
Abstract
Sclerotinia stem rot caused by Sclerotinia sclerotiorum is a major disease of crop brassicas, with inadequate variation for resistance in primary gene pools. We utilized a wild Brassicaceae species with excellent resistance against stem rot to develop a set of B. juncea - B. fruticulosa introgression lines (ILs). These were assessed for resistance using a highly reproducible stem inoculation technique against a virulent pathogen isolate. Over 40% of ILs showed higher levels of resistance. IL-43, IL-175, IL-215, IL-223 and IL-277 were most resistant ILs over three crop seasons. Sequence reads (21x) from the three most diverse ILs were then used to create B. juncea pseudomolecules, by replacing SNPs of reference B. juncea with those of re-sequenced ILs. Genotyping by sequencing (GBS) was also carried out for 88 ILs. Resultant sequence tags were then mapped on to the B. juncea pseudomolecules, and SNP genotypes prepared for each IL. Genome wide association studies helped to map resistance responses to stem rot. A total of 13 significant loci were identified on seven B. juncea chromosomes (A01, A03, A04, A05, A08, A09 and B05). Annotation of the genomic region around identified SNPs allowed identification of 20 candidate genes belonging to major disease resistance protein families, including TIR-NBS-LRR class, Chitinase, Malectin/receptor-like protein kinase, defensin-like (DEFL), desulfoglucosinolate sulfotransferase protein and lipoxygenase. A majority of the significant SNPs could be validated using whole genome sequences (21x) from five advanced generation lines being bred for Sclerotinia resistance as compared to three susceptible B. juncea germplasm lines. Our findings not only provide critical new understanding of the defensive pathway of B. fruticulosa resistance, but will also enable development of marker candidates for assisted transfer of introgressed resistant loci in to agronomically superior cultivars of crop Brassica.
Similar content being viewed by others


Introduction
Sclerotinia sclerotiorum, causing Sclerotinia stem rot, is a serious pathogen of oilseed crops including Brassica juncea (Indian mustard)1,2,3. This necrotrophic fungus attacks mustard crops at all crop growth stages, especially at flowering, causing yield losses of 10–90% and impaired oil quality3,4. Mustard is an important oil seed crop of India and worldwide4,5, but faces increasing challenges to its productivity from this pathogen. Moreover, wide host range of S. sclerotiorum and the long period of survival of its resting sclerotia together limit the option for control through crop rotation or tillage. Fungicide application, at best, provides inconsistent control and is expensive. Hence there is a need to better understand host-pathogen interactions for using host resistance. A plethora of pathogenicity factors; mainly oxalic acid6,7, hydrolases and poly-galacturonases8, small RNAs9 have been reported to facilitate Sclerotinia infection and colonization on rapeseed-mustard, soybean, pea and sunflower. These pathogen-associated molecular patterns act as the initial trigger for altered redox homeostasis of host tissue and switches on the necrotrophic mode of stem rot pathogen after oxidative burst10.
The only sustainable and economical way to manage Sclerotinia stem rot is via genetic improvement to enhance inherent resistance in the host through utilization of conventional/molecular breeding approaches.
Unfortunately, the complex genetic basis of resistance poses challenges in conventional breeding due to involvement of diverse signalling factors (transcription factors, hormones, enzymes, proteins etc.) associated with the host-pathogen interaction11,12,13. Partial resistance to S. sclerotiorum has been reported in some Chinese1,14 and Australian1,15 lines of oilseed rape, but complete genetic resistance to this pathogen does not occur in crop brassicas. However, high levels of disease resistances are present in genetically diverse wild allies of the family Brassicaceae such as Erucastrum gallicum, Capsella bursa-pastoris, E. cardaminoides, Diplotaxis tenuisiliqua, B. fruticulosa and B. oleracea. Many of these have been utilized in recent research into stem rot resistance in B. napus and B. juncea16,17,18. In addition, a number of QTL mapping studies with biparental mapping population have helped to identify chromosomal loci linked to Sclerotinia resistance in B. napus19,20,21,22, soybean23 and sunflower24. Integration analysis of previously known QTLs of the B. napus genome have allowed identification of conserved QTLs and clusters of nucleotide-binding-site, leucine-rich-repeat-containing candidate genes for Sclerotinia resistance on chromosomes A09 and C0625. However, low mapping resolution due to limited recombination events and allelic diversity of these QTL mapping studies constrained prediction of candidate gene(s) and development of marker assisted selection for disease resistance. Fortunately, genome-wide association studies (GWAS) allows improved mapping resolution by exploiting historical recombination events and high allelic diversity present in association panels26,27. Further, GWAS approaches are more cost effective for the identification of the minor QTLs with small effects that underlie many complex traits such as disease resistance20,28,29,30,31. A few success stories for candidate gene association with host infection as well as resistance have been reported in soybean13,32,33,34,35,36 and B. napus37,38 through SNP markers validation by c-DNA microarrays39,40,41,42, RT-seq43 and proteomic44 approaches. GWAS have also revealed association of significant SNPs on chromosomes A08, C04, C06 and C08 in B. napus to Sclerotinia stem rot resistance. These SNPs are close to genes involved in oxidative burst, lignin biosynthesis and jasmonic acid pathway37,38.
In our previous study18, we demonstrated significant stem rot resistance through introgression of B. fruticulosa genomic fragments in to B. juncea. Molecular characterisation of developed ILs was carried out through SSR based linkage mapping and cytogenetic tools18. Although, ten significant marker-trait associations to disease resistance of ILs were discovered consistently over two years of study, there were large mapping intervals as a limited number of transferable SSR markers did not permit precise localization of resistance associated genes. To facilitate finer resolution in search of candidate genes, we now report extension of our previous research studies18through GBSof 88 ILs. This generated large numbers of marker trait associations (MTA’s) and highlighted 49 significant SNPs corresponding to thirteen loci on chromosomes A01, A03, A04, A05, A08, A09 and B05. Many of these MTA’s were consistent across crop seasons and explained significant phenotypic variation (7.34% to 16.04%), and helped to predict 20 candidate genes belonging to major disease resistance protein families. We discuss how discovery of such novel SNP marker associations to Sclerotinia stem rot resistance will not only facilitate novel candidates contributing to mustard immune response, but will also enable development of breeder-friendly markers for assisted introgression of resistant loci into agronomically superior cultivars of crop Brassica against Sclerotinia stem rot.
Results
Phenotypic evaluation of the Sclerotinia stem rot resistance
Phenotypic data for 2011–12 and 2014–15 was the same as published earlier18. Screening of ILs was extended for another year during 2017–18. Continuous phenotypic variations for stem rot resistance confirmed the quantitative nature of Sclerotinia stem rot resistance in B. juncea - B. fruticulosa ILs. Frequency and disease score distribution graphs of 86 introgression lines along with susceptible and resistant checks are presented in Fig. 1(a,b). Analysis of variance (ANOVA) revealed significant differences among the ILs in terms of expression of resistance responses to stem inoculation (Table 1). Variation across years and year × genotype (G × E) effects were also significant. Replication effects were non-significant. Mean values for stem lesion length were: 4.62 cm (season I), 3.60 cm (season II) and 13.23 cm (season III). The corresponding values for the susceptible genotype RLC1 and resistance donor B. fruticulosa were 11.2, 11.8, 10.38, 11.13 and 0.60, 0.30, 1.53, 0.81 cm of lesion length during seasons I, II and III and pooled, respectively. Lesion length scores differed over seasons. Of the ILs investigated, 57 lines fell under the resistant category in season I, 65 in season II and 74 in season III, respectively. Among these, 40% of genotypes were highly resistant with mean lesion length < 2.5 cm during season III, whereas in seasons I and II, proportions of such genotypes were 17% and 31%, respectively (Fig. 1b). AMMI analysis also confirmed variation across environment and genotype × year interactions (Fig. 2). Season II had low interaction, while season I and season III were highly interactive. Season I was rated as a favourable environment but season III was considered an unfavourable environment for disease built up. Out of 88 genotypes, 83 genotypes had lower year × genotype effects and hence they were stable in their resistance responses. Five ILs, namely IL-43, IL-175, IL-215, IL-223 and IL-277, were consistent for their highly resistant responses to stem inoculation over three years of testing.

(a,b): Phenotypic distributions and frequency histogram of stem lesion length in Brassica juncea – B. fruticulosa intro in season I (2012–13), season II (2013–14), season III (2016–17) and pooled across the three seasons.

AMMI biplot analysis of 88 Brassica juncea – B. fruticulosa introgression lines across three environments.
Identification of MTA’s using GWAS
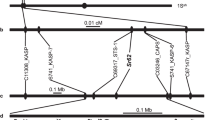
Genotyping by sequencing allowed identification of 31,896,811 SNPs. Only 88,624 SNPs were retained after discarding low quality SNPs and eliminating the ones with minor allele frequency <0.08. These were spread across 18 chromosomes (Supplementary Fig. S1). Heat map of kinship matrix showing genetic relatedness among 88ILs is presented in Supplementary Fig. S2. Horizontal lines on top of heat map show hierarchical clustering of ILs. There were two broad groups, the first group was less diverse and ILs included in this group showed consistently superior resistance responses. Discriminant Analysis of Principal Components (DAPC), however, revealed three major groups (Supplementary Fig. S3). GWAS was performed using five models [GLM, MLM, FarmCPU, GLM(T) and MLM(T)] simultaneously. Goodness of fit of GWAS models was implemented using Quantile-Quantile (Q-Q) plots (Supplementary Fig. S4). We retained only those SNPs that were repeatedly detected over at least two algorithms. Also included were the MTA’s that were consistently detected over at least two seasons. All the SNPs showing strong LD with each other were clumped using LD block as a criterion to define a major QTL. After adopting the threshold -log10(p) value (>3) and clumping of SNPs, thirteen significant loci scattered across seven chromosomes (A01, A03, A04, A05, A08, A09 and B05) were considered (Table 2). Manhattan plots were generated with multi-model plotting (Fig. 3). A01 had the greatest number of five significant loci, followed by three loci on A05 and only one each on A03, A04, A08, A09 and B05. The phenotypic variation explained by these loci ranged from 7.34% to 16.04%. Amongst five peak associated loci detected on chromosome A01 genomic region, A01_11942966-4403 included a cluster of seven SNPs that were in full linkage disequilibrium (LD). These covered a genomic interval of 1437 bp. This lead association signal explained 11.75% of phenotypic variation. Annotation was suggestive of the gene ACX1 in close vicinity (Fig. 4a). This gene encodes acyl CoA oxidase 1 protein. ASNP A01_12606026 was mapped close to the gene LOX-5, encoding Lipoxygenase-1 protein. SNP A01_15152306 was also associated with a similar gene (LOX-1), belonging to the PLAT/LH2 domain-containing lipoxygenase family. A group of five SNPs (A01_23456622-875) clustered in a small genomic region (253 bp) was also identified on A01 with indicative association with SOT18 that encodes desulfoglucosinolate sulfotransferase protein. A SNP on A01 (A01_23756215) explained 8.18% of phenotypic variation. The nearest gene to this MTA was At1g34047, which encodes defensin-like family protein. Single SNP locus identified on chromosome A03 was recorded close to PHOS32 encoding adenine nucleotide alpha hydrolases-like superfamily protein. Also present, at a short genomic distance, was the gene THE1. This gene encodes a Malectin/receptor-like protein kinase family protein. A group of 13 SNPs on chromosome A04 (A04_19791849-2116), covering a small genomic region of 267 bp were present close to the gene MPKKK17. Out of four associated loci, detected on chromosome A05, one locus comprising five SNPs (A05_2460823-1061) and spanning a genomic region of only 238 bp was located adjacent to AT2G43620 and CHI encoding proteins belonging to the chitinase family (Fig. 4b). A genome space (A05_3029963-50244), carrying seven SNPs was also located on chromosome A05. It was proximal to genes ERF113 and HUB1. Three SNPs (9A05_3320578, A05_3320584, A05_3320629) on chromosome A05 appeared associated with the gene CYP76C3 encoding cytochrome p450 protein. SNP locus (A08_3147142) on chromosome A08, shared genome space with the gene LECRK91 encoding Concanavalin A-like lectin protein kinase family protein. Chromosome A09 harboured three SNPs (A09_18223345, A09_18223348, A09_18223407) in a small genomic interval of 62 bp. These were present near to RPS4, AT5G45060 and AT4G19530 belonging to the disease resistance protein family (Fig. 5a). SNP locus (B05_13723719) on chromosome B05 was present close to the gene ERF091 (Fig. 5b). Its protein product is known to be involved in positive regulation of JA responsive defence genes.

Manhattan plots showing GWAS outcomes for Sclerotinia sclerotiorum stem lesion length.

Position of candidate genes near SNP marker spanning 20 kb genomic region on chromosomes (a) A01 (b) A05 of B. juncea. Genes are predicted in 20Kb region using AUGUSTUS tool.

Position of candidate genes near SNP marker spanning 20 kb genomic region on chromosomes (a) A09b) B05 of B. juncea. Genes are predicted in 20Kb region using AUGUSTUS tool.
Linkage disequilibrium
The pairwise LD (r2) of significant and annotated SNP regions was analysed using software Haploview. LD blocks were identified on chromosomes A01, A04 and A05, which showed maximum MTA’s. Region in the LD blocks (black) indicated strong LD between pairs of SNPs (Supplementary Fig. S5). Pairwise LD estimation on chromosome A01 depicted the highest (1 kb) LD in block 1 region. Whereas, significant SNP region on chromosome A04 shows higher r2 value (black), followed by on chromosomes A01 and A05 with less than 1 kb of region.
Validation of identified SNPs
We also compared a set each of susceptible and resistant lines to demonstrate polymorphism for associated SNPs. A majority of associated SNPs were also validated using whole genome sequences (21x) from five advanced generation lines (ADJR_8-Blue, ADJR_8-White, ADJR_8-Black, ADV_6-White and APH64), being bred for Sclerotinia resistance and three susceptible B. juncea germplasm lines (RLC1, NAJR-102B-R2 and DT1). Some of the identified SNPs were in heterozygous state. All this information is included as a supplementary data set (Supplementary Table S1).
Discussion
Incorporation of resistance to Sclerotinia stem rot in a superior agronomic base is a major plant breeding goal in crop brassicas. Although the studies on B. napus21,45 and B. oleracea17,46 have previously helped to identify resistance related QTLs in A, B and C genomes of monogenomic and digenomic Brassica species, these could not be commercially exploited due to poor levels of resistance. While secondary and tertiary gene pools of Brassicaceae exhibit enormous genetic diversity against S. sclerotiorum16, the knowledge of putative gene candidates that contribute to basal/innate immunity remains scarce. B. fruticulosa is one of the wild relatives of Brassica that exhibits high resistance to this devastating pathogen. We successfully utilized next generation strategies of allele mining and candidate gene identification through SNP based approach to characterize genomic introgressions of B. fruticulosa in order to identify potential candidates for stem rot resistance. This study is significant as it mapped 13 significant loci on seven chromosomes (A01, A03, A04, A05, A08, A09 and B05). In addition, annotation of the genomic region around identified SNPs allowed prediction of 20 candidate genes belonging to major disease resistance protein families, including TIR-NBS-LRR class, Chitinase, Malectin/receptor-like protein kinase, defensin-like (DEFL), desulfoglucosinolate sulfotransferase protein and lipoxygenase. Importantly, our findings not only provide critical new understanding of the defensive pathway of B. fruticulosa resistance, but will also enable development of marker candidates for assisted transfer of introgressed resistant loci into agronomically superior cultivars of crop Brassica.
A well-orchestrated network involving diverse defence signalling cascades may arise during plant-pathogen interaction in stem rot (Fig. 6). It initiates with pathogen associated molecular patterns (PAMPs) perceived at host surface; through switching on of PAMP triggered immunity (PTI) via receptor like kinases (RLKs). But if this basal non-race-specific immune response is surpassed by host adapted pathogens, effector triggered immunity (ETI) sets in via release of R proteins (such as SNC1, RPP4). Tight regulation of gene expression for fine tuning defensive responses is necessary, otherwise it may impair the fitness of the host plant47,48. Such reprogramming of immune response is either mediated by pathogen infection or through intracellular signals whose downstream players alter gene expression through chromatin remodelling19, or transcriptional or post translational modification. HUB1, located close to 3 SNPs, identified on chromosome A05, is one such novel gene that is responsible for histone modifications (i.e., methylation and deacetylation). This is a large effect QTL, explaining 14.5% of the phenotypic variation. Mutant studies have revealed that histone 2B monoubiquitinylation through HUB1 activity induces expression of various R genes/R gene complex including SNC1, RPP5 in Arabidopsis against necrotrophic fungi like Botrytiscineria and Alternaria brassicicola49,50. Upstream regulation of HUB1 by hormonal signals (ethylene and salicylic acid) controls chromatin architecture of SUPPRESSOR OF NPR1-1 and CONSTITUTIVE1 (SNC1). These are key regulators of jasmonate-salicylate (JA-SA) crosstalk during systemic and local defences. We found association of 3SNPs on A05 (ERF13) and 1SNP on B05 (ERF 91) with ATERF2 family. ATERF2 are plant specific transcription factors (TFs) that belong to B3 sub-cluster of APETALA2/ethylene response factor (AP2/ERF) family. They play a crucial role in transcriptional regulation of JA-ethylene (ET) responsive defence genes (such as PLANT DEFENSIN1.2/PDF1.2; BASIC CHITINASES/CHI B/PR3) and thus integrate the hormonal crosstalk against necrotrophic pathogens51,52,53. Association of ERF13 with Sclerotinia stem rot resistance in the present study is supported by previous reports on its overexpression with MeJA51 and A.brassicicola51 and chitin54 in Arabidopsis for transcriptional activation of JA-induced disease resistance.

Proposed model to describe transcription factors association and phytohormones crosstalk in resistance responses against Sclerotinia sclerotiorum in Brassica juncea – B. fruticulosa introgression lines. ACX1: Acyl-CoA oxidase 1, ERF: Ethylene response factor, GEF4: Guanine exchange factor4, HUB1: Histone mono-ubiquitination 1, JA: Jasmonic acid, LOX: Lipoxygenase, LECRK91: L-type lectin receptor-like kinase IX.1, MAPKKK: Mitogen-activated protein kinase 17, MEK: MAPK kinases, PDF1.2: Plant Defensin 1.2, PR: Pathogenesis related protein, RPP5: Resistance to Peronospora parasitica5, SA: Salicylic acid, SNC1: Suppressor of npr1-1, constitutive1, THE1: THESEUS1.
Aside, TFs an arsenal of cell surface-localized pattern recognition receptors (PRR) with intracellular kinase domain also fine tune the temporal responses through expression of defensive genes in pathogen-challenged plants. We identified 15SNPs, located to chromosomes A03, A04 and A08 that were strongly associated with Sclerotinia resistance in ILs through kinase signalling cascade. Out of these, 13SNPs formed an LD block on A04 close to the gene encoding mitogen-activated protein kinase, MAPKKK17. However, SNPs corresponding to other receptor kinases LECRK IX.1/LECRK91 and THESEUS1 (THE1) on chromosomes A08 and A03, were also observed. These contributed up to 24.03% of phenotypic variation for disease resistance. Transcriptome studies in B. napus55,56 have emphasized the role of BnaMAPKKK17 (an orthologue of AtMAPKKK17), Bna MAPKKK18 and its downstream associated kinases (MKK3, MKK8 and MKK9) in abiotic and biotic stress tolerance. THE1, a CrRLK1 like kinase may sense cell wall integrity and mediate defence against necrotrophic fungi like B. cinerea in Arabidopsis through elicitation of GEF4 signalling network57. Further, the overexpression of THE1 caused increased deposition of callose, H2O2 accumulation and also the upregulation of PR2 and PDF1.2 transcript levels in B. cinerea treated plants as compared with the wild type57. Studies on LECRK9158,59 have addressed their role in plant innate immunity through cell wall associated defences and jasmonate signalling. Mutation in LECRK91 caused susceptibility to fungal and bacterial pathogens in Arabidopsis, while its overexpression reversed the effect, thereby enhancing immune response by cell death59,60.
In our ILs, 7SNPs in a genomic interval of 1.4Kb on A01 appeared very close to the gene encoding Acyl–CoA oxidase (ACX1) that has a role in JA biosynthesis. These explained 11.75% of phenotypic variation. Three additional SNP markers on chromosome A01 and A03 appeared associated with lipoxygenase LOX1, LOX5 and hydrolases PHOS32. In vitro assays have confirmed phosphorylation of PHOS32 by AtMPK3 and AtMPK3 upon elicitation; and suggested its role in mediating downstream kinase signalling events61. Both LOX1 and LOX5 encoding 9-lipoxygenases, were responsible for hydro-peroxidation of fatty acid component of plant membranes. Increased activity of lipoxygenase has already been reported in a number of plant pathogen systems62,63, including Arabidopsis64, where loss of function of LOX1 and LOX5 mediated resistance occurs against Fusarium head blight. Their hormonal regulation with JA, salicylate (SA) in addition to other stress like wounding suggests that LOX control balance of JA-SA signalling. We observed a major locus with large effect (14.06% phenotypic variation) on chromosome A05, with significant SNPs near to candidate gene of the Chitinase family. Chitinases, also known as pathogenesis related (PR) proteins, are generally induced in the plant system in response to fungal, bacterial or viral infections. The chitinase genes At2g43620 and CHI belong to class IV chitinases and are involved in cell wall biogenesis. These are induced upon pathogen infection65,66 and hormones like ethylene67. We also identified a MTA (A01_23756215) close to another PR protein, defensin. Inductions of these defensins or defensin-like proteins represent a host innate immune response which is triggered upon pathogen infection. These cysteine rich cationic peptides modify fungal membrane permeability and inhibit growth of a broad range of fungal pathogens68.
Overexpression of defensin genes in transgenic B. napus resulted in enhanced resistance to S. sclerotiorum69. Me-JA and SA application reveals complex networking between PR genes and hormonal signals as indicated by their induction in response to biotic stimuli. Identification of genomic region, harbouring 3SNP markers on A09, in proximity of TIR-NBS-LRR class of resistance genes (RPS4; AT5G45250) within a 62Kb region, seemed important. R genes like RPS4 recognise fungal effector-host protein complex and activate ETI against both fungal and bacterial pathogens. RPS4, alone or in pair with RRS1 (R protein complex), can act as a dual resistance system against the hemibiotrophic fungus Colletotrichum higginsianum in Arabidopsis70. Downstream signalling after activation of TIR-NBS-LRR involves genes EDS1 and EDS1 related proteins (PAD4 and SAG 101) with JA-SA regulation of immune responses. Present study also confirmed the role of NIP3-1 as reported previously18. This locus was in strong LD with the SNP A01_ 12606026. Thus search of putative candidates through systematic annotation in identified genomic region following GWAS helped us to identify genomic regions that likely contributed to Sclerotinia resistance in B. juncea-B. fruticulosa ILs. A majority of the significant SNPs could be validated using whole genome sequences (21x) from five advanced generation lines being bred for Sclerotinia resistance as compared to three susceptible B. juncea germplasm lines. We now hope to develop breeder friendly KASPar markers for effective integration of these defensive components into a superior agronomic base, initially in B. juncea and subsequently in B. napus and other vegetable Brassica species.
Methods
Plant materials and resistance screening
Genetic materials and stem inoculation procedures were same as described earlier18. Eighty eight ILs were raised in a randomized block design with two replications over three crop seasons against an aggressive and virulent strain (PAU-4) of S. sclerotiorum at Punjab Agricultural University, Ludhiana, India. Analysis of variance (ANOVA) was undertaken and standard deviation and standard errors were estimated through Multivariate General Linear Model (M-GLM) using software, SPSS17.0.
DNA extraction for library preparation
Genomic DNA of ILs and their parents were isolated from young leaf tissue using a CTAB (cetyltrimethyl ammonium bromide) extraction method71, with the chloroform-isoamyl alcohol purification step repeated twice to assure good quality DNA. RNase was added between the two chloroform: isoamyl alcohol solvent extractions by incubating the samples at 37° Celsius for 15 minutes to allow for a single DNA precipitation step at the end of the protocol. Quality of the extracted DNA was assessed using a NanoDrop® 2000 spectrophotometer and 1% (w/v) agarose gel. Sample DNA having 260/280 absorbance ratio of 1.8-2.0, and with no evidence of substantial band shearing or contamination (either RNA or polysaccharide), was used for library construction.
Resequencing and genotyping by sequencing
Direct resequencing (21x) of four ILs and genotyping by sequencing of 88 ILs were outsourced. For this, high quality DNA of each sample was digested with appropriate combination of restriction enzymes based on in-silico evaluation. This was followed by several rounds of PCR amplification. All samples were then individually pooled and then size-selected for the required fragments to complete the library construction. High quality libraries with appropriate insert sizes were subsequently used for pair-end sequencing on Illumina® HiSeq platform, with the read length of 150 bp at each end. The sequences and corresponding sequencing quality information were stored in a FASTQ file format. The adapter sequences were removed from raw reads using the software Cutadapt. The available reference genome of B. juncea v1.5 was used for reference-based alignments of whole genome sequences (21x) from the four most prominent ILs, using software bowtie2. Initially, one introgression line was aligned into reference genome and SNP called by NGSEP-GBS pipeline. Total SNPs with base quality more 30 were replaced in background genome reference (B. juncea v1.5) using a perl script, pseudomaker.pl implemented in SEG-Map72 to construct the first step of mock-up pseudomolecule(s), which were then used as a reference for subsequent construction of pseudomolecules of sequential replacement of SNPs for remaining three ILs in the same way as described above.
Resultant mock-up reference was then used for aligning sequence tags from 88 ILs. SNPs identification was then carried out by using NGSEP-GBS pipeline73. Resultant marker dataset comprised 31,896,811 SNPs. These were then filtered to include only quality SNPs for further analysis. Filtering parameters were: minimum mapping quality (30), minor allele frequency (0.08), bi-allelic SNVs, minimum number of sample genotyped (70), maximum observed heterozygosity (30%) and maximum missing calls (30%). After filtering, 88,624 SNPs remained. These were imputed using fcGENE and Beagle softwares74.
Genome-wide association analysis (GWAS) based on SNP genotyping
Sclerotinia resistance lesion length (cm) data were first normalized through rank and log transformation. GWAS was carried out using transformed values of lesion lengths and 88,624 SNPs. Kinship matrix and Covariates data was generated through ‘MVP.Data’ function of the software R (MVP-package). Discriminant Analysis of Principal Components (DAPC) was implemented in R software package “adegenet”. PC and DAPC values were then used as covariates in different GWAS analysis algorithms to reduce false positives by accounting for the effects of population stratification. Manhattan plots were generated with multi-model plotting using MVP tools. GLM, MLM and FarmCPU methods were implemented in the software R (MVPpackage) (https://github.com/XiaoleiLiuBio/MVP), using default settings to identify marker trait associations. Identified markers were subsequently confirmed using alternate algorithms as implemented in TASSEL5.0 with DAPC (in adegenet) as covariate for GWAS with model; GLM + DAPC [GLM(T)] and MLM + kinship [MLM(T)]. Visualization of the significant QTLs and SNPs was done using Manhattan plots, generated using the R package qqman75. We used unadjusted p-values < 0.001, to declare significance as the Bonferroni and FDR tests are considered far too conservative for association studies involving thousands of candidate variants at very low minor allele frequencies76. Bonferroni correction also assumes independence of every association test, an assumption which is rarely met due to linkage disequilibrium between SNP markers. We screened 50 kb flanking regions on each side of resistance associated peak SNPs to predict candidate genes using Blast2GO v5.2.5 tool (Gotz77 et al. 2008). Arabidopsis protein database was probed for gene finding as well as blast search. The gene ontologies of Arabidopsis orthologues were used for this analysis as these are better curated than those of B. juncea. Positions of the predicted candidate genes w.r.t. the SNPs, were projected through blast search of their sequences against B. juncea mock-up pseudomolecules. Predicted candidate genes were then verified for their function from literature for their relevance to the target trait.
References
Barbetti, M. J. et al. Comparative genotype reactions to Sclerotinia sclerotiorum within breeding populations of Brassica napus and B. juncea from India and China. Euphytica 197, 47–59 (2014).
Boland, G. J. & Hall, R. Index of plant hosts of Sclerotinia sclerotiorum. Can. J. Pl. Pathol. 16, 93–108 (1994).
Singh, R. et al. Management of Sclerotinia rot of oilseed Brassicas with a focus on India. Brassica 10, 1–27 (2008).
Sharma, P. et al. Sclerotinia sclerotiorum (Lib) de Bary causing Sclerotinia rot in oilseed Brassicas: a review. J. Oilseed Brassica 6, 1–44 (2015).
Kumar, A. Production barriers and technological options for sustainable production of rapeseed-mustard in India. J. Oilseed Brassica 3, 67–77 (2012).
Williams, B. et al. Tipping the balance: Sclerotinia sclerotiorum secreted oxalic acid suppresses host defenses by manipulating the host redox environment. PLoS Pathogens 7, e1002107 (2011).
Kim, K. S., Min, J. Y. & Dickman, M. B. Oxalic acid is an elicitor of plant programmed cell death during Sclerotinia sclerotiorum disease development. Mol. Plant Microbe Interact. 21, 605–612 (2008).
Favaron, F., Sella, L. & D’Ovidio, R. Relationships among endopolygalacturonase, oxalate, pH, and plant polygalacturonase inhibiting protein (PGIP), in the interaction between Sclerotinia sclerotiorum and soybean. Mol. Plant Microbe Interact. 17, 1402 (2004).
Derbyshire, M. C. et al. Small RNAs from the plant pathogenic fungus Sclerotinia sclerotiorum highlight candidate host target genes associated with quantitative disease resistance. BioRxivdoi: https://doi.org/10.1101/354076 (2018).
Kabbage, M., Yarden, O. & Dickman, M. B. Pathogenic attributes of Sclerotinia sclerotiorum: Switching from a biotrophic to necrotrophic lifestyle. Plant Sci. 233, 53–60 (2015).
Wei, W. et al. Genome-wide association mapping of resistance to a Brazilian isolate of Sclerotinia sclerotiorum in soybean genotypes mostly from Brazil. BMC Genomics 18, 849 (2017).
Guo, X. M. & Stotz, H. U. Defense against Sclerotinia sclerotiorum in Arabidopsis is dependent on jasmonic acid, salicylic acid, and ethylene signaling. Mol. Plant Microbe Interact. 20, 1384–1395 (2007).
Calla, B. et al. Gene expression profiling soybean stem tissue early response to Sclerotinia sclerotiorum and in silico mapping in relation to resistance markers. Plant Genome 2, 149–166 (2009).
Li, C. X. et al. New sources of resistance to Sclerotinia stem rot caused by Sclerotinia sclerotiorumin Chinese and Australian Brassica napus and B. juncea germplasm screened under Western Australian conditions. Australas. Plant Pathol. 38, 149–152 (2008).
Barbetti, M. J. et al. New host resistances in Brassica napus and Brassica juncea from Australia, China and India: Key to managing Sclerotinia stem rot (Sclerotinia sclerotiorum) without fungicides. Crop Protect. 78, 127–130 (2015).
Garg, H. et al. High level of resistance to Sclerotinia sclerotiorum in introgression lines derived from hybridization between wild crucifers and the crop Brassica species Brassica napus and B. juncea. Field Crops Res. 117, 51–58 (2010).
Mei, J. et al. Identification of resistant sources against Sclerotinia sclerotiorum in Brassica species with emphasis on B. oleracea. Euphytica 177, 393–399 (2011).
Rana, K. et al. Mapping resistance responses to Sclerotinia infestation in introgression lines of Brassica juncea carrying genomic segments from wild Brassicaceae B. fruticulosa. Sci. Rep. 7, 5904 (2017).
Zhao, J. et al. Quantitative trait loci for resistance to Sclerotinia sclerotiorum and its association with a homeologous non-reciprocal transposition in Brassica napus L. Theor. Appl. Genet. 112, 509–516 (2006).
Wei, D. et al. Quantitative trait loci analyses for resistance to Sclerotinia sclerotiorum and flowering time in Brassica napus. Mol. Breed. 34, 1797–1804 (2014).
Wu, J. et al. Identification of QTLs for Resistance to sclerotinia stem rot and BnaC.IGMT5.a as a candidate gene of the major resistant QTL SRC6 in Brassica napus. PLoS ONE 8, e67740 (2013).
Yin, X. et al. Mapping of QTLs detected in a Brassica napus DH population for resistance to Sclerotinia sclerotiorum in multiple environments. Euphytica 173, 25–35 (2010).
Arahana, V. S. et al. Identification of QTLs for resistance to Sclerotinia sclerotiorum in soybean. Crop Sci. 41, 180–188 (2001).
Micic, Z. et al. Identification and validation of QTL for Sclerotinia mid-stalk rot resistance in sunflower by selective genotyping. Theor. Appl. Genet. 111, 233–242 (2005).
Li, J. et al. Integration analysis of quantitative trait loci for resistance to Sclerotinia sclerotiorum in Brassica napus. Euphytica 205, 483–489 (2015).
Huang, X. H. & Han, B. Natural variations and genome-wide association studies in crop plants. Ann. Rev. Plant Biol. 65, 531–551 (2014).
Rafalski, J. A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 13, 174–180 (2010).
Zhang, D. et al. Association analysis of stem rust resistance in U.S. winter wheat. PLoS ONE 9, e103747 (2014).
Kollers, S. et al. Genome-wide association mapping of tan spot resistance (Pyrenophoratritici-repentis) in European winter wheat. Mol. Breeding 34, 363–371 (2014).
Wang, M. et al. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 196, 125–131 (2012).
FopaFomeju, B. et al. Homoeologous duplicated regions are involved in quantitative resistance of Brassica napus to stem canker. BMC Genom. 15, 498 (2014).
Bastien, M., Sonah, H. & Belzile, F. Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Pl. Genome 7, 1–13 (2014).
Iquira, E., Humira, S. & François, B. Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol. 15, 5 (2015).
Zhao, X. et al. Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J. 82, 245–256 (2015).
Chang, H.-X. et al. Characterization of disease resistance loci in the USDA soybean germplasm collection using genome-wide associations. Phytopathology 106, 1139–1151 (2016).
Moellers, T. C. et al. Main and epistatic loci studies in soybean for Sclerotinia sclerotiorum resistance reveal multiple modes of resistance in multi-environments. Sci. Rep. 7, 3554, https://doi.org/10.1038/s41598-017-03695-9 (2017).
Wu, J. et al. Genome-wide association study identifies new loci for resistance to sclerotinia stem rot in Brassica napus. Front. Plant Sci. 7, 1418, https://doi.org/10.3389/fpls.2016.01418 (2016).
Wei, L. et al. Genome- wide association analysis and differential expression analysis of resistance to Sclerotiniastem rot in Brassica napus. Plant Biotechnol. J. 14, 1368–1380, https://doi.org/10.1111/pbi.12501 (2015).
Liu, R., Zhao, J., Xiao, Y. & Meng, J. Identification of prior candidate genes for Sclerotinia local resistance in Brassica napus using Arabidopsis cDNA microarray and Brassica-Arabidopsis comparative mapping. Sci. China C. Life Sci. 48, 460–470 (2005).
Yang, B., Srivastava, S., Deyholos, M. K. & Kav, N. N. V. Transcriptional profiling of canola (Brassica napus L.) responses to the fungal pathogen Sclerotinia sclerotiorum. Plant Sci. 173, 156–171 (2007).
Zhao, J. et al. Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol. Plant Pathol. 10, 635–649 (2009).
Zhao, J. et al. Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 227, 13–24 (2007).
Yang, B. et al. Characterization of defence signaling pathways of Brassica napus and Brassica carinata in response to Sclerotinia sclerotiorum challenge. Plant Mol. Biol. Rep. 28, 253–263 (2010).
Garg, H., Li, H., Sivasithamparam, K. & Barbetti, M. J. Differentially expressed proteins and associated histological and disease progression changes in cotyledon tissue of a resistant and susceptible genotype of Brassica napus Infected with Sclerotinia sclerotiorum. PLoS ONE 8(6), e65205, https://doi.org/10.1371/journal.pone.0065205 (2013).
Wu, J. et al. Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 6, 19007 (2016).
Mei, J. et al. Identification of genomic regions involved in resistance against Sclerotinia sclerotiorum from wild Brassica oleracea. Theor. Appl. Genet. 126, 549–56 (2013).
Zhu, Y., Du, B., Qian, J., Zou, B. & Hua, J. Disease resistance gene-induced growth inhibition is enhanced by rcd1 independent of defense activation in. Arabidopsis. Plant Physiol. 161, 2005–2013 (2013).
Gou, M. & Hua, J. Complex regulation of an R gene SNC1 revealed by auto-immune mutants. Plant Signal Behav. 7, 213–216 (2012).
Dhawan, R. et al. Histone monoubiquitination1 interacts with a subunit of the mediator complex and regulates defence against necrotrophic fungal pathogens in Arabidopsis. Plant Cell 21, 1000–1019 (2009).
Zou, B. et al. Monoubiquitination of histone 2B at the disease resistance gene locus regulates its expression and impacts immune responses in Arabidopsis. Plant Physiol. 165, 309–318 (2014).
McGrath, K. C. et al. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 139, 949–959 (2005).
Broekgaarden, C. et al. Ethylene: traffic controller on hormonal crossroads to defence. Plant Physiol. 169, 2371–2379 (2015).
Pieterse, C. M. et al. Hormonal modulation of plant immunity. Ann. Rev. Cell Develop. Biol. 28, 489–521 (2012).
Libault, M. et al. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol. Plant Microbe Interact. 20, 900–911 (2007).
Yang, B. et al. Identification and expression analysis of WRKY transcription factor genes in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biology 9, 68 (2009).
Sun, Y. et al. Identification and functional analysis of mitogen-activated protein kinase kinasekinase (MAPKKK) genes in canola (Brassica napus L.). J. Exp. Bot. 65, 2171–2188 (2014).
Qu et al. THESEUS1 positively modulates plant defense responses against Botrytis cinerea through GUANINE EXCHANGE FACTOR4 signaling. J. Integr. Plant Biol. 59, 797–804 (2017).
Bouwmeester, K. et al. The lectin receptor kinase LecRK-I.9 is a novel Phytophthora resistance component and a potential host target for a RXLR effector. PLoS Path. 7, e1001327 (2011).
Wang, Y. et al. Arabidopsis lectin receptor kinases LecRK-IX.1 and LecRK-IX.2 are functional analogs in regulating Phytophthora resistance and plant cell death. Mol. Plant Microbe Interact. 28, 1032–1048 (2015).
Balague, C. et al. The Arabidopsis thaliana lectin receptor kinase LecRK-I.9 is required for full resistance to Pseudomonas syringae and affects jasmonate signalling. Mol Plant Pathol. 18, 937–948 (2016).
Merkouropoulos, G., Andreasson, E., Hess, D., Boller, T. & Peck, S. C. An Arabidopsis protein phosphorylated in response to microbial elicitation, AtPHOS32, is a substrate of MAP kinases 3 and 6. J. Biol. Chem. 283, 10493–10499 (2008).
Melan, M. A. et al. An Arabidopsis thaliana lipoxygenase gene can be induced by pathogens, abscisic acid, and methyl jasmonate. Plant Physiol. 101, 441–450 (1993).
Rance, I., Fournier, J. & Esquerre-Tugaye, M. T. The incompatible interaction between Phytophthora parasitica var nicotianae race 0 and tobacco is suppressed in transgenic plants expressing antisense lipoxygenase sequences. Proc. Natl. Acad. Sci. USA 95, 6554–6559 (1998).
Nalam, V. J. et al. Facilitation of Fusarium graminearum infection by 9-Lipoxygenase in Arabidopsis and wheat. Mol. Plant Microbe Interact. 28, 1142–1152 (2015).
Passarinho, P. A. & de Vries, S. C. Arabidopsis Chitinases: a Genomic Survey. The Arabidopsis Book, https://doi.org/10.1199/tab.0023 (2002).
Rasmussen, U., Bojsen, K. & Collinge, D. B. Cloning and characterization of a pathogen-induced chitinase in Brassica napus. Plant Mol. Biol. 20, 277–287 (1992).
Payton, S., Fray, R. G., Brown, S. & Grierson, D. Ethylene receptor expression is regulated during fruit ripening, flower senescence and abscission. Plant Mol. Biol. 31, 1227–1231 (1996).
Stotz, H. G., Thomson, J. G. & Wang, Y. Plant defensins: Defense, development and application. Plant Signalling Behav. 4(11), 1010–1012 (2009).
Zarinpanjeh, N. et al. Enhanced resistance to Sclerotinia sclerotiorum in Brassica napus by co-expression of defensin and chimeric chitinase genes. J. Appl. Genet. 57, 417–425 (2016).
Narusaka, M. et al. RRS1 and RPS4 provide a dual resistance-gene system against fungal and bacterial pathogen. Plant J. 60, 218–226 (2009).
Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus 12, 13–15 (1990).
Zhao, Q., Huang, X., Lin, Z. & Han, B. SEG-Map: A novel software for genotype calling and genetic map construction from next-generation sequencing. Rice 3, 98–102 (2010).
Duitama, J. et al. An integrated framework for discovery and genotyping of genomic variants from high-throughput sequencing experiments. Nucleic Acids Res. 42, e44 (2014).
Browning, B. L. & Browning, S. R. Genotype imputation with millions of reference samples. Am J Hum Genet. 98(1), 116–26 (2016).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. J. Open Source Softw. 3, 731, https://doi.org/10.21105/joss.00731 (2018).
Bush, W. S. & Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8, e1002822 (2012).
Gotz, S. et al High-throughput funcitonal annoation and data mining with the Blast2Go suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Acknowledgements
Research studies were carried out with the financial assistance from the Department of Biotechnology, Government of India in form of Centre of Excellence and Innovation in Biotechnology “Germplasm enhancement for crop architecture and defensive traits in Brassica”. Initial pre-breeding material was developed with financial support from National Professor Project entitled “Broadening the genetic base of Indian mustard (Brassica juncea) through alien introgressions and germplasm enhancement funded by Indian Council of Agricultural Research (ICAR). S.S.B. is also grateful to ICAR for salary support.
Author information
Authors and Affiliations
Contributions
S.S.B. and M.J.B. designed research. S.S.B. and C.A. constructed the introgression lines. K.R. and P.S.S. and A.G. performed stem inoculations and recorded the disease score. C.A., R.K., A.S. and M.P.S. maintained germplasm and isolated DNA. J.A. carried out bioinformatic analysis. M.M. annotated results. M.G., N.G. and S.S.B. interpreted the data and wrote the paper. M.J.B. edited the manuscript, S.S.B. supervised the whole study. All authors have read and approved the final version of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Atri, C., Akhatar, J., Gupta, M. et al. Molecular and genetic analysis of defensive responses of Brassica juncea – B. fruticulosa introgression lines to Sclerotinia infection. Sci Rep 9, 17089 (2019). https://doi.org/10.1038/s41598-019-53444-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53444-3
This article is cited by
-
Deciphering the genetic architecture of plant height and main shoot length in backcross introgression lines of Sinapis alba + Brassica juncea allohexaploids
Euphytica (2024)
-
Dissection of QTLs conferring drought tolerance in B. carinata derived B. juncea introgression lines
BMC Plant Biology (2023)
-
Assessment of Sclerotinia Stem and Leaf Rot Resistance and its Association with Physical Strength Attributes in Brassicaceae with Special Emphasis on Brassica Juncea
Journal of Plant Growth Regulation (2023)
-
Identification of new resistance source for Sclerotinia stem rot in backcross population of B. juncea + S. alba allohexaploids: key to manage disease through host resistance
Euphytica (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
