
Abstract
Historic calcified dental plaque (dental calculus) can provide a unique perspective into the health status of past human populations but currently no studies have focused on the oral microbial ecosystem of other primates, including our closest relatives, within the hominids. Here we use ancient DNA extraction methods, shotgun library preparation, and next generation Illumina sequencing to examine oral microbiota from 19 dental calculus samples recovered from wild chimpanzees (Pan troglodytes schweinfurthii) who died in Gombe National Park, Tanzania. The resulting sequences were trimmed for quality, analyzed using MALT, MEGAN, and alignment scripts, and integrated with previously published dental calculus microbiome data. We report significant differences in oral microbiome phyla between chimpanzees and anatomically modern humans (AMH), with chimpanzees possessing a greater abundance of Bacteroidetes and Fusobacteria, and AMH showing higher Firmicutes and Proteobacteria. Our results suggest that by using an enterotype clustering method, results cluster largely based on host species. These clusters are driven by Porphyromonas and Fusobacterium genera in chimpanzees and Haemophilus and Streptococcus in AMH. Additionally, we compare a nearly complete Porphyromonas gingivalis genome to previously published genomes recovered from human gingiva to gain perspective on evolutionary relationships across host species. Finally, using shotgun sequence data we assessed indicators of diet from DNA in calculus and suggest exercising caution when making assertions related to host lifestyle. These results showcase core differences between host species and stress the importance of continued sequencing of nonhuman primate microbiomes in order to fully understand the complexity of their oral ecologies.
Similar content being viewed by others

Introduction
The human oral cavity contains an estimated 600 different microbial species1. The oral microbiome also exhibits strong interpersonal and population-specific variation across the globe2,3, while at the same time differentiating between healthy and diseased oral states4. Advances in next generation sequencing and bioinformatic analyses have allowed researchers to study the oral microbiota of modern as well as historic and prehistoric populations through the investigation of dental calculus (calcified plaque). Dental calculus is commonly found in living populations without adequate dental care as well as archaeological skeletal assemblages and has been estimated to contain 200 million cells per milligram5,6 consisting of host cells7, bacteria, viruses, and occasionally dietary information. This biological resource has been used to answer many biological and anthropological questions addressing such topics as Neanderthal diet and behavior8,9, the evolution of antibiotic resistance genes in oral pathogens10, and the bacterial composition of pre-contact Puerto Rican dental calculus11.
Although the oral microbiome has been shown to be associated with host health and disease1 and exhibit incredible diversity across the globe in humans2,12,13,14, little focus has been paid to nonhuman primate oral microbiomes. To date, Weyrich et al.9 is the only study to include a historic oral microbiome sample from Pan troglodytes. As for modern microbiomes, a single study examined modern ape oral ecosystems through saliva, which uncovered a greater similarity between baboon and chimpanzee species (Sierra Leone and Democratic Republic of Congo) when compared to human caretakers from each sanctuary facility15. This research further suggested that a captive environment drastically impacts the primate oral ecology15. Outside of the oral cavity, specifically within the primate gut, clusters known as ‘enterotypes’ show that regardless of geographic origin, gorillas and chimpanzees share a Prevotella-dominated gut signature with modern humans16,17,18. These clusters were generally thought to be associated with the long term dietary practices of the host17. However, the enterotype concept is somewhat controversial and a sole reliance on enterotype clustering classifications may obscure critical microbial variation19. The existence of these enterotype clusters within the human and chimpanzee oral cavity has yet to be explored.
In this study, we characterize the microbiota in the oral cavity of wild chimpanzees using next generation shotgun sequencing of dental calculus. We first focus on differences in abundance between anatomically modern humans (AMH) and chimpanzees at the phylum and genus levels as well as shared types between groups. Second, we address the question of whether chimpanzee oral microbiota adhere to an enterotyping pattern as seen within primate gut microbiomes. Third, we reconstruct a full Porphyromonas gingivalis genome from a single chimpanzee and compare it to previously published genomes. Lastly, since the chimpanzees at Gombe have been observed for more than fifty years and their diet is well documented20,21, we map sequence data indicative of diet to understand whether such methods are useful for inferring lifestyle. This research helps to situate the previously unexplored chimpanzee oral microbiota from dental calculus with other historic and prehistoric human samples in an effort to understand the complexity of microbial diversity across the primate oral ecosystem.
Results
Sequencing statistics and MetaPhlAn2 analyses
For initial analyses we examined data from 19 Gombe chimpanzee calculus samples and two sets of comparative data from a total of 46 individuals. The first set includes 25 historic AMH calculus samples22 and the second set has data from 21 samples including Neanderthals as well as prehistoric, historic, and contemporary AMH, and a nonhuman sample from a historic chimpanzee9 (Table 1). A total of 95% of raw sequence reads passed adapter trimming, merging, and QC > 20 for the data from Gombe chimpanzees reported here. For the previously published datasets, the percentages of reads passing the same quality control thresholds were slightly lower (93% in the AMH dental calculus samples from Mann et al.22, and 69% from the Neanderthal/AMH/chimpanzee samples from Weyrich et al.9).
Oral health in the Gombe chimpanzee population was assessed through examination of both the mandible and maxilla (by R.S.N., with assistance from those mentioned in acknowledgements). A total of 63% (12/19) of chimpanzees exhibited signs of carious and/or abscess lesions with 42% (8/19) possessing afflictions impacting the mandible and 52% (10/19) showing maxillary issues. These numbers represent active caries estimates at the time of death and are likely an underestimate of total lifetime caries, as many teeth were lost throughout the life of the animal. A total of 95% of chimpanzees were observed to have lost at least one tooth across the dental arcade with 74% (14/19) of individuals missing at least one tooth from the mandible and 84% (16/19) of individuals having lost one or more teeth from the maxilla. We compared the presence/absence of caries to genera abundance across chimpanzees and found no significant differences based on presence of active caries/abscesses at time of death. Mann et al. did not report AMH oral health states22 and although Weyrich et al.9 reported some dental information from the historic and prehistoric human samples (which were excluded from further analysis) only a single Neanderthal (El Sidrón 1) was reported to have likely suffered from periodontal disease. Thus, there was not enough dental health information to compare these data to data from the Gombe chimpanzee population.
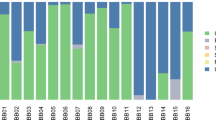
For initial screening purposes, sequences were first compared to the MetaPhlAn2 (metagenomic phylogenetic analysis) database which comprises one million clade-specific marker genes from ~17,000 reference genomes across bacteria, archaea, viruses, and eukaryotes23. In both Gombe chimpanzees and historic AMH from Mann et al.22, samples were dominated by commonly known oral phyla: Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Synergistetes (Fig. 1). Although the average percentage of reads successfully mapped using MetaPhlAn2 was comparable across populations (0.17% for Weyrich et al.9, 0.58% for Mann et al.22, and 0.65% for Gombe chimpanzees), due to the overall low read count of sequences from Weyrich et al.9, we chose to eliminate all samples aside from the Neanderthals (Spy 1, Spy 2, El Sidrón 1, El Sidrón 2) for downstream analyses.

Significant phyla and genera using MALT
Mapping with MALT increased the number of reads that mapped to known species since it uses the NCBI nucleotide (or ‘nt’) database (4.82% for Mann et al.22, and 5.95% for Gombe chimpanzees). Due to the eight (Mann et al.22) and nine (Gombe) fold increase in mapped reads from MALT compared to MetaPhlAn2 and the extensiveness of the ‘nt’ database compared to MetaPhlAn2, we chose to use the MALT results for subsequent analyses. As such, normalized values (~104,000 reads) from chimpanzees and comparative data were used for downstream analyses. The five most dominant bacterial phyla within the chimpanzee calculus (average across all individuals) are Proteobacteria (22%), Actinobacteria (19.6%), Bacteroidetes (18.7%), Fusobacteria (11.4%), and Firmicutes (6.3%) (Fig. 2). The five most dominant bacterial phyla in AMH (average across all individuals) are Proteobacteria (34.3%), Actinobacteria (21.9%), Firmicutes (12.6%), Spirochaetes (7.6%) and Bacteroidetes (5.8%). A total of four phyla (Table 2) are significantly different between AMH and chimpanzee calculus (above 1% abundance cut off). Bacteroidetes and Fusobacteria are significantly more abundant in chimpanzees, while Firmicutes and Proteobacteria are more dominant in AMH calculus (Kruskal-Wallis, p < 0.05). The five most common bacterial genera in chimpanzees (average across all individuals) are Porphyromonas (16.2%), Fusobacterium (12%), Streptomyces (6.8%), Treponema (4%), and Mycobacterium (3.4%) (Fig. 3). The five most common bacterial genera in AMH (average across all individuals) are Treponema (7.9%), Streptomyces (7.3%), Neisseria (7.2%), Streptococcus (6.6%), and Porphyromonas (3.6%). Four genera significantly differed between chimpanzees and historic AMH (above 0.5% abundance cut off) (Table 2). Fusobacterium and Porphyromonas are more abundant within chimpanzees, while Streptococcus and Neisseria are more common in AMH (all p < 0.05). Hits to both Pan and Homo (both likely representing host mitogenomes) are present in the sample sets but are not reported here and have been excluded for enterotype analyses.

Abundance of sequence reads mapped using MALT for phyla. Leftmost samples are chimpanzees (present study) and rightmost reads are previously published data from Mann et al.22.

A box plot indicating genera abundance from chimpanzees using MALT. Those individuals (Porphyromonas in three chimpanzees) exceeding 30% abundance for any given genus were excluded from the figure for space and clarity purposes.
Enterotype analysis
Enterotype analyses (Fig. 4) suggest that chimpanzee and historic AMH samples cluster separately based on the abundance of several core genera. The number of potential clusters for our chosen groupings (AMH/chimpanzees/Neanderthals, chimpanzees only, and AMH only) are estimated using established methods from Arumugam et al.16. These analyses produced the likely number of sample clusters: five for the AMH/chimpanzees/Neanderthals set, two for the AMH set, and two for the chimpanzee set. Anatomically modern human and chimpanzee clusters are driven by the genera previously mentioned as being significant between the two groups: Fusobacterium and Porphyromonas (clusters 1 and 2 respectively in Fig. 4C) for chimpanzees, and Haemophilus and Treponema for AMH (clusters 1 and 2 respectively in Fig. 4B). Neanderthals slightly clustered with historic AMH but the Neanderthal cluster was likely driven by the presence of soil microbiota such as Arthrobacter (either modern or ancient) (cluster 2 in Fig. 4A), a potential contaminant noted previously by the authors9 (which led to the omission of Spy 1 from enterotype analysis). As such, we cannot conclusively state which genera are driving the clustering of the Neanderthal microbiomes and whether these results are genuine or due to environmental contamination.

MEGAN normalized (bacteria and archaea only, all zeroes removed) genus level sequence abundance enterotype clustering. The optimal number of clusters and cluster visualization are displayed for (A) Neanderthals (Spy1 excluded), anatomically modern humans, and chimpanzees, (B) anatomically modern humans only, and (C) chimpanzees only. Results are color coded with orange indicating Neanderthals, blue for anatomically modern humans, and red for chimpanzees.
Neighbor joining analyses for microbiomes
We used normalized MALT outputs in MEGAN to visualize chimpanzee, Neanderthal, and AMH oral microbiome samples in a Bray Curtis neighbor joining tree (Fig. 5). Neanderthals cluster within the AMH population while chimpanzees cluster separately.

A neighbor joining bray curtis tree using all normalized species in MEGAN (bacteria and archaea only). Results are color coded with orange indicating Neanderthals, blue for anatomically modern humans, and red for chimpanzees.
Red complex analysis
A total of 19 chimpanzee samples, 25 AMH samples22, and four Neanderthal samples9 were examined for the red complex (using MALTn, normalized in MEGAN) (Fig. 6). Normalized abundance in chimpanzee calculus was an average of 16.2% for P. gingivalis compared to 3.4% in AMH, which was significant at the p < 0.05 level. Conversely, T. denticola was more dominant in AMH (7.8%) compared to chimpanzees (4.1%), and this was also significant at the p < 0.005 level. Neanderthal samples showed low read counts of all three members of the red complex, and thus, they were not included in Kruskal-Wallis significance tests. Although we did observe differences in abundances between MetaPhlAn2 and MALT both showed low abundance of T. forsythia in chimpanzees, which was also shown in a previous study of human dental calculus to be in very high abundance (using MALT)24. Additionally, for degraded material, MALT (using BLASTn) was found to be the most accurate method for determining taxonomic information from shotgun sequences25.

Box plots of normalized species abundance from MEGAN for all three red complex bacteria across Neanderthals, anatomically modern humans, and chimpanzees. Those individuals (P. gingivalis in three chimpanzees) exceeding 30% abundance for a microbial species were excluded from the figure for space and clarity purposes.
Genome reconstruction and phylogenetic tree building
We used bwa to map dental calculus sequencing reads from the Gombe chimpanzee 17C to the Porphyromonas gingivalis genome (NC_010729.1). Out of a total of 29,144,776 merged sequence reads, 838,334 (Q > 30, duplicates removed) reads mapped to P. gingivalis genome (Supplementary Fig. 1). The GC content of the mapped sequence is slightly less than that of the reference sequence (47.6% compared to 48.4%). A total of 2,118 annotated genes within the P. gingivalis genome were used for Circos mapping26. A total of 2,167,869 bp out of a possible 2,354,886 bp mapped to the reference genome (92.1%). The genome was visualized in 250 bp windows, with a minimum of 0x coverage, a maximum of 123.4x coverage, and an average coverage of 29.2x. This reference aligned genome was compared to 58 previously published P. gingivalis genomes including three outgroups (T. forsythia, T. denticola, P. asaccharolytica) (Supplementary Table 2). The genome from Gombe did not cluster specifically with those recovered from humans from any one geographic region, with the samples phylogenetically closest originating in Romania, United Kingdom, and United States (Fig. 7).

A neighbor joining (500 bootstraps, pairwise deletion) alignment of 58 previously published genomes along with three outgroups: Porphyromonas asaccharolytica, Tannerella forsythia, Treponema denticola.
Dietary reconstruction
To determine the extent to which DNA sequences recovered from dental calculus showed evidence of host dietary practices (at Q > 20)27,28, we used bwa, samtools, and mapDamage 2.029,30. We used 14 full and partial genomes associated with diet analyzed in Weyrich et al.9 with an additional six genomes from chimpanzee food sources commonly found at Gombe National Park. (Supplementary Tables 3 and 4). In particular, after initial mapping with bwa, we created consensus sequences from five of the seven Neanderthals and from chimpanzee samples. These consensus sequences spanned 11 of the selected dietary genomes (for a total of 22 specific cases of evidence of diet). Our results show that some reads from each individual did map to these dietary reference genomes (0 to 1,355 reads) (Supplementary Table 3). We also examined sequences from our initial MALT analysis that matched each of these species of plants, animals, and fungi (Supplementary Table 4) and found evidence suggesting that some Neanderthal calculus (Spy1 and Spy2) contained traces of Ovis aries (sheep) and calculus from one chimpanzee (13C) contained DNA sequences potentially belonging to Elaeis guineensis (African palm).
Discussion
We detected five bacterial phyla in the dental calculus of Gombe chimpanzees (Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Proteobacteria) which are also commonly found within historic AMH samples. We also found significant differences in abundance between AMH and chimpanzees across four phyla. Previous human calculus studies8,10 showed a high abundance of Firmicutes and Proteobacteria, and we report that these phyla are significantly reduced in the mouths of chimpanzees. Conversely, chimpanzees had significantly higher Bacteroidetes and Fusobacteria compared to historic AMH. Actinobacteria, another phylum reported as being abundant in the human oral cavity8,10 was also abundant in chimpanzees, but not to a significant degree over historic AMH. Additionally, we report a number of abundant genera in chimpanzee dental calculus including Fusobacterium, Porphyromonas, and Streptomyces (>5% average abundance). Both Fusobacterium and Porphyromonas abundance was significantly higher in chimpanzees compared to historic AMH (p < 0.05). The significance of Fusobacterium within the oral cavity is not fully understood. In some studies it was reported that Fusobacterium could be either a pathogen or commensal31, while others show associations with oral disease and systemic issues throughout the body32. It should be noted that the mere presence of a genus that contains pathogenic species does not mean the species found here play a pathogenic role in the oral cavity. Our analysis of chimpanzee oral health based on caries and tooth loss did not find a significant association between one particular genus and the presence of caries or the absence of teeth. In addition to questions about the role of these bacteria in health states, it should also be noted that these differences seen between AMH and chimpanzees may stem from environmental differences. Comparative AMH samples from Mann et al.22 were from several locations across Asia, Europe, and North America, while our chimpanzee data only represent Gombe National Park, one location in Eastern Africa. Future studies sampling historic nonhuman primates and human populations in Africa may show similar oral microbiome signatures to those recovered from wild chimpanzees from Gombe National Park.
Our data show that oral microbiomes from AMH, chimpanzees, and Neanderthals did not adhere to an enterotype clustering pattern reminiscent of the gut microbiome. A global study of human gut metagenomes found that individuals cluster into three robust enterotype groups that are independent of body mass index, age, gender, and geographic location16. Our results however, do not necessarily cluster randomly as seen in previous studies16, but somewhat along host species lines, with most chimpanzees clustering together, most AMH clustering together, and a with smaller group of AMH and Neanderthals set slightly apart. The driving genera are those noted as being significantly different between AMH and chimpanzees. Specifically, AMH enterotypes are driven by Treponema, which has strong associations with periodontal disease33 and Haemophilus which can be commonly found in human plaque34 and has been associated with a healthy human mouth35. However, species of Haemophilus also exhibit pathogenic properties throughout the body36. Secondary drivers of these AMH enterotypes include both Streptococcus, which has been identified as both a genus including commensal and pathogenic species37, and Neisseria, which also exhibits both pathogenic and non-pathogenic strains in humans38. The signature in the Neanderthal calculus seems to be driven by Arthrobacter, which is a common soil microbe39 but has also associated with skin lesions in humans40. Chimpanzee enterotypes were driven by both Fusobacterium and Porphyromonas, both of which are considered by some to be causative agents in periodontal disease41. Unfortunately, we do not have oral health data from the archaeological samples sequenced by Mann et al.22, and there was not a significant difference in abundance of Fusobacterium and Porphyromonas related to caries or tooth loss in chimpanzees. Independent of health states, the partitioning of these enterotypes by host species echoes what was observed in previous studies of human and chimpanzee salivary microbiomes15. In the years since enterotypes were first proposed, they were found to be associated with long-term diet42 and population43, with some studies suggesting enterotypes are not as distinct as first documented44 and others questioning the existence of discrete clusters completely45. For example, a subsequent study examined how sample processing and data analysis can alter enterotype recovery, but note that enterotypes are still beneficial for exploring overall microbial composition19. Here we use the original definition of enterotypes to investigate primate dental calculus microbiomes and show that they mainly adhere to a two-group system (based on host species). We posit that both AMH and chimpanzee clusters are likely driven by long term unhealthy oral states within the host as reflected in the increased abundance of known pathogens belonging to the genera Porphyromonas and Fusobacterium in chimpanzees and Haemophilus and Treponema in AMH.
A known cause of oral dysbiosis within humans is attributed to periodontal disease46. This disease is commonly associated with pathogenic microbiota collectively referred to as the red complex (Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia (formerly Bacteroides forsythus)). Initially the detection of red complex bacteria was linked to poor oral health5 but it is by no means the only indicator of periodontal disease47. Observable traits in skeletal remains including, tooth loss, tooth wear, and abscesses are manifestations of periodontal infection and have been documented in captive and wild great apes48,49, but the connection between these and the red complex bacteria in the Pan oral cavity is not known. Studies have shown both positive and negative correlations between the presence of P. gingivalis and oral disease states50,51,52 yet others suggest their abundance is independent of disease and more closely related to host weight53 and age54,55,56. However, species of Porphyromonas likely have different roles within the mouth at different times57 with P. gingivalis acting as a late colonizer which inhabits the top layer of already formed biofilms58 and a species such as P. catoniae occupying the mouths of infants prior to tooth eruption59. In longitudinal studies, the abundance of T. denticola and P. gingivalis are linked together as indicators of chronic periodontitis progression60. However, our results suggest that their increased abundance is not always linked, due to the low presence of T. denticola across chimpanzees. Low abundance of Tannerella was also reported in the oral cavity of another nonhuman primate, Rhesus macaques (Macaca mulatta)61 from the Caribbean Primate Research Center in Puerto Rico. Although we observed caries and abscesses within the dental arcade of several chimpanzees, we cannot make statements regarding the role of any single microbe or any group of microbes as causative agents of disease. It is likely a very complex process involving many elements, as dental calculus recovered from healthy human teeth and those afflicted with periodontal disease do not significantly differ in microbial, protein, and metabolomic profiles62. As such, it is imperative to continue to characterize oral microbiomes from modern and historic primates with varying health states in order to further comprehend the factors that drive these complex ecosystems.
The Porphyromonas gingivalis genome recovered from one of the Gombe chimpanzees was selected for analysis because it was the most complete genome observed with the highest level of total coverage. The phylogenetic analyses of a P. gingivalis genome assembled from a single chimpanzee individual (17C) did not distinctly separate it from previously published genomes. However, research suggests that P. gingivalis strains likely undergo frequent recombination with other strains63 which may obscure phylogeography. These DNA exchange events generates diverse phenotypes among microbial communities64. In P. gingivalis, the high mosaicity arises from an increase in the likelihood of recombination events due to the use of carbon from exogenous DNA as sources of energy63,65. Considering that P. gingivalis has a complex genome that readily recombines, it would be beneficial in the future to isolate, culture, and sequence this microbe in chimpanzee plaque using traditional laboratory methods in order to understand the nuanced differences in genotypes and phenotypes of this strain.
Because the chimpanzees at Gombe have been subject to decades of observation20,21,66,67, their diet is known and this can be used to assess whether dental calculus preserves genetic material from plants and animals indicative of dietary habits. We searched for evidence of dietary DNA sequences in five Neanderthal samples and two Gombe chimpanzees using full and partial genome reference data from fourteen organisms (Weyrich et al.9) and an additional six associated with the environment in Gombe National Park. Although some short sequences mapped to possible dietary sources (Supplementary Table 3), an additional screening of the initial MALT results show only two cases in which dietary DNA may be present: sheep sequences in the Spy Neanderthals and palm DNA in one of the Gombe chimpanzees. Although it is not out of the realm of possibility that dietary DNA is present in these and Weyrich et al.9 calculus samples, due to the very nature of ancient and degraded historic DNA (short fragments), the lack of high sequencing depth, and the presence of only highly conserved regions in 16S ribosomal RNA genes and chloroplast DNA in most reference databases, we hesitate to conclude that these sequences definitively originate from the hosts’ diet. We suggest that future dietary analyses use proteomics and phytoliths along with genome capture in order to confirm shotgun DNA sequence data. Additionally, we stress using caution when interpreting ‘shared’ oral microbial genera as being indicative of ‘interaction’ between individuals, in agreement with other authors68.
In conclusion, our results present an important piece of the puzzle in understanding the composition and evolution of the primate oral microbiome. Chimpanzee and AMH oral microbes differ significantly but it is still unclear as to the underlying causes of these differences: diet, geography, host genomes, or factors unknown. Future studies should continue to integrate bioarchaeological, observational, and cultural evidence into studies of historic microbiomes whenever possible in order to establish the most complete picture of primate oral ecologies.
Materials and Methods
Sample collection and extraction
A total of 19 calculus samples were removed from Gombe chimpanzee skeletal remains. The source of the chimpanzee skeletal remains is the long-term non-invasive study led by Dr. Jane Goodall. No chimpanzees were harmed to obtain these skeletal remains. Bodies of chimpanzees that died from natural causes were recovered and either buried or kept in a container until soft tissues had decayed69. Due to the lack of abundant calculus across the dental arcade of Gombe chimpanzees, samples were collected opportunistically and pooled together for each single individual. When available, calculus was sampled from at least one tooth on both the mandibular and maxillary sides (<15 mg total). Overall dental health was also assessed at the time of sampling (Supplementary Table 1). Teeth were counted as having a carious lesion if the enamel was infiltrated and not caused by a clear breakage (many of the teeth are discolored, making a true assessment of cavities difficult). Teeth with abscesses also qualified as carious lesions. Tooth loss was classified as a clear resorption of bone and not caused by postmortem damage (marked with ‘O’ for adult teeth and ‘dO’ for deciduous teeth).
Samples were shipped to a UV-equipped, class 10,000 HEPA filtration ancient DNA facility at Arizona State University. Throughout the preparation and extraction of specimens, full ancient lab precautions were utilized including full length sterile suits, hairnets, facemasks, and eye protection. Calculus samples were pulverized using a sterile hammer and UV-ed in a DNA crosslinker for 2 minutes on each side (5– 15 mg). Samples were transferred to a 2 mL tube and washed using 1 mL of 0.5 M EDTA (Ambion) on a rotating nutator for 15 minutes at room temperature (RT). They were then centrifuged at 13.2 k rpm for 3 minutes and the supernatant was removed and discarded. Fresh EDTA (1 mL) was added to the pellet and resuspended by vortexing and placed on a rotating nutator overnight at RT. A total of 100 µL of Proteinase K (Qiagen) was added to the 2 mL tube and set on a rotating nutator at 37 °C for 8 hours. Samples were left to rotate overnight at RT once more. The next day samples were centrifuged at 13.2 k rpm for 3 minutes and the supernatant was kept at 4 °C. Fresh EDTA was added to the pellet along with 50 µL more of Proteinase K. Samples were left to rotate overnight one final time at RT. Samples were centrifuged at 13.2 k rpm for 3 minutes and both supernatants were added to a total of 12 mL of PB Buffer (Qiagen) in a Zymo reservoir attached to a MinElute PCR Purification kit (Qiagen) silica column (within a 50 mL Falcon tube). Samples were spun for 6 k rpm for 4 minutes, rotated 180° and spun another 2 minutes. The MinElute column was washed according to manufacturer specifications and eluted into 30 µL.
Shotgun build, amplification, and sequencing
Extracts for calculus samples underwent double stranded shotgun builds. For initial blunt end repair, a total of 20 µL (~800 ng) of DNA was added to 5.0 µL NEB Buffer, 0.50 µL dNTP mix (2.5 mM), 4.0 µL BSA (10 mg/mL), 5.0 µL ATP (10 mM), 2.0 µL T4 PNK, 0.40 µL T4 Polymerase, and 13.10 µL ddH2O was incubated at 15 °C for 15 minutes followed by 25 °C for 15 minutes. The solution was then purified using a MinElute according to manufacturer protocol and eluted into 18 µL EB buffer. For adapter ligation, 18 µL of template DNA was added to 20 µL Quick Ligase Buffer, 1.0 µL Solexa Mix70, and 1.0 µL Quick Ligase and incubated at room temperature for 20 minutes. The solution was then purified again using a MinElute according to manufacturer protocol and eluted into 20 µL EB buffer. For the final fill in portion of the shotgun build, 20 µL of template DNA was added to 4.0 µL Thermo pol buffer, 0.50 µL dNTP mix (2.5 mM), 2.0 µL Bst polymerase, and 13.50 µL ddH2O was incubated at 37 °C for 20 minutes followed by 80 °C for 20 minutes. Following shotgun preparation, samples were amplified using Amplitaq Gold DNA Polymerase (Thermo Fisher Scientific) to a total of 10 cycles. Shotgun libraries were split into four identical PCR reactions which contained 9.0 µL of DNA, 9.27 µL PCR Buffer II (10x), 9.27 µL MgCl2 (25 mM), 3.68 µL dNTP mix (10 nM), 2.21 µL BSA (10 mg/mL), 2.0 µL P5 primer, 2.0 µL P7 primer, 61.09 µL of ddH2O, and 1.48 µL of Amplitaq Gold enzyme. The PCR conditions were as follows: initial denaturation at 95 °C for 15 minutes, followed by cycling of 95 °C for 30 seconds, 58 °C for 30 seconds, and 72 °C for 45 seconds, with a final elongation of 72 °C for 10 minutes. Each P5 and P7 primer pair used for the four samples had a unique set of barcodes71 in order to separate the individual samples from the pooled material bioinformatically. Samples were purified using the MinElute according to manufacturer protocol and eluted into 30 µL of EB buffer. After checking concentration using a DNA1000 Bioanalyzer chip (Agilent) samples were pooled in equimolar amounts and pooled on a single Illumina HiSeq. 2500 2 × 100 pe (Rapid Mode) lane at the Yale Center for Genome Analysis (YCGA). Two of the chimpanzee samples were sequenced deeper (13C and 17C) with chimpanzee exome captures a sequencing run with the same specifications at YCGA.
Sequence processing and data analysis
Samples for this publication were returned as de-multiplexed reads from YGCA and paired end samples from comparative studies were downloaded from the Online Ancient Gene Repository (OAGR) under the project title “Reconstructing Neanderthal behavior, diet, and disease using ancient DNA from dental calculus” (https://www.oagr.org.au/experiment/view/65/) for Weyrich et al.9 and the NCBI Short Read Archive (SRA) under the Bioproject accession PRJNA445215 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA445215/) for Mann et al.22. For the chimpanzee sample set in the present study, Weyrich et al.9, and Mann et al.22, paired end files were unzipped, adapters were removed, and paired ends were merged using SeqPrep72 with a minimum overlap of 30 bp and a minimum quality threshold of 20. Taxonomic abundances of phyla and genera were inferred using MetaPhlAn2.073, as used in previous publications74. Additionally, reads were mapped to the NCBI nucleotide database using MALT (BLASTn (February 2017), 85% sequence similarity, minimum support percent of 0.01, top percent value of 1.0)75 and analyzed in MEGAN76. MALT analyses were carried out using XSEDE77. MEGAN allowed the data to be normalized and grouped into shared species using a bray Curtis neighbor joining method (only bacteria and archaea selected). We used normalized abundance (Table 1) from MEGAN to determine the totals of phyla and genera across samples. We used Kruskal-Wallis within R to determine significant phyla and genera between human and chimpanzee groups78. For enterotyping, we used normalized count data from all three groups (Neanderthals, AMH, and chimpanzees) and used methods from a previous publication16 to call clusters and generate figures within R. Spy 1 was removed from Fig. 4A due to contamination concerns presented by Weyrich et al.9
Prior to mapping, raw reads from 17C were adapter trimmed and merged using seqprep (>Q30)72. Reads were mapped to the Porphyromonas gingivalis ATCC 33277 genome (NC_010729.1)79 using BWA v. 0.7.527 following recommendations by Schubert et al.80. Mapped reads were quality filtered (>Q30), duplicates were removed, and sequences with multiple mappings were removed using Samtools v. 0.1.1928. The program mapDamage 2.0 was used to rescale BAM files and characterize damage patterns29,30. The full genome was visualized in Geneious 981 (https://www.geneious.com/) which was used to export a consensus sequence. The consensus sequence was visualized using Circos26 with gray bars indicating 25x to 125x coverage (intervals of 25) and each green line extending outward representing a 250 bp window of base pair coverage. Total coverage is represented by the inner green coloration (250 bp windows), and GC content represented by a second green circle (250 bp windows) with a gray line representing average GC content.
A total of 58 full and partially assembled genomes from P. gingivalis (ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/bacteria/Porphyromonas_gingivalis/latest_assembly_versions/) were downloaded from Genbank and the sequences were aligned to the reference genome using previously published methods82 (Supplementary Table 1). In brief, for each previously published complete or partial genomes, we used similar methods to those reported for 17C (using BWA v. 0.7.527 and Samtools v. 0.1.1928 but not mapDamage 2.029,30). Then using Picard83, a sequence dictionary was created with the aforementioned reference genome for Porphyromonas gingivalis. Lastal84 and Samtools v. 0.1.1928 were used to convert each mapped genome to sam and bam files, and bcftools85 was used to create a VCF file. GATK86 was then used to combine variants from all files and custom scripts were used to create a VCF variant table and finally a FASTA alignment. The resulting file was used to create a neighbor joining tree (500 bootstraps) using MEGA787.
Previously published full and partial genomes indicative of diet (Supplementary Tables 3 and 4) were downloaded from NCBI. We mapped two chimpanzee samples 13C and 17C (due to their high sequencing depth) and four samples from Spy and El Sidrón (including an additional deeper sequenced El Sidrón 1 sample labelled merely ‘ELSIDRON’) against 15 indicators of diet present in Weyrich et al.9 along with six additional indicators of diet that documented in observational data compiled from Gombe National Park20,21. We selected several commonly eaten items, but it should be noted that some foods are eaten during restricted fruiting seasons and not necessarily year round20,21. We used identical methods to those used to map the 17C P. gingivalis genome but reduced the quality filtering during seqprep and mapping to 20. The number of reads that mapped to their dietary species are reported in Supplementary Table 3. Additionally, we compiled raw reads from the original MALT analysis that matched these dietary sources and reported those values in Supplementary Table 4.
Data availability
Raw data sequences have been deposited into the NCBI Short Read Archive (SRA) under the BioProject ID PRJNA531027 (SAMN11408660-SAMN11408678).
References
Dewhirst, F. E. et al. The Human Oral Microbiome. Journal of Bacteriology 192, 5002–5017, https://doi.org/10.1128/jb.00542-10 (2010).
Nasidze, I., Li, J., Quinque, D., Tang, K. & Stoneking, M. Global diversity in the human salivary microbiome. Genome Research 19, 636–643, https://doi.org/10.1101/gr.084616.108 (2009).
Mason, M. R., Nagaraja, H. N., Camerlengo, T., Joshi, V. & Kumar, P. S. Deep Sequencing Identifies Ethnicity-Specific Bacterial Signatures in the Oral Microbiome. Plos One 8, e77287, https://doi.org/10.1371/journal.pone.0077287 (2013).
Yang, F. et al. Saliva microbiomes distinguish caries-active from healthy human populations. The ISME Journal 6, 1–10, https://doi.org/10.1038/ismej.2011.71 (2012).
Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C. & Kent, R. L. Microbial complexes in subgingival plaque. Journal of Clinical Periodontology 25, 134–144, https://doi.org/10.1111/j.1600-051X.1998.tb02419.x (1998).
Socransky, S. S. & Haffajee, A. D. Periodontal microbial ecology. Periodontology 2000 38, 135–187, https://doi.org/10.1111/j.1600-0757.2005.00107.x (2005).
Ozga, A. T. et al. Successful enrichment and recovery of whole mitochondrial genomes from ancient human dental calculus. American Journal of Physical Anthropology 160, 220–228, https://doi.org/10.1002/ajpa.22960 (2016).
Adler, C. J. et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat Genet 45, 450–455, https://doi.org/10.1038/ng.2536 (2013).
Weyrich, L. S. et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544, 357–361, https://doi.org/10.4225/55/584775546a409 (2017).
Warinner, C. et al. Pathogens and host immunity in the ancient human oral cavity. Nature genetics 46, 336–344, https://doi.org/10.1038/ng.2906 (2014).
Santiago-Rodriguez, T. M., Narganes-Storde, Y., Chanlatte-Baik, L., Toranzos, G. A. & Cano, R. J. Insights of the dental calculi microbiome of pre-Columbian inhabitants from Puerto Rico. PeerJ 5, e3277, https://doi.org/10.7717/peerj.3277 (2017).
Nasidze, I. et al. High diversity of the saliva microbiome in Batwa Pygmies. PLoS One 6, e23352, https://doi.org/10.1371/journal.pone.0023352 (2011).
Contreras, M. et al. The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology 156, 3282–3287, https://doi.org/10.1099/mic.0.043174-0 (2010).
Ozga, A. T. et al. Oral microbiome diversity among Cheyenne and Arapaho individuals from Oklahoma. American Journal of Physical Anthropology, https://doi.org/10.1002/ajpa.23033 (2016).
Li, J. et al. The saliva microbiome of Pan and Homo. BMC Microbiology 13, 204–204, https://doi.org/10.1186/1471-2180-13-204 (2013).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180, https://doi.org/10.1038/nature09944 (2011).
Moeller, A. H. et al. Chimpanzees and Humans Harbor Compositionally Similar Gut Enterotypes. Nature communications 3, 1179–1179, https://doi.org/10.1038/ncomms2159 (2012).
Moeller, A. H. et al. Stability of the Gorilla Microbiome Despite SIV Infection. Molecular ecology 24, 690–697, https://doi.org/10.1111/mec.13057 (2015).
Costea, P. I. et al. Enterotypes in the landscape of gut microbial community composition. Nature Microbiology 3, 8–16, https://doi.org/10.1038/s41564-017-0072-8 (2018).
van Lawick-Goodall, J. The behaviour of free-living chimpanzees in the Gombe Stream Reserve. Animal Behaviour Monographs 1, 161–311 (1968).
Wrangham, R. W. In Primate Ecology: Studies of feeding and ranging behaviour in lemurs, monkeys and apes (ed. Clutton-Brock, T. H.) Ch. 17, 503–538 (Academic Press, 1977).
Mann, A. E. et al. Differential preservation of endogenous human and microbial DNA in dental calculus and dentin. Scientific Reports 8, 9822, https://doi.org/10.1038/s41598-018-28091-9 (2018).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature Methods 12, 902–903, https://doi.org/10.1038/nmeth.3589 (2015).
Willmann, C. et al. Oral health status in historic population: Macroscopic and metagenomic evidence. PLoS ONE 13, e0196482, https://doi.org/10.1371/journal.pone.0196482 (2018).
Eisenhofer, R. & Weyrich, L. S. Assessing alignment-based taxonomic classification of ancient microbial DNA. PeerJ 7, e6594–e6594, https://doi.org/10.7717/peerj.6594 (2019).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645, https://doi.org/10.1101/gr.092759.109 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Ginolhac, A., Rasmussen, M., Gilbert, M. T., Willerslev, E. & Orlando, L. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27, https://doi.org/10.1093/bioinformatics/btr347 (2011).
Jonsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. & Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684, https://doi.org/10.1093/bioinformatics/btt193 (2013).
Signat, B., Rogues, C., Poulet, P. & Duffaut, D. Fusobacterium nucleatum in periodontal health and disease. Current issues in molecular biology 13, 25–36 (2011).
Han, Y. W. Fusobacterium nucleatum: a commensal-turned pathogen. Current opinion in microbiology 0, 141–147, https://doi.org/10.1016/j.mib.2014.11.013 (2015).
Dashper, S. G., Seers, C. A., Tan, K. H. & Reynolds, E. C. Virulence factors of the oral spirochete Treponema denticola. Journal of dental research 90, 691–703, https://doi.org/10.1177/0022034510385242 (2011).
Liljemark, W. F. et al. Distribution of oral Haemophilus species in dental plaque from a large adult population. Infection and immunity 46, 778–786 (1984).
Bik, E. M. et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J 4, 962–974, https://doi.org/10.1038/ismej.2010.30 (2010).
King, P. Haemophilus influenzae and the lung (Haemophilus and the lung). Clinical and translational medicine 1, 10–10, https://doi.org/10.1186/2001-1326-1-10 (2012).
Abranches, J. et al. Biology of Oral Streptococci. Microbiology Spectrum 6, https://doi.org/10.1128/microbiolspec.GPP3-0042-2018 (2018).
Liu, G., Tang, C. M. & Exley, R. M. Non-pathogenic Neisseria: members of an abundant, multi-habitat, diverse genus. Microbiology 161, 1297–1312, https://doi.org/10.1099/mic.0.000086 (2015).
Mongodin, E. F. et al. Secrets of Soil Survival Revealed by the Genome Sequence of Arthrobacter aurescens TC1. PLOS Genetics 2, e214, https://doi.org/10.1371/journal.pgen.0020214 (2006).
Huang, Y. et al. Arthrobacter scleromae sp. nov. isolated from human clinical specimens. Journal of clinical microbiology 43, 1451–1455, https://doi.org/10.1128/JCM.43.3.1451-1455.2005 (2005).
Han, Y. W. et al. Interactions between Periodontal Bacteria and Human Oral Epithelial Cells: Fusobacterium nucleatum Adheres to and Invades Epithelial Cells. Infection and immunity 68, 3140–3146, https://doi.org/10.1128/iai.68.6.3140-3146.2000 (2000).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108, https://doi.org/10.1126/science.1208344 (2011).
Ou, J. et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. The American Journal of Clinical Nutrition 98, 111–120, https://doi.org/10.3945/ajcn.112.056689 (2013).
Huse, S. M., Ye, Y., Zhou, Y. & Fodor, A. A. A Core Human Microbiome as Viewed through 16S rRNA Sequence Clusters. PLoS ONE 7, e34242, https://doi.org/10.1371/journal.pone.0034242 (2012).
Knights, D. et al. Rethinking “Enterotypes”. Cell host & microbe 16, 433–437, https://doi.org/10.1016/j.chom.2014.09.013 (2014).
Sundqvist, G. & Figdor, D. Life as an endodontic pathogen. Endodontic Topics 6, 3–28, https://doi.org/10.1111/j.1601-1546.2003.00054.x (2003).
Arora, N., Mishra, A. & Chugh, S. Microbial role in periodontitis: Have we reached the top? Some unsung bacteria other than red complex. Journal of Indian Society of Periodontology 18, 9–13, https://doi.org/10.4103/0972-124x.128192 (2014).
Legge, S. S. Dentoalveolar abscess variation among three groups of chimpanzees (Pan troglodytes schweinfurthii, Pan troglodytes troglodytes, and Pan paniscus). International Journal of Paleopathology 2, 1–9, https://doi.org/10.1016/j.ijpp.2012.06.002 (2012).
Lowenstine, L. J., McManamon, R. & Terio, K. A. Comparative Pathology of Aging Great Apes: Bonobos, Chimpanzees, Gorillas, and Orangutans. Veterinary Pathology 53, 250–276, https://doi.org/10.1177/0300985815612154 (2016).
Frandsen, E. V. G., Poulsen, K., Curtis, M. A. & Kilian, M. Evidence of Recombination in Porphyromonas gingivalis and Random Distribution of Putative Virulence Markers. Infection and immunity 69, 4479–4485, https://doi.org/10.1128/iai.69.7.4479-4485.2001 (2001).
Rôças, I. N., Siqueira, J. F. Jr., Santos, K. R. N., Coelho, A. M. A. & de Janeiro, R. “Red complex” (Bacteroides forsythus, Porphyromonas gingivalis, and Treponema denticola) in endodontic infections: A molecular approach. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontics 91, 468–471, https://doi.org/10.1067/moe.2001.114379 (2001).
Ximenez-Fyvie, L. A., Haffajee, A. D. & Socransky, S. S. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol 27, 648–657, https://doi.org/10.1034/j.1600-051x.2000.027009648.x (2000).
Matsushita, K. et al. The novel association between red complex of oral microbe and body mass index in healthy Japanese: a population based cross-sectional study. Journal of Clinical Biochemistry and Nutrition 57, 135–139, https://doi.org/10.3164/jcbn.15-19 (2015).
Savitt, E. D. & Kent, R. L. Distribution of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis by Subject Age. Journal of periodontology 62, 490–494, https://doi.org/10.1902/jop.1991.62.8.490 (1991).
Umeda, M. et al. Risk Indicators for Harboring Periodontal Pathogens. Journal of periodontology 69, 1111–1118, https://doi.org/10.1902/jop.1998.69.10.1111 (1998).
Papaioannou, W. et al. The microbiota on different oral surfaces in healthy children. Oral microbiology and immunology 24, 183–189, https://doi.org/10.1111/j.1399-302X.2008.00493.x (2009).
Darveau, R. P. The Oral Microbial Consortium’s Interaction with the Periodontal Innate Defense System. DNA and Cell Biology 28, 389–395, https://doi.org/10.1089/dna.2009.0864 (2009).
Zijnge, V. et al. Oral Biofilm Architecture on Natural Teeth. PLoS One 5, e9321, https://doi.org/10.1371/journal.pone.0009321 (2010).
Könönen, E., Kanervo, A., Takala, A., Asikainen, S. & Jousimies-Somer, H. Establishment of Oral Anaerobes during the First Year of Life. Journal of dental research 78, 1634–1639, https://doi.org/10.1177/00220345990780100801 (1999).
Byrne, S. J. et al. Progression of chronic periodontitis can be predicted by the levels of Porphyromonas gingivalis and Treponema denticola in subgingival plaque. Oral microbiology and immunology 24, 469–477, https://doi.org/10.1111/j.1399-302X.2009.00544.x (2009).
Kirakodu, S., Chen, J., Gonzalez Martinez, J., Gonzalez, O. A. & Ebersole, J. Microbiome Profiles of Ligature-Induced Periodontitis in Nonhuman Primates Across the Lifespan. Infection and immunity, IAI.00067–00019, https://doi.org/10.1128/iai.00067-19 (2019).
Velsko, I. M. et al. Microbial differences between dental plaque and historic dental calculus are related to oral biofilm maturation stage. Microbiome 7, 102, https://doi.org/10.1186/s40168-019-0717-3 (2019).
Tribble, G. D. et al. Natural Competence Is a Major Mechanism for Horizontal DNA Transfer in the Oral Pathogen Porphyromonas gingivalis. mBio 3, https://doi.org/10.1128/mBio.00231-11 (2012).
Tribble, G. D., Kerr, J. E. & Wang, B.-Y. Genetic diversity in the oral pathogen Porphyromonas gingivalis: molecular mechanisms and biological consequences. Future microbiology 8, 607–620, https://doi.org/10.2217/fmb.13.30 (2013).
Acuña-Amador, L., Primot, A., Cadieu, E., Roulet, A. & Barloy-Hubler, F. Genomic repeats, misassembly and reannotation: a case study with long-read resequencing of Porphyromonas gingivalis reference strains. BMC Genomics 19, 54, https://doi.org/10.1186/s12864-017-4429-4 (2018).
White, F. J. & Wrangham, R. W. Feeding competition and patch size in the chimpanzee species Pan paniscus and Pan troglodytes. Behaviour 105, 148–164 (1988).
Goodall, J. The Chimpanzees of Gombe: Patterns of Behavior. (Harvard University Press, 1986).
Charlier, P., Gaultier, F. & Héry-Arnaud, G. Interbreeding between Neanderthals and modern humans: Remarks and methodological dangers of a dental calculus microbiome analysis. Journal of Human Evolution 126, 124–126, https://doi.org/10.1016/j.jhevol.2018.06.007 (2018).
Kirchhoff, C. A. et al. Infanticide in chimpanzees: Taphonomic case studies from Gombe. American Journal of Physical Anthropology 165, 108–122, https://doi.org/10.1002/ajpa.23335 (2018).
Meyer, M. & Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harbor Protocols 2010, pdb.prot5448, https://doi.org/10.1101/pdb.prot5448 (2010).
Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic acids research 40, e3, https://doi.org/10.1093/nar/gkr771 (2012).
SeqPrep, https://github.com/jstjohn/SeqPrep.
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature Methods 9, 811, https://doi.org/10.1038/nmeth.2066 (2012).
Tung, J. et al. Social networks predict gut microbiome composition in wild baboons. eLife 4, e05224, https://doi.org/10.7554/eLife.05224 (2015).
Herbig, A. et al. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv, https://doi.org/10.1101/050559 (2016).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Research 17, 377–386, https://doi.org/10.1101/gr.5969107 (2007).
Towns, J. et al. XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering 16, 62–74, https://doi.org/10.1109/MCSE.2014.80 (2014).
R: A Language and Environment for Statistical Computing (R Foundationg for Statistical Computing, Vienna, Austria, 2014).
Naito, M. et al. Determination of the Genome Sequence of Porphyromonas gingivalis Strain ATCC 33277 and Genomic Comparison with Strain W83 Revealed Extensive Genome Rearrangements in P. gingivalis. DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes 15, 215–225, https://doi.org/10.1093/dnares/dsn013 (2008).
Schubert, M. et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nature Protocols 9, 1056, https://doi.org/10.1038/nprot.2014.063 (2014).
Geneious_9, https://www.geneious.com, https://www.geneious.com
Honap, T. P. et al. Mycobacterium leprae genomes from naturally infected nonhuman primates. PLoS neglected tropical diseases 12, e0006190, https://doi.org/10.1371/journal.pntd.0006190 (2018).
PicardTools (2018).
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P. & Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research 21, 487–493, https://doi.org/10.1101/gr.113985.110 (2011).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford, England) 27, 2987–2993, https://doi.org/10.1093/bioinformatics/btr509 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
We acknowledge members of the Gombe research team including Anthony Collins, Dismas Mwacha, Deus Mjungu, and Joel Bray for their assistance in sample collection and dental documentation. We thank Dr. Jane Goodall for her contributions at Gombe and the initial preservation of these samples. We also acknowledge Ana Duggan for her assistance with the genome visualization software Circos and Claudio Ottoni for his helpful comments on the manuscript. We would also like to thank the reviewers for their insights into improving this manuscript. Funding for this research has been provided by the Strategic Initiative Funds, Office of the President, Arizona State University to the Institute of Human Origins’ DNA and Human Origins at Arizona State University project. Funding was also provided to A.T.O. by the Center for Evolution and Medicine postdoctoral fellowship at Arizona State University. Funding was also provided by The Leakey Foundation. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. The authors also acknowledge Research Computing at Arizona State University for providing resources contributing to the analysis and storage of results reported within this manuscript.
Author information
Authors and Affiliations
Contributions
A.T.O. planned the project. I.G., A.P., M.L.W. and R.S.N. provided the samples. A.T.O. carried out laboratory experiments and analyzed the data. A.C.S. provided partial funding and laboratory space/equipment. A.T.O. wrote the manuscript with critical input from A.C.S. and additional input from all other coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ozga, A.T., Gilby, I., Nockerts, R.S. et al. Oral microbiome diversity in chimpanzees from Gombe National Park. Sci Rep 9, 17354 (2019). https://doi.org/10.1038/s41598-019-53802-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53802-1
This article is cited by
-
Characterization of the Oral Microbiota in Captive Sichuan Golden Snub-nosed Monkeys (Rhinopithecus roxellana) of Different Ages
International Journal of Primatology (2023)
-
Comparative analysis of oral-gut microbiota between captive and wild long-tailed macaque in Thailand
Scientific Reports (2021)
-
Metagenomic analysis of dental calculus in ancient Egyptian baboons
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
