Welsh Government and Higher Education Funding Council for Wales (HEFCW) AquaWales Project through the Sêr Cymru National Research Network for Low Carbon Energy and Environment (NRN-LCEE). DR, MP-L and RX were supported by FCT under the Programa Operacional Potencial Humano – Quadro de Referência Estratégico Nacional funds from the European Social Fund and Portuguese Ministério da Educação e Ciência (DR doctoral grant SFRH/BD/117943/2016; MPL: IF/00764/2013; RX: IF/00359/2015).
Author information
Authors and Affiliations
Contributions
D.R., R.X. and R.S. designed the research. R.S. collected the samples. D.R. performed laboratory work and analyzed the results. M.P.-L. contributed to the statistical analysis. R.X., J.C., M.P.-L. and F.T. supervised and provided intellectual content. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosado, D., Xavier, R., Severino, R. et al. Effects of disease, antibiotic treatment and recovery trajectory on the microbiome of farmed seabass (Dicentrarchus labrax). Sci Rep 9, 18946 (2019). https://doi.org/10.1038/s41598-019-55314-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55314-4
This article is cited by
-
Association of microbial community structure with gill disease in marine-stage farmed Atlantic salmon (Salmo salar); a yearlong study
BMC Veterinary Research (2024)
-
When the host’s away, the pathogen will play: the protective role of the skin microbiome during hibernation
Animal Microbiome (2023)
-
Sex-dependent effects of mechanical delousing on the skin microbiome of broodstock Atlantic salmon (Salmo salar L.)
Scientific Reports (2023)
-
Hybrid of Metapenaeus dobsoni lectin and platinum nanoparticles exert antimicrobial and immunostimulatory effects to reduce bacterial bioburden in infected Nile tilapia
Scientific Reports (2023)
-
Bacterial networks in Atlantic salmon with Piscirickettsiosis
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
