
Abstract
During the Late Pleistocene, narrow-headed voles (Lasiopodomys gregalis) inhabited Eurasia’s vast territories, frequently becoming the dominant small mammal species among steppe-tundra communities. We investigated the relationship between this species’ European and Asiatic populations by sequencing the mtDNA genomes of two extant specimens from Russia and 10 individuals from five Central European sites, dated to the post-LGM period. Phylogenetic analyses based on a large portion of mtDNA genomes highly supported the positioning of L. gregalis within Arvicolinae. The phylogeny based on mtDNA cytochrome b sequences revealed a deep divergence of European narrow-headed voles from Asiatic ones and their sister position against the extant L. gregalis and L. raddei. The divergence of the European lineage was estimated to a minimum 230 thousand years ago. This suggest, contrary to the current biogeographic hypotheses, that during the interglacial periods narrow-headed vole did not retreat from Europe but survived the unfavourable conditions within the refugial areas. Based on this result, we propose to establish a cryptic species status for the Late Pleistocene European narrow-headed vole and to name this taxon Lasiopodomys anglicus.
Similar content being viewed by others
Introduction
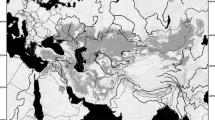
Although the extant narrow-headed vole (Lasiopodomys gregalis) inhabits Asia’s vast territories, its range is disjunct into several areas of tundra in Arctic Russia from Northern Dvina River, Yamal Peninsula, to Kolyma River, forest-tundra in Central Yakutia and steppe and forest-steppe belt from Kama River East to Amur River, and from south Siberia, Tian Shan to interfluve of rivers Huang He and Yangtze in East China1 (Fig. 1).

The Late Pleistocene and extant ranges of narrow-headed voles. The extant distribution (light gray) is given according to Shenbrot and Krasnov1 and the Late Pleistocene one (dark gray) is compiled from the literature. The southern range in Asia is marked tentatively. Circles denote sampling localities of extant narrow-headed voles from previous studies16 while squares indicate the Late Pleistocene sites. Color corresponds to the main mtDNA lineages of L. gregalis (red – A; blue – B; green – C) and L. raddei (D - yellow). Violet denote the newly recognized lineage E. The source map was made using public domain data from www.naturalearthdata.com. Range of the extant narrow-headed vole was downloaded from iucn.org.
During the Late Pleistocene, its habitat range was much larger, including nearly the whole Northern Eurasia. In Europe, it reached the Northern2 or even the Central Spain3, as well as the British Isles4. Its southern distribution has its limits in Northern5 or even Central Italy6,7, Serbia8, Northern Bulgaria9, Crimea10 and the area near Azov Sea in the European Russia11. Throughout the Late Pleistocene, the narrow-headed vole was either the dominant or subdominant small mammal species, within steppe-tundra environments of many regions in the European Plain, from southern France12 through Benelux Countries13, Central Europe14 to Ukraine and Russia15 (Fig. 1). The extinction of European populations and the fragmentation of the Asiatic habitat range probably occurred during the Holocene, with the expansion of forests16.
Among extant Asiatic populations, genetic variation is well recognised, comprising four main mtDNA lineages (A-D) with parapatric distributions. Lineage A covers most of the species range and is further subdivided into six sublineages (A1-A6) confined to specific geographic regions16. The main lineages’ divergence was estimated to occur between 0.75 Ma and 0.25 Ma years ago, with the most divergent Transbaikal lineage D being recently raised to the status of cryptic species Lasiopodomys raddei17. It was suggested that despite the Holocene’s range contraction, narrow-headed vole populations maintained a high genetic diversity16. This was supported by analyses of ancient DNA from Late Glacial and Holocene specimens from Middle and Northern Ural sites which showed increase of effective narrow-headed vole population size during the LGM (Last Glacial Maximum) and only slight decrease during the Late Glacial warming18. High morphological variation accompanies genetic variability. For example, vole populations from Polar Urals’ Eastern slopes, both subfossil and recent, exhibit high variability in terms of m1 length and morphotypes, comparable to those observed within the whole species range, but without any evident time trends nor continuity throughout the Late Pleistocene, Holocene until modern times11.
The relationship between Asiatic and European fossil populations has not been intensively studied. After taxonomic revision of Sutcliffe and Kowalski4 all authors accepted Microtus gregalis as a correct available name for the European narrow-headed voles. Only Rekovets19, on the basis of material from the LGM site Novgorod-Severskiy, classified Late Pleistocene narrow-headed voles from Ukraine as the separate subspecies M. g. kriogenicus, which went extinct at the beginning of the Holocene. Other studies showed that the high morphological variation of m1 teeth of Late Pleistocene European voles20,21 is within the range of variation of voles from North-eastern Europe and Western Siberia11,22. Differences between Late Pleistocene populations lie mainly in the proportion of different molar morphotypes (m1), although classification is sometimes difficult due to the presence of many intermediate forms. It was also shown that, in narrow-headed voles, high morphological similarity may be accompanied by high genetic differentiation11.
To better understand the relationship between European and Asiatic populations of narrow-headed voles we sequenced mtDNA genomes from 10 specimens excavated from Late Pleistocene sites in Central Europe (Fig. 1).
Results
We de novo assembled mtDNA genomes of two present-day narrow-headed voles from the Southern Ural region, which resulted in 16,292 bp long sequences with the typical mammal gene order. One such sequence was subsequently used as a reference for mapping the reads obtained from the sequencing of Late Pleistocene samples. Ten samples from five sites yielded over 70% of the mtDNA genome sequences covered at least twice, which were used for phylogenetic analyses (Table 1). All samples exhibited excess of damage at the ends of the DNA molecules, typical of ancient DNA (Table 1).
Using 9.09 kb of mtDNA, we reconstructed a phylogeny of all species from the Microtus, Neodon, Alexandromys and Lasiopodomys genera with available mtDNA genome sequences (Fig. 2). The resulting tree in general reassembled phylogenetic relationships previously inferred from mtDNA cytochrome b sequences23. The recovered tree consisted of two main branches, the first of which comprised the subgenera Microtus (Microtus arvalis, Microtus mystacinus (= levis)) and Terricola (Microtus subterraneus), which formed a sister clade to Agricola (Microtus agrestis) and Iberomys (Microtus cabrerae) with Microtus ochrogaster being the basal species. The second branch consisted of narrow-headed voles (L. gregalis) from both the extant and the Late Pleistocene, which clustered with L. mandarinus forming together a sister clade to the genus Alexandromys represented here by A. fortis, A. oeconomus and A.kikuchii. All the above-mentioned species were in a sister relationship to the Asiatic Neodon lineage (N. irene and N. sikimensis). The deep divergence between present-day narrow-headed voles from Russia and Late Pleistocene European ones is noteworthy. The total divergence (Dxy) between these two groups was 8.6%, higher than between the Northern and both the Portuguese and the Mediterranean lineages of Microtus agrestis (5.89% and 5.72%, respectively), as well as between Microtus arvalis and Microtus mystacinus (=levis) (5.82%) (Fig. 2).

Bayesian phylogeny of Arvicolini based on the 9,090 bp fragment of mtDNA. The samples obtained in this study are written in bold. Nodes with posterior probabilities above 0.95 and bootstrap supports above 95 are marked with black dots. Otherwise posterior/bootstraps are displayed on the node. The percent of pairwise divergence between two branches is presented in front of the selected nodes (see text for details).
To better characterise the relationships between the Late Pleistocene European and extant Asiatic narrow headed-vole populations we reconstructed the phylogeny based on mtDNA cytochrome b sequences of 179 specimens. It revealed five well supported lineages (Fig. 3). Four of them correspond to the previously described lineages A – D16,17, whereas the fifth, designated as E, was basal with respect to all others and included the narrow-headed voles from Europe. The divergence between lineage E and the others ranged between 8.8% (E-A) and 10.5% (E-B), which was slightly lower than the divergence between lineage D and the others (range: 10.4% (D-A) −11.5% (D-B)).

Bayesian phylogeny of extant and the Late Pleistocene narrow-headed voles based on the 889 bp fragment of mtDNA cytochrome b. The posterior probabilities of main nodes higher than 0.95 are indicated by black dots. Numbers below nodes are the estimated tMRCAs and the grey bars represent the 95% highest posterior density intervals of node ages. Node colours corresponds to those on Fig. 1.
The phylogeny was calibrated using the substitution rate of 4 × 10−7 substitutions/site/year−1, without additional fossil calibration. This resulted in much younger divergence estimates of mtDNA lineages than previously reported16. Most of the genetic diversity of the extant Asiatic populations coalesced between 15 and 10 ka. Within lineage A, sublineage divergence was estimated as ranging between 65 and 35 ka. The divergence of lineages A, B and C was estimated as ranging between 125 and 90 ka. The divergence between lineage D, which was recently established as Lasiopodomys raddei and lineages A-C was estimated as ca. 190 ka, whereas the divergence of newly recognised lineage E was estimated as ca. 230 ka (Fig. 3).
To include in the analysis sequences of the 21 Late Pleistocene narrow-headed voles from Urals18 we reconstructed also the phylogeny based on a shorter fragment of mtDNA cytochrome b (461 bp). The resulting tree had the same topology as the one reconstructed from the longer fragment, only the divergence times of the main lineages were slightly older (Fig. S1).
Discussion and Conclusions
For many years, the narrow-headed vole was included in the genus Microtus within the separate subgenus Stenocranius, based on skull shape characteristics24,25. From the 1980s, far before the application of molecular markers to vole systematics and phylogeny, palaeontologists concluded that the narrow-headed vole is one of the earliest evolutionary offshoots of rootless voles derived from the Allophaiomys stock. As a consequence, Stenocranius was thereafter considered to represent a distinct phylogenetic lineage of Microtus s. l.26,27. It is represented by an ancestor-descendant array of species „hintoni” – „gregaloides”– „gregalis” first recognised by Fejfar and Horáček28. However, this point of view was not widely accepted until the 1990s, when the morphological feature of the first lower molar called “Pitymys-like rhombus”, for which the early forms of the clade were traditionally regarded as “Pitymys hintoni” and “Pitymys gregaloides”4,20, appeared merely as a common ancestral feature in several genera lineages, rather than a prove of their taxonomic relation26,29,30.
Mezhzherin et al.31 were perhaps the first to reveal a close relationship between Microtus gregalis and Lasiopodomys brandtii (together with A. oeconomus, A. middendorffii, and A. fortis) based on allozyme studies. The first application of molecular tools to explore species limits in Microtus, especially inference based on mtDNA cytochrome b analysis32 did not clarify the taxonomic status of the narrow-headed vole. The species M. gregalis was the most divergent Microtus species, occupying an unstable phylogenetic position. Results from further studies performed in recent years, in particular the evidence gathered from nuclear genes33,34, have supported the sister relationship of Stenocranius with representatives of the Lasiopodomys (L. mandarinus and L. brandtii) genus. Its taxonomic status within Lasiopodomys is currently accepted by taxonomists35. Our phylogeny, based on a large portion of mtDNA genome, is consistent with those findings. The position of L. gregalis together with L. mandarinus is well supported by both used phylogenetic methods. However, the Lasiopodomys clade is not differentiated from all other Microtus groups but is in a sister relationship with the Asiatic lineages Alexandromys and Neodon as suggested from previous allozyme31 and mtDNA cytochrome b studies23.
MtDNA cytochrome b-based phylogeny provided a more accurate characterisation of the relationship between European and Asiatic narrow-headed voles, and confirmed a high genetic divergence between them. However, the estimated divergence times of Asiatic lineages were much younger than in the previous study16 where the phylogeny was calibrated using combined heterochronous genetic and fossil data. It was suggested that due to the time-dependence of molecular rates, fossil calibration might overestimate the timing of intraspecific events36. The substitution rates of mtDNA cytochrome b were recently estimated for a range of small mammal species using either heterochronous data or biogeographic events. All estimations yielded similar mutation rates: 3.27 × 10−7 (substitutions/site/year−1) for Microtus arvalis37, 3.88 × 10−7 and 4.57 × 10−7 for Microtus agrestis38,39, 1.53 × 10−7 and 4.63 × 10−7 for Dicrostonyx torquatus40,41 and 5.38 × 10−7 for Ctenomys sociabilis41,42. It was shown that, across mammals, mtDNA mutation rates correlate with several traits such as lifespan, body mass and generation turnover rate43. Horn et al.44 found a correlation between rodents’ rates and lifespan. Therefore, an mtDNA cytochrome b substitution rate around 4 × 10−7 substitutions/nucleotide/year−1 used here is possibly universal for the Microtus genus and other rodents with similar life history. Interestingly, most of the genetic diversity of the extant Asiatic populations coalesce between 15 and 10 ka (Fig. 3), suggesting that it arose mostly during the Late Glacial and the Holocene, and that climate changes at the end of the Pleistocene and the Pleistocene to Holocene transition might have exerted a larger impact on narrow-headed vole populations than previously thought.
The divergence of lineage E, which includes all European specimens, was estimated at around 230 ka. The used substitution rates may be applied to estimate the divergence of younger branches of the narrow-headed voles’ tree, although it is not clear whether they are still valid to estimate the age of highly divergent lineages, such as D and E. Although, “intraspecific” substitution rates were recently used to estimate the ages of highly divergent lineages and species within the M. agrestis and M. arvalis species groups45, this estimation should be interpreted with caution and regarded as minimal. Lasiopodomys gregalis appeared for the first time in the European fossil record in the Middle Pleistocene, probably in MIS 17, an equivalent of Cromerian Interglacial II27, or in MIS 15, correlated with Muchkap Stage Interglacial46, ca. 0.7–0.5 Ma. This may be regarded as the maximal estimate of lineage E divergence.
Both phylogenetic reconstructions clearly showed that European post-LGM narrow-headed voles represent a highly divergent lineage from contemporary and the Late Pleistocene Asiatic ones. This new evolutionary unit does not match those of any currently recognised haplogroups of extant species of Lasiopodomys, such as L. gregalis16 or L. raddei17. The observed divergence from the extant Asiatic populations (8.8–10.4%) is higher than between the lineages within the Microtus agrestis (3–6%)45 or Microtus arvalis (4–9%) groups47, currently considered by most specialists as a separate species or close to a separate species35.
Interestingly, the results of Ecological Niche Modelling showed little or no suitable narrow-headed voles’ habitats in Central and Western Europe during the LGM18. It was proposed that Late Pleistocene European populations may have had a broader or different ecological niche than the Asiatic ones, possibly as a consequence of local adaptations18. This suggests that the divergence of the European narrow-headed voles might have been a consequence of habitat fragmentation and isolation of the European population in the refugial area during one of the interglacials, possibly Holsteinian (MIS 11). This could have led to the rapid evolutionary change and shift of the ecological niche of the narrow-headed voles. Similar mechanism was recently suggested for the extant Norwegian lemmings which survived the LGM in the isolated refugium in Scandinavia48.
Given the lack of clear morphological differentiation, possible different ecological niche and especially the phylogenetic placement of European narrow-headed voles as a sister taxon against L. gregalis and L. raddei, we suggest that it should be considered as a cryptic species within the Lasopodomys gregalis species group.
Species designations follow nomenclatural priorities, which also apply to genetically distinct lineages. In Europe, the Late Pleistocene fossil narrow-headed vole was first recognised by Nehring (1875)49 who found “Arvicola gregalis” in Thiede near Wolfenbuttel, Central Germany. Woldřich50 reported this species from rich fossil assemblages in Sudslavice, South Bohemia, and later based on multiple records from Moravian and Austrian caves’ deposits, identifying it as an index fossil of glacial communities50. Newton51, simultaneously described this species as Microtus (=Arvicola) gregalis based on specimens from Ightham Fissures, Kent, England. This species was later described by Hinton52 as a new one - Microtus anglicus. Among the numerous collections from Ightham Fissures, Hinton52 designated the type specimen as a nearly complete adult skull, essentially similar to the extant representatives of Stenocranius. In the same description he also indicated a greatly reduced fourth outer angle in the first lower molar. This was the basis on which Microtus anglicus was later synonymised with Microtus gregalis by most authors4,20,28. Thus, in terms of the taxonomic nomenclature the name Lasiopodomys anglicus has priority in designation of Late Pleistocene narrow-headed vole lineage from Europe as a species.
Among the presented results, the most interesting aspect is their incompatibility with the available paleobiogeographic hypotheses. The traditional view on the history of the European narrow-headed voles predicts its complete disappearance during the interglacials, with woodland vegetation, and mass re-expansion during glacial stages from the core distribution area beyond the limits of Europe. Its distribution throughout the Weichselian fits that hypothesis quite well, as the species first appears as a rare element by the end of MIS 4, becoming a dominant form only during MIS 312,21,53,54. The present results suggest no occurrence of distant migrations, requiring the species to survive interglacial stages in refugia within Europe (supposedly in the alpine mountains or in Scandinavia). However, to our knowledge, no reliable records are available that support this hypothesis. Lasiopodomys anglicus has been an index species and the dominant component of glacial communities throughout the Middle and Late Pleistocene of Central Europe. The uncertainties on its paleobiogeography’s real dynamics indicate that our knowledge of the Quaternary past remains incomplete, requiring urgent further research.
Materials and Methods
Samples
Narrow-headed vole remains were obtained from palaeontological sites in Poland (Obłazowa cave WE, Nad Tunelem cave, Komarowa cave), Czech Republic (Býčí Skala cave) and Slovakia (Peskö cave). The samples were collected from layers dated to the Latest Pleistocene, between 20 and 11 ka: layer III from Obłazowa WE, layer B from Komarowa, layer 8a from Býčí Skala and layer 4 from Peskö55,56,57 (Fig. 1). Four samples from Nad Tunelem cave were radiocarbon dated in the Poznań Radiocarbon Laboratory to determine the layer’s age (Supplementary Table S1). Radiocarbon ages were calibrated with Oxcal 4.258 using IntCal13 calibration curve59. Ethanol-preserved tissue fragments of two present-day narrow-headed voles were obtained from specimens caught in Ural Mountains.
DNA extraction, enrichment and sequencing
Prior to laboratory procedures, all samples were photographically documented. All experimental procedures were performed in a laboratory dedicated to ancient DNA work in the Laboratory of Paleogenetics and Conservation Genetics, Centre of New Technologies, the University of Warsaw. Strict precautions were taken to avoid contamination. Teeth were thoroughly rinsed with water, submerged in extraction buffer (0.5 M EDTA pH = 8.0; 0.5% N-Laurylsarcosine; 0.1 mg Proteinase K) and crushed with a pipette tip. DNA was extracted according to a protocol optimised for retrieval of short DNA molecules60. After overnight incubation, one part of extraction buffer was mixed with thirteen parts of binding buffer (5 M guanidine hydrochloride, 40% isopropanol) and eluted through MinElute silica column (Qiagen). Silica suspension was washed twice with 750 µl of PE buffer (Qiagen) and DNA was eluted twice from columns, each time using 30 µl of pre-warmed EB buffer. 20 µl of DNA extracts were converted into double-stranded, double-indexed sequencing libraries according to a previous protocol61 with the following modifications: after blunt-end repair, instead of undergoing SPRI clean-up, enzymes were heat-inactivated for 20 min at 75 °C. In the ligation step, sample cross-talk during sequencing was minimised using P5 and P7 adaptors with 7 bp barcodes at the ends, in addition to standard Illumina indexes, as previously described62. Final indexing PCR was performed in three parallel reactions using 19 cycles and AmpliTaqGold MasterMix (Thermo Fisher Scientific). Sequencing libraries were enriched for L. gregalis mtDNA, as previously described63. Hybridisation DNA bait was produced using ethanol-preserved muscle fragments of two present-day L. gregalis specimens. DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) and mtDNA was amplified as a set of different length amplicons designed on the M. arvalis (NC_038176.1) and M. mystacinus (=levis) (NC_008064.1) mtDNA genome sequences. PCR products were mixed in equimolar ratios and sheared to the length of ca. 200 bp using the Covaris S220 sonicator. Shared DNA was converted into bait as previously described63. Two hybridisation runs were performed on library pools from up to five specimens. Enriched libraries were amplified for 15 cycles after each round, quantified with qPCR (Illumina Library Quantification kit, KAPA), pooled and sequenced with other libraries on the NextSeq platform using the 2 × 75 bp paired end mode. Additionally, 10 µl of shared mtDNA of modern L. gregalis were also transformed into sequencing libraries as described above but with 12 cycles of indexing PCR, after which they were sequenced.
Sequencing reads were demultiplexed using Bcl2fastq. Reads containing the appropriate barcode were filtered with Sabre script, after which the AdapterRemoval v.264 was used to collapse overlapping reads. mtDNA genome sequences of two present-day L. gregalis were assembled de novo using NOVOPlasty65 and annotated using MITOS66. The resulting mtDNA sequence was used as a reference to map the reads from Late Pleistocene samples. Mapping was performed using the mem algorithm in bwa with option -B 1 to allow mapping of divergent reads. Duplicates were removed; variants and consensus sequences were called using samtools and bcftools67. Only reads with mapping quality over 30 and longer than 30 bp were retained and only positions with minimum 2x coverage were called. Each bam alignment was manually inspected using Tablet68. The presence of excessive DNA damage at the molecules’ ends was checked with mapDamage v.269.
Phylogenetic analyses
Phylogenetic analyses were performed on two datasets; the phylogenetic position of European narrow-headed voles within Arvicolini was investigated using the dataset comprising mtDNA genome sequences of 15 species from genus Microtus, Lasiopodomys and Neodon, together with sequences of two extant and ten Late Pleistocene L. gregalis obtained in this study (Table 1). To include data pertaining to several recently published species, the alignment length was limited to 9.09 kb of mtDNA.
Phylogeny was reconstructed using the Maximum Likelihood (ML) and Bayesian approaches. For both analysis the partitioning scheme and substitution model were chosen using PartitionFinder270 (Supplementary Table S2). The ML tree was reconstructed using RaxML v.871, the best tree was selected from 20 ML searches, and the support was assessed with 100 rapid bootstraps. The Bayesian tree was reconstructed using ExaBayes 1.572. Two runs, each with four coupled chains were run for two million generations sampled every 500 generations. Default values were used for tuning and branch swap parameters. Convergence and sampling were assessed in Tracer 1.773. For all parameters, the EES values were above 200.
The relationships between European and Asiatic populations of narrow-headed voles were investigated using a dataset comprising 179 previously published partial cytochrome b mtDNA sequences (889 bp) of Lasiopodomys gregalis (n = 28; KF751077–KF751104), (n = 133; KJ192239–KJ192327), (n = 2; AF163895, AY513803), (n = 2; GQ352466- GQ352467), (n = 1; AF429817), as well as those obtained in the present study.
Bayesian phylogeny was reconstructed using the Beast 1.8.474 and the best fitting substitution model was selected using jModeltest-2. The phylogeny was calibrated using a substitution rate of 4 × 10−7 substitutions/site/year−1. Late Pleistocene samples were dated by stratigraphic layers, the ages of which was estimated using 14C dating. Each sample was assigned an age corresponding to the mean form the age range formed by the youngest and oldest values of the 95.4% probability distributions of all calibrated radiocarbon dates known for the layer (Table 1). Marginal Likelihood Estimation (MLE) was used to test which clock model and tree prior best fitted the data75. The constant population sizes were compared to SkyGrid tree priors76 and strict versus relaxed lognormal clock models. For each tree prior/clock model combination analysis was run for 100 million generations with trees sampled every 10,000 generations. MLEs were estimated using path sampling and stepping stone sampling analyses, both with 1,000 steps run for 100,000 generations sampled every 1,000 generations. Bayes factors strongly favoured the SkyGrid tree prior combined with the relaxed lognormal clock model over all other combinations (logBF > 10). The selected model was used to run additional analysis using the same MCMC settings. The trees from two runs were combined in logcombiner and the Maximum Clade Credibility tree was selected from among the 9,000 trees using the treeannotator.
To include in the analysis also the Late Pleistocene and Holocene (n = 32; KC295791- KC295822) narrow-headed voles from the Ural Mountains published earlier18 we also reconstructed phylogeny based on 461 bp mtDNA cytb fragment. The phylogeny was reconstructed in Beast 1.8.4 using identical settings as for the longer fragment.
The genetic divergence (Dxy) between sequence groups was estimated in DnaSP v677.
Data availability
Sequences obtained in this study was deposited in GenBank under accession no. MN199169 – MN199180.
References
Shenbrot, G. I. & Krasnov, B. R. An atlas of the geographic distribution of the Arvicoline rodents of the world. (Pensoft, 2005).
López-García, J. M. et al. Palaeoenvironmental and palaeoclimatic reconstruction of the Latest Pleistocene of L’Arbreda Cave (Serinyà, Girona, northeastern Iberia) inferred from the small-mammal (insectivore and rodent) assemblages. Palaeogeogr. Palaeoclimatol. Palaeoecol. 435, 244–253 (2015).
Laplana, C. et al. Cold-climate rodent indicators for the Late Pleistocene of Central Iberia: New data from the Buena Pinta Cave (Pinilla del Valle, Madrid Region, Spain). C.R. Palevol 15, 696–706 (2016).
Suttcliffe, A. J. & Kowalski, K. Pleistocene rodents of the British Isles. Bull. Br. Museum (Natural Hist.) Geol. 27, 31–147 (1976).
López-García, J. M., Luzi, E. & Peresani, M. Middle to Late Pleistocene environmental and climatic reconstruction of the human occurrence at Grotta Maggiore di San Bernardino (Vicenza, Italy) through the small-mammal assemblage. Quat. Sci. Rev. 168, 42–54 (2017).
Luzi, E. Morphological and Morphometric Variations in Middle and Late Pleistocene Microtus arvalis and Microtus agrestis Populations: Chronological Insight, Evolutionary Trends and Palaeoclimatic and Palaeoenvironmental Inferences. PhD thesis. Universitat Rovira i Virgili, Tarragona. 322 pp. (2018)
López-García, J. M., Berto, C. & Peresani, M. Environmental and climatic context of the hominin occurrence in northeastern Italy from the late Middle to Late Pleistocene inferred from small-mammal assemblages. Quaternary Science Reviews 216, 18–33 (2019).
Bogićević, K., Nenadić, D. & Mihailović, D. Late Pleistocene voles (Arvicolinae, Rodentia) from the Baranica Cave (Serbia). Geol. Carpathica 63, 83–94 (2012).
Popov, V. In The Pleistocene, Geography, Geology and Fauna (eds. Huard, G. & Gareau, J.) 109–236 (Nova Science Publishers, Inc., 2018).
Markova, A. K. Small mammals from Palaeolithic sites of the Crimea. Quat. Int. 231, 22–27 (2011).
Ponomarev, D. & Puzachenko, A. Changes in the morphology and morphological diversity of the first lower molar of narrow-headed voles (Microtus gregalis, Arvicolinae, Rodentia) from northeastern European Russia since the Late Pleistocene. Quat. Int. 436, 239–252 (2017).
Royer, A. et al. Investigating the influence of climate changes on rodent communities at a regional-scale (MIS 1-3, Southwestern France). PLoS One 11, e0145600 (2016).
López-García, J. M. et al. Palaeoenvironmental and palaeoclimatic reconstruction of the Middle to Late Pleistocene sequence of Scladina Cave (Namur, Belgium) using the small-mammal assemblages. Hist. Biol. 29, 1125–1142 (2017).
Nadachowski, A. Origin and history of the present rodent fauna in Poland based on fossil evidence. Acta Theriol. (Warsz). 34, 37–53 (1989).
Rekovets, L. I. & Krokhmal, A. I. Pleistocene small mammal localities of the Ukraine and adjacent areas(in Russian). (National Academy of Sciences of the Ukraine, 2010).
Petrova, T. V., Zakharov, E. S., Samiya, R. & Abramson, N. I. Phylogeography of the narrow-headed vole Lasiopodomys (Stenocranius) gregalis (Cricetidae, Rodentia) inferred from mitochondrial cytochrome b sequences: an echo of Pleistocene prosperity. J. Zool. Syst. Evol. Res. 53, 97–108 (2015).
Petrova, T. V., Tesakov, A. S., Kowalskaya, Y. M. & Abramson, N. I. Cryptic speciation in the narrow-headed vole Lasiopodomys (Stenocranius) gregalis (Rodentia: Cricetidae). Zool. Scr. 45, 618–629 (2016).
Prost, S. et al. Losing ground: past history and future fate of Arctic small mammals in a changing climate. Glob. Chang. Biol. 19, 1854–64 (2013).
Rekovets, L. I. New subspecies of narrow-headed vole (Microtus gregalis) from the Upper Pleistocene deposits of Ukraine. Dokl. AN USSR. Ser. Biol., 6, 559–563 (in Ukrainian) (1978).
Chaline, J. Les rongeures du pléistocène moyen et supérieur de France:(systématique, biostratigraphie, paléoclimatologie). Cahiers Paléontologie (CNRS, 1972).
Nadachowski, A. Late Quaternary rodents of Poland with special reference to morphotype dentition analysis of voles. (Państwowe Wydawnictwo Naukowe, Warszawa – Kraków, 1982).
Smirnov, N. G., Bolshakov, V. N. & Borodin, A. V. Pleistocene rodents of northern Western Siberia (in Russian). (Nauka, 1986).
Bannikova, A. A. et al. Molecular phylogeny and evolution of the Asian lineage of vole genus Microtus (Rodentia: Arvicolinae) inferred from mitochondrial cytochrome b sequence. Biol. J. Linn. Soc. 99, 595–613 (2010).
Hinton, M. A. C. Monograph of the Voles & Lemmings (Microtinae) Living and Extinct. (British Museum (Natural History), 1926).
Ellerman, J. R. & Morrison-Scott, T. C. S. Checklist of Palaearctic and Indian Mammals 1758 – 1946. (British Museum (Natural History), 1966).
Rekovets, L. & Nadachowski, A. Pleistocene voles (Arvicolidae) of the Ukraine. Paleontol. i Evol. 28–29, 145–245 (1995).
Maul, L. C. & Markova, A. K. Similarity and regional differences in Quaternary arvicolid evolution in Central and Eastern Europe. Quat. Int. 160, 81–99 (2007).
Fejfar, O. & Horáček, I. Zur Entwicklung der Kleinsäugerfaunen im Villanium und Alt-Biharium auf dem Gebiet der ČSSR. Schriftenr. für geologische Wissenschaften 19/20, 111–207 (1983).
Nadachowski, A. Lower Pleistocene rodents of Poland: faunal succession and biostratigraphy. Quartärpaläontologie 8, 215–223 (1990).
Markova, A. K. Eastern European rodent (Rodentia, Mammalia) faunas from the Early-Middle Pleistocene transition. Quat. Int. 131, 71–77 (2005).
Mezhzherin, S. V., Zykov, A. E. & Morozov-Leonov, S. Y. Biochemical variation and genetic divergence of Palearctic voles (Arvicolidae). Meadow voles Microtus Schrank, 1798, snow voles, Chionomys Miller, 1908, water voles, Arvicola Lacepede, 1799. Genetica 29, 28–41 (1993).
Jaarola, M. et al. Molecular phylogeny of the speciose vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 33, 647–663 (2004).
Abramson, N. I., Lebedev, V. S., Tesakov, A. S. & Bannikova, A. A. Supraspecies relationships in the subfamily Arvicolinae (Rodentia, Cricetidae): An unexpected result of nuclear gene analysis. Mol. Biol. 43, 834–846 (2009).
Martínková, N. & Moravec, J. Multilocus phylogeny of arvicoline voles (Arvicolini, Rodentia) shows small tree terrace size. Folia Zool. 61, 254–267 (2012).
Pardiñas, U. et al. in Handbook of the Mammals of the World - Volume 7 Rodents II (eds. Wilson, D. E., Lacher, T. E. & Mittermeier, R. A.) 204–279. (Lynx, 2017).
Ho, S. Y. W. et al. Time-dependent rates of molecular evolution. Mol. Ecol. 20, 3087–3101 (2011).
Martínková, N. et al. Divergent evolutionary processes associated with colonization of offshore islands. Mol. Ecol. 22, 5205–5220 (2013).
Herman, J. S. & Searle, J. B. Post-glacial partitioning of mitochondrial genetic variation in the field vole. Proc. R. Soc. B Biol. Sci. 278, 3601–3607 (2011).
Herman, J. S. et al. Land-Bridge calibration of molecular clocks and the post-glacial colonization of Scandinavia by the Eurasian field vole Microtus agrestis. PLoS One 9, e103949 (2014).
Palkopoulou, E. et al. Synchronous genetic turnovers across Western Eurasia in Late Pleistocene collared lemmings. Glob. Chang. Biol. 22, 1710–1721 (2016).
Molak, M., Lorenzen, E. D., Shapiro, B. & Ho, S. Y. W. Phylogenetic estimation of timescales using ancient DNA: the effects of temporal sampling scheme and uncertainty in sample ages. Mol. Biol. Evol. 30, 253–62 (2013).
Chan, Y. L., Anderson, C. N. K. & Hadly, E. A. Bayesian estimation of the timing and severity of a population bottleneck from ancient DNA. PLoS Genet. 2, e59 (2006).
Welch, J. J., Bininda-Emonds, O. R. & Bromham, L. Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol. Biol. 8, 53 (2008).
Horn, S. et al. Mitochondrial genomes reveal slow rates of molecular evolution and the timing of speciation in beavers (Castor), one of the largest rodent species. PLoS One 6, e14622 (2011).
Paupério, J. et al. Cryptic speciation in the field vole: A multilocus approach confirms three highly divergent lineages in Eurasia. Mol. Ecol. 21, 6015–6032 (2012).
Markova, A. K. Paleolandscape reconstruction of Likhvin Interglacial by the data of Eastern European small mammals. Izv. RAN, Ser. Geogr. 2, 39–51 (2004).
Mahmoudi, A., Darvish, J., Aliabadian, M., Moghaddam, Y. & Kryštufek, B. New insight into the cradle of the grey voles (subgenus Microtus) inferred from mitochondrial cytochrome b sequences. Mammalia 81, 583–593 (2017).
Lagerholm, V. K. et al. On the origin of the Norwegian lemming. Mol. Ecol. 23, 2060–2071 (2014).
Nehring, A. Fossile Lemminge und Arvicolen aus den Diluviallehm von Thiede bei Wolfebüttel. Zeitschrift für die Gesammten. Naturwissenschaften 45, 1–28 (1875).
Woldřich, J. N. Uebersicht der Wirbelthierfaunades ‘Böhmischen Massivs’ während der anthropozoischen Epoche. Jahrb. d. k. k. geol. Reichsanstalt 47, 393–428 (1897).
Newton, E. T. The vertebrate fauna collected by Mr Lewis Abbot from the fissure near Ightham, Kent. Q. Jl. Geol. Soc. Lond. 50, 188–210 (1894).
Hinton, M. A. C. Some new late Pleistocene voles and Lemmings. Ann. Mag. Nat. Hist. 6, 34–39 (1910).
Horáček, I. & Sánchez-Marco, A. Comments on the Weichselian small mammal assemblages in Czechoslovakia and their stratigraphical interpretation. Neues Jahrb. für Geol. und Paläontologie 9, 560–576 (1984).
van Kolfschoten, T. in Sesselfelsgrotte VI. Natuwissenschaftliche Untersuchungen (eds. Frend, G. & Reisch, L.) 27–118 (Franz Steiner Verl, 2014).
Horáček, I. & Sazelova, S. in Mesolithic of Northern Bohemia II 39–48 (Institute of Archeology of the CAS, 2017).
Nadachowski, A. & Valde-Nowak, P. New Late Pleistocene faunal assemblages from Podhale Basin, Western Carpathians, Poland: preliminary results. Acta Zool. cracoviensia 58, 181–194 (2015).
Nadachowski, A. et al. Late Pleistocene environment of the Częstochowa Upland (Poland) estimated from faunistic evidence of archaeological cave sites. (Institute of Systematics and Evolution of Animals, PAS, 2009).
Ramsey, C. B. & Lee, S. Recent and planned developments of the Program OxCal. Radiocarbon 55, 720–730 (2013).
Reimer, P. J. et al. Intcal13 and marine13 radiocarbon age calibration curves 0 – 50,000 years cal bp. Radiocarbon 55, 1869–1887 (2013).
Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 110, 15758–63 (2013).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 5, t5448 (2010).
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Partial uracil-DNA-glycosylase treatment for screening of ancient. DNA. Philos. Trans. R. Soc. B Biol. Sci. 370, 20130624–20130624 (2014).
Horn, S. In Ancient DNA. Methods and Protocols. (eds. Shapiro, B. & Hofreiter, M.) 840, 189–95 (Humana Press, 2012).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 1–7 (2016).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, gkw955 (2016).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Milne, I. et al. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 14, 193–202 (2013).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, msw260 (2016).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–3 (2014).
Aberer, A. J., Kobert, K. & Stamatakis, A. ExaBayes: Massively Parallel Bayesian Tree Inference for the Whole-Genome Era. Mol. Biol. Evol. 31, 2553–2556 (2014).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Baele, G. et al. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29, 2157–67 (2012).
Gill, M. S. et al. Improving bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724 (2013).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Acknowledgements
This study was supported by Polish National Science Centre grants no. 2015/19/D/NZ8/03878 to MB and 2017/25/B/NZ8/02005 to AN. We are grateful to L.E. Yalkovskaya from Institute of Plant & Animal Ecology UrB RAS, Ekaterinburg, Russia who provided tissues of the extant L. gregalis.
Author information
Authors and Affiliations
Contributions
M.B. and A.N. conceived and supervised the study; M.B., K.B. and D.P. performed laboratory procedures and analysed the data; A.N., A.L. and I.H. assembled palaeontological materials and provided information on palaeontological context; M.B. and A.N. wrote the manuscript with the input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baca, M., Popović, D., Lemanik, A. et al. Highly divergent lineage of narrow-headed vole from the Late Pleistocene Europe. Sci Rep 9, 17799 (2019). https://doi.org/10.1038/s41598-019-53937-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53937-1
This article is cited by
-
First asserted record of the house mouse in Morocco: application of a multidisciplinary approach to the site of Rirha (5th − 1st c. BC)
Archaeological and Anthropological Sciences (2024)
-
Paleoclimatic and paleoenvironmental reconstructions based on the small vertebrates from the Middle Paleolithic of Hohle Fels Cave, SW Germany
Archaeological and Anthropological Sciences (2022)
-
Population dynamics and demographic history of Eurasian collared lemmings
BMC Ecology and Evolution (2022)
-
Signatures of genetic isolation of the three lineages of the narrow-headed vole Lasiopodomys gregalis (Cricetidae, Rodentia) in a mosaic steppe landscape of South Siberia
Mammalian Biology (2021)
-
Incisor microwear of Arctic rodents as a proxy for microhabitat preference
Mammalian Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
