
Abstract
The World Health Organization recently listed snakebite envenoming as a Neglected Tropical Disease, proposing strategies to significantly reduce the global burden of this complex pathology by 2030. In this context, effective adjuvant treatments to complement conventional antivenom therapy based on inhibitory molecules for specific venom toxins have gained renewed interest. Varespladib (LY315920) is a synthetic molecule clinically tested to block inflammatory cascades of several diseases associated with elevated levels of secreted phospholipase A2 (sPLA2). Most recently, Varespladib was tested against several whole snake venoms and isolated PLA2 toxins, demonstrating potent inhibitory activity. Herein, we describe the first structural and functional study of the complex between Varespladib and a PLA2-like snake venom toxin (MjTX-II). In vitro and in vivo experiments showed this compound’s capacity to inhibit the cytotoxic and myotoxic effects of MjTX-II from the medically important South American snake, Bothrops moojeni. Crystallographic and bioinformatics analyses revealed interactions of Varespladib with two specific regions of the toxin, suggesting inhibition occurs by physical blockage of its allosteric activation, preventing the alignment of its functional sites and, consequently, impairing its ability to disrupt membranes. Furthermore, based on the analysis of several crystallographic structures, a distinction between toxin activators and inhibitors is proposed.
Similar content being viewed by others
Introduction
Venomous snakes of medical importance are widely distributed in tropical and sub-tropical countries from Africa, Asia, Latin-America and Oceania, being responsible for an estimated 420,000 to 1,800,000 envenomings every year1,2. The most affected victims are usually agricultural workers from rural areas, hunters and children who do not have adequate protective clothing, secure housing or education about other protective measures that can help prevent snakebites3. In 2017, the World Health Organization (WHO) added snakebite envenoming to its list of neglected tropical diseases, with a focus on strategies to reduce the burden and control the effects of these envenomings4,5. Further, toxin-specific treatments that could be administered at the time of a bite, or where conventional serum therapies are not adequately effective, remain elusive6.
In Latin America, Bothrops species are responsible for the majority of snakebite envenomings, followed by Crotalus species7,8,9. Accidents involving the former are characterized by drastic local effects, often due to the action of myotoxic proteins causing muscle necrosis and, in severe cases, tissue loss, or even limb amputation and disability of the victim10,11,12.
Venoms from Bothrops snakes are composed of a set of proteins that have diversified functions13,14,15. Among venom components, several variants of secreted phospholipases A2 (PLA2s) are common in these venoms. Asp49-PLA2s display catalytic activity, and the basic variants are typically myotoxic, in contrast to their acidic counterparts which generally lack myotoxic activity. On the other hand, the Lys49-PLA2-like proteins lack catalytic activity, but induce myotoxicity. By acting in synergy between themselves16 and with proteinases17, myotoxic Asp49-PLA2s and Lys49-PLA2-like proteins are the main venom components responsible for local myonecrosis in Bothrops envenomings. Asp49-PLA2 toxins are calcium-dependent small proteins (~13 kDa) with secondary structure composed by three α-helices, one anti-parallel β-sheet and a calcium binding loop18,19. Their myotoxic activity depends on the hydrolysis of membrane phospholipids, as chemical modification of the catalytic His48 abolishes both catalysis and myotoxicity16. On the other hand, Lys49-PLA2-like myotoxins share the same general scaffold as Asp49-PLA2s, but display myotoxicity in the absence of catalysis. A hypothesis on the mechanism of myotoxicity induced by Lys49-PLA2-like toxins was recently presented, and involves several steps. It is proposed that initiation occurs by the binding of a fatty acid into the hydrophobic channel of the toxin, followed by allosteric activation and alignment of functional sites. These functional sites interact with the cell membrane via a membrane docking site (MDoS), and by the disruption of membrane integrity by the action of a membrane disrupting site (MDiS), ultimately resulting in cell necrosis20,21,22.
The cornerstone for the treatment of snakebite envenomings is antivenom administration, but scarce availability and considerable costs frequently limit the access to these life-saving antidotes in many impoverished rural areas of the world12. Moreover, since antivenoms have to be administered in health facilities, there is often a delay in antivenom infusion owing to the usual difficulties of patients to reach these facilities in many rural settings. In search of alternative and effective adjuvant treatments to complement the conventional antivenom therapy, in vitro and in vivo studies have tested a number of inhibitors against diverse crude venoms, or isolated toxins such as PLA2s23,24,25,26,27,28,29,30,31,32, monoclonal antibodies33,34,35,36 and synthetic molecules37,38,39,40,41,42,43,44,45,46,47,48. Ideally, these novel antidotes could be used in the field rapidly after the onset of envenoming, hence halting the deleterious action of venom toxins in the tissues. In order to understand how these inhibitors block the action of toxins, protein crystallography has been employed as a powerful tool to understand the inhibitory mechanisms of a variety of small ligands toward PLA2 toxins6,21,41,44,45,47,49,50.
Among a wide variety of molecules capable of inhibiting PLA2 enzymes51,52, one potent inhibitor of human secreted group IIA PLA2s is Varespladib (LY315920)53. This synthetic molecule was developed and clinically tested for the purpose of blocking inflammatory cascades of several diseases associated with elevated sPLA2 levels such as rheumatoid arthritis, sepsis and acute coronary syndrome54. Partly on the basis of homology between the human group IIA PLA2 and PLA2 toxins found in snake venoms, Varespladib was tested against a large panel of whole venoms from medically important snakes from different continents and potent inhibition of their PLA2 activity was found42. Inhibition has been also studied using several isolated PLA2 toxins, including a myotoxin isolated from the venom of Bothrops asper48.
In this communication, we describe the crystal structure of MjTX-II, a PLA2-like toxin isolated from Bothrops moojeni55,56 co-crystallized with Varespladib, revealing two inhibitor molecules interacting with the hydrophobic channel of the dimeric assembly of this toxin, and a comprehensive analysis of other crystal structures of bothropic PLA2-like toxins/inhibitor complexes using bioinformatics approaches. Furthermore, we performed in vitro and in vivo studies to assess the inhibition of toxic effects of MjTX-II by Varespladib. Taken together, the data presented hereby provide a molecular basis to understand such inhibition. This comparative analysis of crystallographic structures of PLA2-like toxins/inhibitors contributes to organize and classify the different inhibition models for toxic effects of PLA2-like toxins by different molecules into three main classes.
Results
Varespladib inhibits the in vivo myotoxicity and in vitro cytotoxicity of MjTX-II
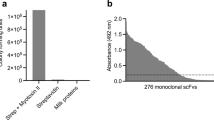
As typical of Lys49 PLA2-like toxins, the intramuscular injection of 50 µg of MjTX-II in mice caused a prominent elevation of plasma creatine kinase activity, indicative of skeletal muscle necrosis (Fig. 1A). This increment was reduced by nearly 50% when the toxin was preincubated with Varespladib, a statistically significant (p < 0.05) difference. Preincubation with Varespladib was also able to significantly inhibit the cytotoxic action of MjTX-II on C2C12 myoblasts in vitro, reducing the release of lactate dehydrogenase to the medium by nearly 75% in comparison to the effect of toxin alone (Fig. 1B).

Myotoxic and cytotoxic activities of MjTX-II in mice and cultured C2C12 myoblasts, respectively, and their inhibition by preincubation with Varespladib. (A) Mice were injected by intramuscular route with toxin alone (50 μg, in 100 μL of PBS), or preincubated for 15 min with Varespladib (VAR) at a final concentration of 400 μM. Control groups were injected with PBS alone or Varespladib alone, respectively. After 3 hr, blood was obtained and the plasma creatine kinase (CK) activity was determined, as described in Methods. Each bar represents the mean ± SD of 4–5 mice per group. (B) Cells were exposed to the toxin alone (20 μg, in 150 μL of medium), or preincubated for 15 min with Varespladib (VAR) at a final concentration of 400 μM. After incubating the cells for 3 hr at 37 °C, an aliquot of supernatant was assayed for lactate dehydrogenase (LDH) activity, as described in Methods. Cytotoxicity is expressed as percentage, considering the LDH activity of cells exposed to medium with 0.1% Triton X-100, or to medium alone, as 100% and 0%, respectively. Each bar represents the mean ± SD of three replicates. Statistically significant (p < 0.05) differences between values obtained with the toxin alone or the toxin preincubated with Varespladib are indicated by an asterisk.
Crystallographic structure of MjTX-II/Varespladib
MjTX-II/Varespladib crystallographic structure has similar folding compared to group II PLA2s proteins previously solved, showing a dimeric configuration with seven disulfide bridges in each protomer18,19.
MjTX-II/Varespladib crystals belong to P21 space group and diffracted up to 1.75 Å resolution. Refined data converge to an Rcryst of 22.42% (Rfree = 23.73%) with a dimeric final model composed of two Varespladib molecules, two DMSO molecules and 93 solvent molecules (Fig. 2). Due to the lack of electron density, atoms from side chains of following residues are not modeled in chain A: Lys7, Lys16, Lys53, Lys69, Lys78, Lys80, Lys116, Lys128, and in chain B: Lys7, Lys36, Lys53, Lys69, Lys70, Lys78, Lys128 and Lys129.

Crystal structure of the complex MjTX-II/Varespladib. (A) The overall structure of the complex is depicted as cartoon representation (cyan) and the inhibitor molecules are represented as sticks (yellow). (B) Omit electron density map (coefficients 2|Fobs| − |Fcalc|) corresponding to Varespladib bound to monomer A and (C) Varespladib bound to monomer B. The maps corresponding to inhibitor molecules are contoured at 1.0 σ.
Refinement statistics information for complex crystal structure is described in Table 1. The final model of the complex MjTX-II/Varespladib was deposited in RCSB PDB data bank, under the code 6PWH.
Molecular dynamics simulations
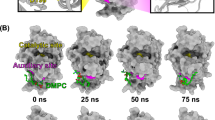
The MjTX-II/Varespladib complex presented a high stability as shown by MD simulations, evidenced by the RMSD calculations of MjTX-II backbone and Varespladib non-hydrogen atoms which present average values of 1.94 ± 0.27 Å and 1.76 ± 0.18 Å, respectively (Fig. 3A). In agreement with this, RMSF calculations of the backbone atoms of MjTX-II showed values below 5 Å (Fig. 3B).

Molecular dynamic simulation of the MjTX-II/Varespladib complex. (A) RMSD for MjTX-II backbone atoms (black line) and Varespladib non-hydrogen atoms (red line). (B) Backbone atoms RMSF for MjTX-II chains A (dark blue line) and B (cyan line).
Table 2 shows the prevalence of contacts of the toxin’s residues with Varespladib molecules below 4.5 Å. The majority of the residues presented interactions above 80% of the time simulation, except residues Gly32 and Tyr52 from both monomers that interacted between 50% and 80% of the time simulation. Furthermore, Tyr121 residue of both monomers and Leu122 of monomer B presented the lowest values, interacting with Varespladib less than 20% of the time simulation.
It is interesting to note that Varespladib molecules interacted with residues considered important for the myotoxic activity of this class of proteins, including Helix-I, MDiS, and the hydrophobic channel. The Helix-I residues Leu2 and Leu5 from both monomers were in contact with Varespladib molecules during the entire simulation time, while the Phe126 residue of both monomers interacted almost 100% of the time. Furthermore, Varespladib molecules showed a maximum percentage interaction with the His48 residues from both monomers of the MjTX-II. The binding of Varespladib to MjTX-II partially buried its MDiS region, as shown by the comparison between SASA (solvent-accessible surface area) values of MjTX-II/fatty acid (MjTX-II/myristic acid, PDB id 6B80) (1040.76 Å2) and MjTX-II/Varespladib (834.56 Å2) structures. In addition, the stability of the complex was also evidenced by the favorable ΔG of −44.98 ± 7.17 kcal/mol predicted by MM-PBSA calculation.
Discussion
Inhibition of PLA2-like toxins by varespladib
Varespladib has been reported to inhibit the enzymatic and toxic actions of a variety of snake venom PLA2s42,57, including a number of enzymes that display myotoxic activity48. Varespladib was able to reduce muscle damage when preincubated with the myotoxic enzymes before injection, as well as when the drug was administered soon after enzyme injection48. However, no information was available until now on the possible inhibition of catalytically-inactive PLA2-like myotoxins by this synthetic compound. In the present study, functional assays performed in mice and in cell culture demonstrated that Varespladib interacts with MjTX-II leading to a significant inhibition of its myotoxic and cytotoxic effects. In the absence of catalytic activity by MjTX-II, this finding suggests that Varespladib is able to interfere with the interaction of MjTX-II with the membrane or that it reduces its ability to disrupt plasma membrane integrity in muscle cells.
Varespladib appeared to be more effective in neutralizing the cytotoxic effect in vitro (75%) than the myotoxic action in vivo (50%), at the same inhibitor concentration (400 µM, selected from previous studies on the inhibition of catalytically-active PLA2s)48. It is likely that MjTX-II has a higher affinity for its target on mature muscle cells, compared to the myoblast cell line in culture, since an increase in susceptibility to the action of Lys49-PLA2-like myotoxins has been previously demonstrated to occur during the differentiation of the C2C12 myogenic cell line58. Therefore, differences in the affinity of MjTX-II to membrane sites in mature muscle cells in vivo and myoblasts in vitro may explain the inhibition results obtained. Our observations for inhibition of myotoxicity by Varespladib led us to focus on elucidating the molecular basis of this neutralizing interaction by using co-crystallization and MD simulation approaches, which are valuable tools to explore the mechanisms of toxicity by PLA2-like proteins, and to unravel the diverse modes of inhibition exerted by different small compounds. In the light of the high abundance in many viperid snake venoms of PLA2-like toxins devoid of enzymatic activity, but capable of inducing myonecrosis, our findings underscore that Varespladib is not only effective in the inhibition of catalytically-active toxic PLA2s, but also of these PLA2-like proteins, hence expanding the potential therapeutic usefulness of this inhibitor.
Structural evidences for inhibition of PLA2-like toxins by varespladib
A mechanism to explain the myotoxic activity of Lys49 PLA2-like toxins was proposed recently. This model underscores the importance of interaction of fatty acid molecules in the hydrophobic channel for the initial steps of the mechanism. Essentially, after fatty acids location into the hydrophobic channel, the monomers are reoriented, resulting in toxin activation and, subsequently, the toxin docks and disrupts the integrity of the plasma membrane20,21,59. In this type of toxin, membrane disruption is completely independent of phospholipids enzymatic hydrolysis.
The crystal structure of the complex MjTX-II/Varespladib reveals the presence of inhibitor molecules in the hydrophobic channel of the toxin (Fig. 4), interacting particularly with His48 and Lys49 residues. The structural importance of these residues was also observed by MD simulation, in which they were found to interact with the ligand during almost the whole time (Table 2). Therefore, binding of Varespladib into the toxin’s hydrophobic channel is likely to prevent the membrane’s fatty acids binding and, consequently, precludes the structural alignment of the functional MDoS and MDiS regions, resulting in reduction of toxicity. Another structural feature observed was the particular dimeric assembly of MjTX-II/Varespladib when compared to other PLA2-like toxins. The superposition between dimeric proteins reveals a high distortion in their dimeric assembly, expressed by the high value of the Euler roll angle21 and RMSD values (Table 3). When compared to other PLA2-like toxins, the distorted conformation observed in the crystal structure of MjTX-II/Varespladib is also observed in the crystal structure of MjTX-II/rosmarinic acid (MjTXII/RA) and MjTX-II/acetylsalicylic acid (MjTX-II/ASA)50, suggesting that this structural conformation may be related to the inactive structure of MjTX-II and also shedding light on how the inhibitors can influence the structure of the toxin.

Schematic representation of the interaction of Varespladib molecules with MjTX-II. (A) Interaction of Varespladib with monomer A. (B) Contacts of Varespladib with monomer B. The figure was made using Lidia extension from Coot v.0.8.967.
Previously, MD simulations using the complexes MjTX-II/RA and MjTX-II/ASA showed that the distorted quaternary structural conformation of MjTX-II has some structural aspects that might be related to the myotoxic activity. The inhibitor RA remained bound to MjTX-II MDiS after 100 ns of MD simulation, preventing the access of fatty acids to the hydrophobic channel. In contrast, the ASA molecules showed an unstable interaction with MjTX-II, and left the hydrophobic channel of the toxin rapidly, allowing the further interaction of fatty acids molecules to the binding site50. In the present work, MD simulation of the MjTX-II/Varespladib complex showed high stability (Fig. 3A) and affinity of the inhibitor in the binding site on hydrophobic channel (Table 2), which suggests that Varespladib prevents the interaction of fatty acids with the toxin by competitive inhibition, and also preserves the distorted structure of the toxin.
The comparison of ΔG values obtained by MD simulations for MjTX-II/Varespladib and other two MjTX-II/inhibitors complexes from the three different classes (RA and suramin inhibitors - Table 3) also revealed interesting results. Although ΔG values for Varespladib and RA are in the same order of magnitude (−17.16 ± 11.67 and −44.98 ± 7.17 kcal/mol, respectively for RA e Veraspladib), the lower value for Varespladib is according to structural environment of both inhibitors. RA is partially exposed to solvent and binds superficially to MjTX-II while Varespladib binds into the hydrophobic channel of the toxin, indicating that Varespladib is an efficient inhibitor candidate for PLA2s-like toxins. Indeed, the very low ΔG value of MjTX-II/Suramin complex (−148.16 +/− 14.64 kcal/mol) reflects the large number of interactions and between the highly charged inhibitor and the MjTX-II.
The crystallographic structure and MD simulations analyses show that Varespladib interacts with the same site as fatty acids. Thus, the question emerges on how these molecules can bind to this site and induce contrasting effects on MjTX-II? The answer may be associated with the length of the bound molecule. Fatty acids are long chain molecules, where their tails can interact with residues from Helix-I (residues from 1 to 10) and residues from MDiS (Leu122 and Phe126 residues), hence filling the entire hydrophobic channel, and collaborating with the alignment and exposure of this functional site to the solvent (Fig. 5). On the other hand, Varespladib maintains strong interactions with the same regions (Helix-I: Leu2, Leu5, Gly6 and Ile9 residues; MDiS: Leu122 and Phe126 residues; and residues His48 and Lys49), as observed in the crystal structure (Fig. 2) and MD simulations (Table 2), occupying the hydrophobic channel internally (Fig. 5). The interaction of Varespladib with residues of the hydrophobic channel forces MDiS to remain in contact with the toxin, favoring the distorted conformation. Indeed, SASA of the MDiS region of the MjTX-II/Varespladib is about 20% lower compared to the MjTX-II/myristic acid structure (PDB id 6B80). Therefore, Varespladib may inhibit the myotoxin by two different mechanisms: (i) hydrophobic channel blockage and (ii) preventing the MDiS exposure to the solvent.

Interaction of Varespladib and myristic acid (FA14) with side chain of residues from Helix-I, hydrophobic channel and MDiS residues. (A) Overall crystal structure of MjTX-II/Varespladib (cyan), zoomed region of contacts of inhibitor to side chain of aminoacids from protein and the same region after 90° of rotation. (B) Overall of crystal structure of MjTX-II/FA14 (green), zoomed region from the interaction of fatty acid molecule with the protein and the same region after 90° of rotation.
PLA2-like toxins inhibitors
The activation of PLA2-like toxins involves important structural changes in their oligomeric structures with particular features after the binding of fatty acids into the solvent-exposed hydrophobic channel: (i) alignment of the membrane docking site (MDoS), where the distance between positive clusters that comprise the MDoS (Lys16, Lys20, Lys115 and Arg118) from both monomers must be ≤16 Å; (ii) Euler Roll angle >160° and; (iii) changing the distance of the MDiS hydrophobic residues Leu121 (122 for MjTX-II) and Phe125 ≤ 5 Å21,60. These structural features were observed in recent studies by analyses of several crystallographic structures of PLA2-like toxins in native form and bound with inhibitory molecules6,25,37,41,44,45,46,49,50,61.
Crystallographic structures of PLA2-like toxins isolated from Bothrops snake venoms in complex with different inhibitory ligands showed that these inhibitors may interact with the toxin in distinct regions (Table 3). Basically, three regions were identified, which coincidentally are involved in the myotoxic effects according to the structural studies: hydrophobic channel, MDoS, and MDiS regions20,21,37,45. To date, eight different inhibitors have been studied in complex with different bothropic PLA2-like toxins. Their binding region(s) are next described: (a) bound into the hydrophobic channel (bound to His48): p-bromophenacyl bromide (BPB)41,61, Zinc ions37, Suramin44 and Varespladib (present work); (b) bound in the entrance of hydrophobic channel: Chicoric acid25 and Rosmarinic acid49; (c) bound to the membrane disrupting site (MDiS): Zinc ions37, Aristolochic acid6, Chicoric acid25 and Rosmarinic acid50; (d) bound with MDoS residues: Caffeic acid6; (e) bound simultaneously to MDoS and MDiS regions and also induces protein oligomerization: Suramin45,46.
In summary, different inhibitors may bind to different regions of PLA2-like toxins of Bothrops sp snake venoms but may be classified into three different classes related to different mechanisms of inhibition: (1) Inhibitors that block the access to the hydrophobic channel for fatty acid molecules (inhibitors a and b, previous paragraph), impairing the natural movement between the monomers of the dimeric structure; (2) inhibitors that bind to functional sites of the proteins and block MDoS and MDiS interaction with cell membrane (inhibitors c and d), and (3) inhibitors that can induce protein oligomerization, leading to the combination of the previous mechanisms (inhibitor e) (Fig. 6; Table 3).

Different forms of inhibition of Lys49 PLA2-like toxins by inhibitors described in literature. The monomer A is showed as light gray and the monomer B is represented as dark gray (A) Class 1 inhibitors (yellow), which prevent the binding of fatty acids (yellow sticks - activator) to hydrophobic channel (cyan) of toxin. (B) Class 2 inhibitors (blue) which bind to functional MDoS (green) and MDiS (red) and prevent the interaction of toxin to membrane (orange). (C) Class 3 inhibitors (magenta) which induce protein oligomerization.
Conclusion
We have described functional and structural studies involving a toxin isolated from Bothrops moojeni venom, MjTX-II, and the inhibitor Varespladib. The results obtained by in vitro and in vivo experiments reveal that this synthetic compound is able to inhibit the myotoxic and cytotoxic effects of MjTX-II, a well characterized member of the Lys49-PLA2-like myotoxins, widespread among many crotaline snake species. Given that the safety of Varespladib has been already confirmed in clinical trials, present results strengthen the importance of this compound as a repurposed drug candidate to aid in the treatment of snakebite envenomings. The crystal structure and MD simulations described the interaction of Varespladib with a site similar to that of fatty acids binding and with the MDiS site, suggesting that the inhibition of MjTX-II by Varespladib is due to physical blockage of the interaction with allosteric activator molecules in the hydrophobic channel, and by restricting the movement of the MDiS of these toxins, preventing the alignment of its functional sites and, consequently, impairing its ability to disrupt the integrity of membranes. Furthermore, based on the crystallographic structure analyses, the distinction between activators and inhibitors is discussed, and the three classes of inhibition mechanisms for PLA2-like proteins are proposed.
Methods
Toxin and varespladib
The isolation of MjTX-II from crude venom of B. moojeni was previously described50,56. Varespladib (compound LY315920) in free acid form was kindly provided by Ophirex. Inc. (original source: ChemieTek, Indianapolis, Indiana, USA; 99.99% pure by NMR and HPLC).
In vivo myotoxicity
Myotoxic activity assays were carried out in groups of five CD-1 mice (18–20 g body weight), which had access to food and water ad libitum, as described previously62. One group of mice was injected in the gastrocnemius muscle with 50 µg of toxin dissolved in 100 µL of phosphate-buffered saline (PBS, pH 7.2; 0.12 M NaCl, 0.04 M sodium phosphate). A mixture of 50 µg of toxin and 400 mM Varespladib (in 4% dimethyl sulfoxide; DMSO) was preincubated for 15 min at room temperature, and subsequently injected in another group of mice under otherwise identical conditions. Control groups of four mice were injected with either 100 µL of PBS, or 100 µL of 4% DMSO in PBS, respectively. After 3 h, blood was collected from the tip of the tail into a heparinized capillary and centrifuged. Then, plasma creatine kinase (CK) activity, expressed in U/L, was determined using a UV kinetic assay (CK-NAC UV, Wiener Lab). Animal experiments were approved by ‘Comité Institucional para el Cuidado y Uso de los Animales (CICUA, permit #084-17), Universidad de Costa Rica. After the experiments, mice were sacrificed by CO2 inhalation. For comparison of mean values from more than two groups, ANOVA was used followed by Tukey-Kramer tests, and differences were considered statistically significant when p < 0.05.
In vitro cytotoxicity on C2C12 myoblasts
Murine C2C12 skeletal muscle myoblast cells (CRL-1772; American Type Culture Collection) were used in cytotoxicity assays, as previously described16,48. The assays were performed using a stock solution of Varespladib dissolved at 10 mM in dimethyl sulfoxide (DMSO). This solution was diluted prior to each assay with the corresponding buffers to a final concentration of 400 µM, decreasing the DMSO concentration to 4%. Control tests performed preliminarily showed that this concentration of DMSO did not result in a cytotoxic effect in the assay system. The C2C12 cell cultures were maintained as undifferentiated myoblasts at subconfluent levels in Dulbecco’s modified Eagle’s medium (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS), L-glutamine and penicillin/streptomycin. Cells (approximately 105/200 µL) were added to each well of a 96-well plate and incubated at 37 °C, with a 7% CO2 humidified atmosphere. When cultures reached 80–90% confluence, myoblasts were incubated with either toxin alone or toxin that had been pre-incubated with Varespladib (400 µM) for 15 min at room temperature. After 3 h of cell exposure at 37 °C, an aliquot of 60 µL of each supernatant was obtained to quantify the activity of lactate dehydrogenase (LDH) release from damaged cells, using a commercial assay (LDH-P UVAA - Wiener Lab). The LDH activity in supernatants of cells exposed to DMEM alone, or DMEM containing 0.1% Triton X-100, was considered as 0% and 100% reference points, respectively. The assays were performed in triplicate cell cultures and the results are presented as mean ± SD. The statistical significance of differences between means of two groups was determined by the Student’s t-test, where values of p < 0.05 were considered significant.
Crystallization and X-ray data collection
Crystallization trials for MjTX-II/Varespladib complex were performed using the purified toxin concentrated up to 10 mg.mL−1 (diluted in 20 mM ammonium bicarbonate pH 8.0) and Varespladib (diluted in 100% DMSO) added to obtain the molar ratio of 1:10 respectively. Crystals of the MjTX-II/Varespladib complex were obtained by conventional hanging drop vapor-diffusion method63 at 291 K from a drop mixture of 0.7 µL of protein, 0.3 µL Varespladib and 1 µL reservoir solution equilibrated against 500 µL reservoir. The reservoir cocktail was composed by 30% v/w polyethylene glycol (PEG) 4000, 0.1 M TrisHCl pH 8.5 and 0.2 M lithium sulfate.
A dataset was obtained using a synchrotron radiation source (MX2 station, Laboratório Nacional de Luz Sincrotron (LNLS), Campinas, Brazil) and a PILATUS 2 M detector (Dectris) using a wavelength of 1.459 Å (at 100 K). The X-ray diffraction data were collected using crystal-to-detector distance of 100 mm, oscillation of 1° per frame resulting in 250 frames. The processing was executed using the HKL2000 v.1.8.4 program package64 as described Table 1.
Structure determination and refinement
The crystal structure of MjTX-II/Varespladib was solved by the molecular replacement method using the program PHASER65 from PHENIX package v.1.1266. The coordinates of the monomer A from MjTX-II (PDB access code 4KF3) was used as search model. The manual modeling and refinement process of the protein, insertion of Varespladib, DMSO and solvent molecules were performed using program Coot v.0.8.967. Structural automated refinement and the general quality check of models were performed using PHENIX package v.1.1266 and MolProbity program (http://molprobity.biochem.duke.edu/)68.
Structural comparative analysis
For the structural comparisons, the structures of the MjTX-II/Varespladib presented here and structures presented in Table 3 were used. Molecular comparisons of structures were performed using Coot v.0.8.967 and PyMOL v.1.8.669 programs. All structural figures were generated using PyMOL v.1.8.669 program.
Molecular dynamics (MD) simulations
MD simulations of MjTX-II in complex with Varespladib were carried out using GROMACS (Groningen Machine for Chemical Simulation) v.5.0.570 under the CHARMM36 force field71. All the initial input parameters and Varespladib topology were generated by CHARMM-GUI webserver72 and the protonation of the residues was set to pH 7.0 determined by PROPKA3 server73. The complex was placed in a cubic box with 5 Å from the farthest atom, solvated with TIP3P water molecules and equilibrated with 100 mM of NaCl. Further, the system was minimized until reaching an energy below 100 kJ/mol/nm using the Steepest Descent algorithm. A 1-ns NVT ensemble was performed generating the velocities randomly according to Maxwell-Boltzmann distribution at temperature of 300 K using the V-rescale thermostat74 followed of a 1-ns NPT ensemble with Berendsen barostat75 at 1 bar. Both steps were performed restraining the backbone and hydrogen atoms of the protein and Varespladib, respectively. A following step of an unrestrained 100 ns NPT step was performed using the Nose-Hoover thermostat76,77 and Parrinello-Rahman barostat78. Short-range cutoffs for electrostatic and Van der Waals interactions were set to 12 Å with a force-switch function from 10 to 12 Å and hydrogen bonds were constrained using LINCS algorithm79.
The binding energy (ΔG) of the MjTX-II/Varespladib complex was predicted using the MM-PBSA method implemented in g_mmpbsa software80, collecting the frames every 500 ps for the last 10 ns of MD simulation. As MjTX-II has many charged residues, the solute dielectric constant (εsolute) was set to 8. In order to compare the ΔG value of Varespladib with other molecules bound to the same toxin, inhibitors from classes 1, 2 and 3 (Table 3) were selected: (i) Rosmarinic Acid (PDB ID: 6MQD)50, and (ii) Suramin (PDB ID: 4YV5)45, then ΔG values were calculated using the same protocol as for Varespladib.
The prevalence of contacts of the MjTX-II residues and Varespladib molecules were calculated selecting the residues that interacted with the ligand below a cutoff of 4.5 Å from the crystallographic structure. Further, these residues were analyzed according to the minimal distance to Varespladib molecules for each frame from MD simulations considering an interaction only when below the cutoff, then these interactions were converted to percentage and classified as weak (<50%), moderate (<80% and >50%) or strong (>80%). Root-Mean-Square Deviation (RMSD), Root-Mean-Square-Fluctuations (RMSF) and Solvent-Accessibly Surface Area (SASA) calculations were performed using built-in tools provided by GROMACS.
Ethical statement
Experiments in mice were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUA), Universidad de Costa Rica (permit #084-17). Animal procedures were in accordance with the guidelines for animal care prepared by the Committee on Care and Use of Laboratory Animal Resources, National Research Council, USA.
References
Kasturiratne, A. et al. The global burden of snakebite: a literature analysis and modelling based on regional estimates of envenoming and deaths. PLoS Med 5, e218 (2008).
Longbottom, J. et al. Vulnerability to snakebite envenoming: a global mapping of hotspots. Lancet 392, 673–684 (2018).
World Health Organization. Snakebite envenoming, http://www.who.int/news-room/fact-sheets/detail/snakebite-envenoming (2018).
World Health Organization. Neglected tropical diseases, http://www.who.int/neglected_diseases/en/ (2018).
Williams, D. J. et al. Strategy for a globally coordinated response to a priority neglected tropical disease: Snakebite envenoming. PLoS Negl Trop Dis 13, e0007059 (2019).
Fernandes, C. A. et al. Structural Basis for the Inhibition of a Phospholipase A2-like Toxin by Caffeic and Aristolochic Acids. PLoS One 10, e0133370 (2015).
de Oliveira, R. C. W. F. H. & Sifuentes, D. N. In Animais peçonhentos do Brasil: biologia, clínica e terapêutica dos envenenamentos (eds J. L. C.; França Cardoso, F. O. S.; Wen, F. H.; Malaque, C. M. S.; Haddad Jr., V.) 6–21 (Sarvier, 2009).
Gutierrez, J. M. Envenenamientos por mordeduras de serpientes en América Latina y el Caribe: Una visión integral de carácter regional. Boletín de Malariología Y Salud Ambiental L1 (2011).
Chippaux, J. P. Incidence and mortality due to snakebite in the Americas. PLoS Negl Trop Dis 11, e0005662 (2017).
Gutierrez, J. M. & Ownby, C. L. Skeletal muscle degeneration induced by venom phospholipases A2: insights into the mechanisms of local and systemic myotoxicity. Toxicon 42, 915–931 (2003).
Lomonte, B. & Gutierrez, J. M. Phospholipases A2 from viperidae snake venoms: how do they induce skeletal muscle damage? Acta Chim Slov 58, 647–659 (2011).
Warrell, D. A. Snake bite. Lancet 375, 77–88 (2010).
Calvete, J. J. et al. Snake population venomics and antivenomics of Bothrops atrox: Paedomorphism along its transamazonian dispersal and implications of geographic venom variability on snakebite management. J Proteomics 74, 510–527 (2011).
Nunez, V. et al. Snake venomics and antivenomics of Bothrops atrox venoms from Colombia and the Amazon regions of Brazil, Peru and Ecuador suggest the occurrence of geographic variation of venom phenotype by a trend towards paedomorphism. J Proteomics 73, 57–78 (2009).
Rodrigues, V. M. et al. Geographic variations in the composition of myotoxins from Bothrops neuwiedi snake venoms: biochemical characterization and biological activity. Comp Biochem Physiol A Mol Integr Physiol 121, 215–222 (1998).
Mora-Obando, D., Fernandez, J., Montecucco, C., Gutierrez, J. M. & Lomonte, B. Synergism between basic Asp49 and Lys49 phospholipase A2 myotoxins of viperid snake venom in vitro and in vivo. PLoS One 9, e109846 (2014).
Bustillo, S. et al. Synergism between baltergin metalloproteinase and Ba SPII RP4 PLA2 from Bothrops alternatus venom on skeletal muscle (C2C12). cells. Toxicon 59, 338–343 (2012).
Arni, R. K. & Ward, R. J. Phospholipase A2-a structural review. Toxicon 34, 827–841 (1996).
Kini, R. M. Excitement ahead: structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 42, 827–840 (2003).
Fernandes, C. A., Borges, R. J., Lomonte, B. & Fontes, M. R. A structure-based proposal for a comprehensive myotoxic mechanism of phospholipase A2-like proteins from viperid snake venoms. Biochim Biophys Acta 1844, 2265–2276 (2014).
Borges, R. J., Lemke, N. & Fontes, M. R. M. PLA2-like proteins myotoxic mechanism: a dynamic model description. Sci Rep 7, 15514 (2017).
dos Santos, J. I., Soares, A. M. & Fontes, M. R. Comparative structural studies on Lys49-phospholipases A2 from Bothrops genus reveal their myotoxic site. J Struct Biol 167, 106–116 (2009).
Aung, H. T., Furukawa, T., Nikai, T., Niwa, M. & Takaya, Y. Contribution of cinnamic acid analogues in rosmarinic acid to inhibition of snake venom induced hemorrhage. Bioorg Med Chem 19, 2392–2396 (2011).
Borges, M. H. et al. Neutralization of proteases from Bothrops snake venoms by the aqueous extract from Casearia sylvestris (Flacourtiaceae). Toxicon 39, 1863–1869 (2001).
Cardoso, F. F. et al. Structural basis of phospholipase A2-like myotoxin inhibition by chicoric acid, a novel potent inhibitor of ophidian toxins. Biochim Biophys Acta Gen Subj 1862, 2728–2737 (2018).
Carvalho, B. M. et al. Snake venom PLA2s inhibitors isolated from Brazilian plants: synthetic and natural molecules. Biomed Res Int 2013, 153045 (2013).
Cavalcante, W. L. et al. Neutralization of snake venom phospholipase A2 toxins by aqueous extract of Casearia sylvestris (Flacourtiaceae) in mouse neuromuscular preparation. J Ethnopharmacol 112, 490–497 (2007).
Hage-Melim, L. I., Sampaio, S. V., Taft, C. A. & Silva, C. H. Phospholipase A2 inhibitors isolated from medicinal plants: alternative treatment against snakebites. Mini Rev Med Chem 13, 1348–1356 (2013).
Lindahl, M. & Tagesson, C. Flavonoids as phospholipase A2 inhibitors: importance of their structure for selective inhibition of group II phospholipase A2. Inflammation 21, 347–356 (1997).
Samy, R. P., Thwin, M. M., Gopalakrishnakone, P. & Ignacimuthu, S. Ethnobotanical survey of folk plants for the treatment of snakebites in Southern part of Tamilnadu, India. J Ethnopharmacol 115, 302–312 (2008).
Soares, A. M. et al. Medicinal plants with inhibitory properties against snake venoms. Curr Med Chem 12, 2625–2641 (2005).
Ticli, F. K. et al. Rosmarinic acid, a new snake venom phospholipase A2 inhibitor from Cordia verbenacea (Boraginaceae): antiserum action potentiation and molecular interaction. Toxicon 46, 318–327 (2005).
Lomonte, B. & Kahan, L. Production and partial characterization of monoclonal antibodies to Bothrops asper (terciopelo) myotoxin. Toxicon 26, 675–689 (1988).
Fernandez, G. P. et al. Neutralization of Bothrops mattogrossensis snake venom from Bolivia: experimental evaluation of llama and donkey antivenoms produced by caprylic acid precipitation. Toxicon 55, 642–645 (2010).
Prado, N. D. et al. Inhibition of the Myotoxicity Induced by Bothrops jararacussu Venom and Isolated Phospholipases A2 by Specific Camelid Single-Domain Antibody Fragments. PLoS One 11, e0151363 (2016).
Tanjoni, I. et al. Toxicon 42, 801–808 (2003). Snake venom metalloproteinases: structure/function relationships studies using monoclonal antibodies.
Borges, R. J. et al. Functional and structural studies of a Phospholipase A2-like protein complexed to zinc ions: Insights on its myotoxicity and inhibition mechanism. Biochim Biophys Acta 1861, 3199–3209 (2017).
Arruda, E. Z., Silva, N. M., Moraes, R. A. & Melo, P. A. Effect of suramin on myotoxicity of some crotalid snake venoms. Braz J Med Biol Res 35, 723–726 (2002).
de Oliveira, M. et al. Antagonism of myotoxic and paralyzing activities of bothropstoxin-I by suramin. Toxicon 42, 373–379 (2003).
Fathi, B., Harvey, A. L. & Rowan, E. G. Suramin inhibits the early effects of PLA2 neurotoxins at mouse neuromuscular junctions: A twitch tension study. J Venom Res 2, 6–10 (2011).
Fernandes, C. A. et al. Comparison between apo and complexed structures of bothropstoxin-I reveals the role of Lys122 and Ca+2-binding loop region for the catalytically inactive Lys49-PLA2s. J Struct Biol 171, 31–43 (2010).
Lewin, M., Samuel, S., Merkel, J. & Bickler, P. Varespladib (LY315920) Appears to Be a Potent, Broad-Spectrum, Inhibitor of Snake Venom Phospholipase A2 and a Possible Pre-Referral Treatment for Envenomation. Toxins (Basel) 8 (2016).
Lin-Shiau, S. Y. & Lin, M. J. Suramin inhibits the toxic effects of presynaptic neurotoxins at the mouse motor nerve terminals. Eur J Pharmacol 382, 75–80 (1999).
Murakami, M. T. et al. Inhibition of myotoxic activity of Bothrops asper myotoxin II by the anti-trypanosomal drug suramin. J Mol Biol 350, 416–426 (2005).
Salvador, G. H. et al. Structural and functional evidence for membrane docking and disruption sites on phospholipase A2-like proteins revealed by complexation with the inhibitor suramin. Acta Crystallogr D Biol Crystallogr 71, 2066–2078 (2015).
Salvador, G. H. M. et al. Structural and functional characterization of suramin-bound MjTX-I from Bothrops moojeni suggests a particular myotoxic mechanism. Sci Rep 8, 10317 (2018).
Zhou, X. et al. Structural characterization of myotoxic ecarpholin S from Echis carinatus venom. Biophys J95, 3366–3380 (2008).
Bryan-Quiros, W., Fernandez, J., Gutierrez, J. M., Lewin, M. R. & Lomonte, B. Neutralizing properties of LY315920 toward snake venom group I and II myotoxic phospholipases A2. Toxicon 157, 1–7 (2019).
dos Santos, J. I. et al. Structural and functional studies of a bothropic myotoxin complexed to rosmarinic acid: new insights into Lys49-PLA2 inhibition. PLoS One 6, e28521 (2011).
Salvador, G. H. M. et al. Search for efficient inhibitors of myotoxic activity induced by ophidian phospholipase A2-like proteins using functional, structural and bioinformatics approaches. Sci Rep 9, 510 (2019).
Kokotou, M. G., Limnios, D., Nikolaou, A., Psarra, A. & Kokotos, G. Inhibitors of phospholipase A2 and their therapeutic potential: an update on patents (2012–2016). Expert Opin Ther Pat 27, 217–225 (2017).
Nikolaou, A., Kokotou, M. G., Vasilakaki, S. & Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 941–956 (2019).
De Luca, D. et al. Varespladib inhibits secretory phospholipase A2 in bronchoalveolar lavage of different types of neonatal lung injury. J Clin Pharmacol 52, 729–737 (2012).
Varespladib. Am J Cardiovasc Drugs 11, 137–143, (2011).
Lomonte, B. et al. Isolation of basic myotoxins from Bothrops moojeni and Bothrops atrox snake venoms. Toxicon 28, 1137–1146 (1990).
Soares, A. M. et al. A rapid procedure for the isolation of the Lys-49 myotoxin II from Bothrops moojeni (caissaca) venom: biochemical characterization, crystallization, myotoxic and edematogenic activity. Toxicon 36, 503–514 (1998).
Lewin, M. R. et al. Delayed LY333013 (Oral) and LY315920 (Intravenous) Reverse Severe Neurotoxicity and Rescue Juvenile Pigs from Lethal Doses of Micrurus fulvius (Eastern Coral Snake) Venom. Toxins (Basel)10 (2018).
Angulo, Y. & Lomonte, B. Inhibitory effect of fucoidan on the activities of crotaline snake venom myotoxic phospholipases A2. Biochem Pharmacol 66, 1993–2000 (2003).
Fernandes, C. A. et al. Structural bases for a complete myotoxic mechanism: crystal structures of two non-catalytic phospholipases A2-like from Bothrops brazili venom. Biochim Biophys Acta 1834, 2772–2781 (2013).
Salvador, G. H. M., Dos Santos, J. I., Borges, R. J. & Fontes, M. R. M. Structural evidence for a fatty acid-independent myotoxic mechanism for a phospholipase A2-like toxin. Biochim Biophys Acta 1866, 473–481 (2017).
Marchi-Salvador, D. P., Fernandes, C. A., Silveira, L. B., Soares, A. M. & Fontes, M. R. Crystal structure of a phospholipase A2 homolog complexed with p-bromophenacyl bromide reveals important structural changes associated with the inhibition of myotoxic activity. Biochim Biophys Acta 1794, 1583–1590 (2009).
Lomonte, B. & Gutierrez, J. M. A new muscle damaging toxin, myotoxin II, from the venom of the snake Bothrops asper (terciopelo). Toxicon 27, 725–733 (1989).
McPherson, A. Introduction to Macromolecular Crystallography. (Wiley - Blackwell, 2009).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography Pt A276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, (213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010).
Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1 (2010).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Huang, J. & MacKerell, A. D. Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR. data. J Comput Chem 34, 2135–2145 (2013).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865 (2008).
Olsson, M. H., Sondergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of Chemical Theory and Computation 7, 525–537 (2011).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J Chem Phys 126, 014101 (2007).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 81 (1984).
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A Gen Phys 31, 1695–1697 (1985).
Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Molecular Physics 52, 255–268 (1983).
Parrinello, M., Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 52 (1981).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry 18, 1463–1472 (1997).
Kumari, R., Kumar, R. & Lynn, A. g_mmpbsa - a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54, 1951–1962 (2014).
Acknowledgements
Funding was provided by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Vicerrectoría de Investigación, Universidad de Costa Rica (Grant no. VI-741-B5-602). Ophirex, Inc. is acknowledged for kindly providing materials.
Author information
Authors and Affiliations
Contributions
G.H.M.S. performed functional assays, crystallographic and structural comparative analyses under supervision of B.L. and M.R.M.F. A.A.S.G. performed molecular dynamics simulations under supervision of M.R.M.F. W.B.Q. performed functional assays under supervision of B.L. M.R.M.F., G.H.M.S., J.F., M.R.L., J.M.G. and B.L. designed and analysed the experiments. G.H.M.S., M.R.M.F., A.A.S.G. and B.L. wrote the manuscript with inputs of other authors.
Corresponding author
Ethics declarations
Competing interests
M.R.L. has an ownership stake in Ophirex, Inc, a Public Benefit Corporation. The other authors have declared that no competing interests exist.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salvador, G.H.M., Gomes, A.A.S., Bryan-Quirós, W. et al. Structural basis for phospholipase A2-like toxin inhibition by the synthetic compound Varespladib (LY315920). Sci Rep 9, 17203 (2019). https://doi.org/10.1038/s41598-019-53755-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53755-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
