
Abstract
The design of near-IR materials is highly relevant to energy and pharmaceutical sciences due to the high proportion of near-IR irradiation in the solar spectrum and the high penetration of near-IR light in biological samples. Here, we show the one-step synthesis of hexacoordinated ruthenium and iron complexes that exhibit a main absorption band in the near-IR region. For that purpose, novel tridentate ligands were prepared by condensation of two diimines and four cyanoaryl derivatives in the presence of ruthenium and iron template ions. This method was applied to a wide variety of cyanoaryl, diimine, and metal ion combinations. The relationship between the structure and the optical and electrochemical properties in the resulting complexes was examined, and the results demonstrated that these compounds represent novel near-IR materials whose physical properties can be controlled based on rational design guidelines. The intense absorption bands in the 700–900 nm region were assigned to metal-to-ligand charge transfer (MLCT) transitions, which should allow applications in materials with triplet excited states under irradiation with near-IR light.
Similar content being viewed by others

Introduction
Light-harvesting materials play a prominent role in various fields of materials sciences, such as solar energy conversion1,2, artificial photosynthesis3,4, environmental analysis5, and therapeutics6,7. Organic materials present advantages over inorganic materials in terms of the diversity of the structures available, their flexibility, and the relatively low cost of their synthesis. Light is typically classified by its wavelength, and visible-light-harvesting materials usually exhibit bright colors, and have thus attracted great interest for a long time. On the other hand, near-IR light (especially the 700–1000 nm region) is “invisible” and its practical applications have remained undeveloped. However, the characteristics of near-IR light are attractive for applications in advanced functional materials. For instance, half the density of solar energy is composed of near-IR light. The high penetration of human tissue of light in this spectral region (the so-called therapeutic windows) moreover suggests relatively low toxicity and high selectivity for biological applications. Hence, a wide range of potential applications of near-IR materials has been proposed, such as in dye-sensitized solar cells (DSSCs)8,9, organic photovoltaics (OPVs)10,11, photocatalysts12,13,14, photosensitizing agents15,16, n-type semiconductors17,18, biological imaging19,20,21,22, and cancer treatment23,24,25.
Herein, we report the synthesis and characterization of a series of Temari (a traditional hand-made Japanese ball)-shaped metal complexes that absorb in the near-IR window (Fig. 1). For the development of near-IR materials, a synthetically facile organic “platform” with robust, finely tunable, and predictable properties would be highly desirable; however, so far, this puzzle piece remains missing in organic materials chemistry. Phthalocyanine (Pc)-metal complexes present highly symmetrical D4h structures with an intense absorption band in the visible region (the so-called Q band), whose properties can be tuned by modifications on the Pc skeleton26. Heavy-atom effects of the metal center are also important for optical materials that exhibit triplet excited states. Currently, Pc derivatives are employed in optical and electronic materials under visible light in various fields27,28. On the other hand, the typical Pc Q band can be assigned to π–π* transitions in the Pc macrocycle, indicating that orbital effects of the central heavy metal are relatively small. Ruthenium polypyridine complexes are well-known as efficient optical materials29. Metal-to-ligand charge transfer (MLCT) transitions in the lower energy region are often essential for the unique optical properties of such Ru(II) complexes. Although a number of Ru(II) complexes have been designed to reduce the energy of MLCT transitions, which is commensurate with a bathochromic shift of the absorption8,30,31,32, a general strategy toward compounds able to exploit the near-IR light (>700 nm) with high efficiency (ε > 104 M−1 cm−1) remains elusive. Various ligands for Ru(II) complexes have been synthesized, most of them by literature procedures. We believe that structurally novel organic ligands synthesized by newly developed methods will provide the opportunity to overcome the aforementioned unresolved problems. Recently, several automated processes for the development of new organic synthetic methods have been proposed, albeit that they require expert chemistry knowledge and specialized training33,34. Regardless, the design of unusual reactions and compounds by synthetic chemistry experts remains an important topic in synthetic organic chemistry.

General properties of azaporphyrins and the metal complexes in this work.
Results
Synthesis of hexacoordinated complexes of Fe and Ru
Pc ruthenium complexes (RuPc) bearing one or two axial ligands were synthesized by a direct condensation or by ligand-substitution reactions using suitable Pc precursors35. We have previously reported the unique optical properties of octaaryl-substituted tetraazaporphyrin (TAP) phosphorus(V) complexes36,37, and thus TAP ruthenium complexes (RuTAPs) were chosen as our initial synthetic targets. A RuTAP complex bearing an axial pyridyl ligand (RuTAP) was synthesized from the corresponding pyrroline-diimine derivatives, ruthenium trichloride, and pyridine, following a modified literature procedure38. On the other hand, the synthesis of RuTAP complexes bearing an axial cyanophenyl ligand was unsuccessful when pyrroline-diimine and RuCl3 were condensed in the presence of benzonitrile instead of pyridine39. Interestingly, an unexpected moss-green compound (1a) was isolated instead of RuTAP. A detailed structural characterization (vide infra) revealed that 1a exhibits a novel ball-shaped structure composed of one octahedral hexacoordinated ruthenium and two tridentate ligands (Fig. 2). The tridentate ligand consists of one diimine and two benzonitrile moieties; therefore, 1a was generated in a seven-component one-step reaction. The robustness of this structure was confirmed by changing the ligands and central metal. Figure 3 illustrates the generality of the synthesis, and the optimization of the reaction conditions is summarized in Supplementary Table 4. Both cyanoaryl (Ar1 position) and diimine (Ar2 position) compounds were successfully transformed. When 4-cyanopyridine was used, only 1c was obtained, while 4-cyanopyridine-coordinated RuTAP was not observed. Moreover, benzoannulated complexes 2a–c were obtained from the corresponding (un)substituted diiminoisoindoline derivatives. This synthetic method can also be applied to the synthesis of iron complexes. Both non-benzoannulated (3) and benzoannulated (4) complexes were obtained using FeCl3 as the metal template.

Synthesis of RuTAP and 1a.

General scope of the seven-component condensation reaction between cyanoaryls, diimines, and metal salts.
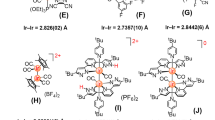
These compounds display excellent solubility in common organic solvents, and were fully characterized by NMR spectroscopy, HR-MALDI-TF-ICR mass spectrometry and elemental analysis. For instance, the 1H NMR signals derived from the cyanoaryl (Ar1) and peripheral aryl groups (Ar2) of the diimine units of 1a appear as sharp peaks in the aromatic region (7–8 ppm), supporting the notion that these compounds exhibit a four-fold symmetric structure and diamagnetic character (Supplementary Fig. 1). A downfield singlet ( > 9 ppm) could be assigned to the imine proton of the cyanoaryl moiety. The structures of Temari-shaped complexes 1d (Ru) and 3 (Fe) were unambiguously determined by single-crystal X-ray diffraction analysis (Fig. 4). The symmetry of their unit cells is tetragonal with a highly symmetrical S4 structure. No residual peaks remained after the signals for the complex and the solvent were assigned, indicating that these complexes are neutral. Considering the results of the NMR and X-ray diffraction measurements suggests that the octahedral-hexacoordinated central metals are in the oxidation state + II. The chelation angles weakly deviate from perfect octahedral coordination due to the unsymmetric nature of the ligand. The ligands in the Ru and Fe complexes present similar bond lengths (Supplementary Fig. 2). Although the structure of the auxiliary ligand is rigid and planar, significant bond length alternation was observed. The bond length between the central metal and nitrogen is consistent with typical M–X bonds of hexacoordinated metal complexes40,41. Therefore, other metals that favor octahedral hexacoordinated structures might also be used for the synthesis of corresponding Temari-shaped complexes.

X-ray crystal structure of (a) ruthenium complex 1d and (b) iron complex 3. Thermal ellipsoids are shown at 50% probability, and hydrogen atoms as well as solvent molecules have been omitted for clarity.
Near-IR optical and electrochemical properties
The absorption and magnetic circular dichroism (MCD) spectra of RuTAP and 1a are shown in Fig. 5. The Q bands of typical Pc and TAP ruthenium complexes appear in the range of 600–700 nm35. Interestingly, the sharp (full width at half maximum (fwhm) = 680 cm−1) and intense (ε = 3.5 × 104 M−1 cm−1) absorption band of 1a appears in the near-IR region beyond 700 nm (e.g. 767 nm in chloroform), despite its non-cyclic structure. The MCD intensity indicates changes in the orbital angular momentum between the ground and excited states42. For instance, the intense dispersion-type Faraday A terms (604 and 590 nm) in the Q band region of RuTAP indicate a large angular momentum change (±9) derived from π–π* transitions in the TAP macrocycle. On the other hand, a very weak MCD signal (776 nm) was found for 1a in the near-IR region, indicating a small change in angular momentum. Hence, it may be concluded that the intense absorption peak does not correspond to a simple π–π* transition in the ligand. Small solvent effects were also observed in the absorption spectra of 1a in various solvents (Supplementary Fig. 3). A detailed assignment is provided below. Despite the intense absorption in the near-IR region, the photostability of the complexes is very high. A CHCl3 solution of 1a showed no photobleaching ( < 5%) after irradiation for 22 h (Supplementary Fig. 4).

UV-vis-NIR MCD (top) and absorption (bottom) spectra of RuTAP and 1a in CHCl3.
Figure 6 shows the absorption spectra of the Temari-shaped complexes synthesized in this work. Structural effects were rationalized in terms of the three types of components: (1) Peripheral substituent effects (Ar1 and Ar2 moieties), (2) benzoannulation effects, and (3) central metal effects (Ru vs Fe). The intense near-IR band was maintained (760–780 nm) upon changing the peripheral substituents to both electron-donating (1d) and electron-withdrawing (1b, 1e) groups, while electron-deficient pyridyl-substituted 1c showed a blue-shifted band (732 nm). This trend resembles the peripheral substituent effects of typical TAP metal complexes43,44, since these groups do not participate in the π-conjugation of the ligands. Although the Q band of typical azaporphyrin derivatives is red-shifted upon benzoannulation45, the near-IR band of the Temari-type complexes is blue-shifted upon benzoannulation (Fig. 6c). The introduction of electron-donating groups at suitable positions of Pc is an efficient approach to red-shift the Q band (by ca. 100 nm)46,47, while substituent effects on the benzoannulated complexes are relatively small (2a vs 2c: 11 nm, 220 cm−1, 2b vs 2c: 39 nm, 740 cm−1). The effects of changing the central metal are more significant than those arising from changing the peripheral substituents or from benzoannulation (Fig. 6d). The Fe complexes with both non-benzoannulated (3) and benzoannulated (4) units exhibit a red-shift of ca. 110 nm (ca. 1700 cm−1) compared to the bands of the corresponding Ru complexes (1a and 2a). Moreover, the shoulder of the absorption band of 3 reached 900 nm. This central-metal effect clearly indicates that the orbitals of the metal contribute to these intense near-IR bands.

UV-vis-NIR absorption spectra in CHCl3 of Ru complexes upon (a) changes at the Ar1 position, (b) changes at the Ar2 position, (c) benzoannulation, and (d) of iron complexes.
The cyclic voltammograms (CVs) of RuTAP, 1a, 1c, 1d, 2a, 2b, and 3 were measured in THF, and representative curves are shown in Fig. 7. It is well established for azaporphyrins that the potential difference between the first oxidation and reduction potentials (E1ox − E1red) is correlated to the lowest transition energy48. All complexes present one or two reduction and oxidation processes. The reduction waves of some complexes are partly irreversible, so that their redox potentials were assigned by using corresponding differential pulse voltammograms (DPVs) (Supplementary Fig. 5). The value of E1ox − E1red decreases in the order RuTAP > 2a ~ 2b > 1c > 1a > 3, supporting the position of the intense absorption band in the visible-to-near-IR region. An anodic shift (0.18 V) of the oxidation potential was observed upon changing the Ar1 substituent (from 1a to 1c), while changing the Ar2 group (from 1a to 1d) only marginally influenced the oxidation potential. Upon benzoannulation (from 1a to 2a), the first reduction couple of 2a exhibits a cathodic shift of 0.19 V compared to the first reduction couple (−1.57 V) of 1a, i.e., the LUMO is destabilized by the benzoannulation. On the other hand, the first oxidation couple of 3 shows a cathodic shift of 0.08 V compared to the first oxidation couple (−0.19 V) of 1a. Hence, the red-shift of the near-IR band of Fe complexes can be assigned to a destabilization of the HOMO.

Cyclic voltammograms of RuTAP, 1a, 1c, 1d, 2a, 2b, and 3; [analyte] = 0.5 mM; solvent: THF; supporting electrolyte: 0.1 M [nBu4N][ClO4]. All potentials are referenced to the ferrocene/ferrocenium couple. †In CHCl3.
Rationalizing the structure–property relationships
Based on the experimental optical and electrochemical results, we attempted to rationalize the structure–property relationships in these Temari-shaped complexes by performing molecular-orbital (MO) calculations. Model complexes RuTAP’ (tetraazaporphyrin), 1a’ (non-benzoannulated), and 2a’ (benzoannulated) were used, where the peripheral p-tert-butylphenyl substituents were replaced by phenyl groups in the interest of simplicity. The optimized structures were consistent with those determined from the experimental spectroscopy measurements (Supplementary Fig. 6). The partial MO energy diagrams of the model structures and calculated absorption spectra are shown in Fig. 8a,b, and the results of time-dependent density functional theory (TD-DFT) calculations are summarized in Supplementary Table 5. The calculated intense absorption bands in the longer wavelength regions for all compounds correspond to HOMO–LUMO transitions. The frontier orbitals of RuTAP’ follow the Gouterman “four-orbital” model49, i.e., these orbitals correspond to a1u-, egy-, and egx orbitals. Therefore, the calculated transition at 508 nm can be assigned to the experimental Q band as a π–π* transition, while the contribution of the central ruthenium atom is small.

(a) Partial molecular energy diagram and orbitals of RuTAP’, 1a’, and 2a’ as well as (b) their calculated absorption spectra. Blue and red plots indicate occupied and unoccupied MOs, respectively. (c) Magnified HOMO of 1a’. (d) Magnified LUMO of 1a’. (e) Magnified HOMO of 2a’. (f) Magnified LUMO of 2a’. (g) Qualitative MO diagrams of the frontier orbitals of 1a’ and 2a’ and the HOMO of 1,3-butadiene. The MO isovalues are 0.030 in panels a and g, and 0.010 in panels c–f. Calculations were carried out at the ωB97XD/631SDD//B3LYP/631SDD level of theory, using the polarizable continuum model (PCM) that mimicked the solvation effect of CHCl3 (for details, see the Supplementary Information).
The calculated transition of 1a’ at 633 nm supports the intense absorption band of 1a appearing at a longer wavelength than that of RuTAP (the calculated absorption bands for RuTAP’ are 515 and 512 nm). The HOMO of 1a’ is located on the d-orbital of the central ruthenium atom, while the LUMO is localized on the ligand. Hence, the HOMO–LUMO transition can be assigned to an MLCT transition. The MCD and CV results support this conclusion. The weak MCD signal indicates that the transition has CT character with small changes in the angular momentum37. Since the oxidation potential contributes to the HOMO energy level, the oxidation potential of Fe complex 3 shifts cathodically with a smaller E1ox − E1red gap.
Although the peripheral substituent effects were relatively small, these effects were clarified by careful inspection of the MOs of model complexes. In terms of the electronic contributions to the HOMO of 1a’, a weak contribution from the Ar1 position was observed (Fig. 8c). In the case of 1c, the electron-deficient pyridyl groups stabilize the HOMO of the complex. Therefore, the band gap of 1c increases and a blue-shifted absorption band is observed. The first oxidation couple in the voltammogram of 1c (−0.01 V) appears more anodically shifted than that of 1a, confirming that the heterocycles influence the HOMO of the complexes. The calculated absorption spectrum of the model complex 1c’ also supported the differences (Supplementary Fig. 7). On the other hand, the contribution from the Ar2 positions is small for both the HOMO and LUMO (Fig. 8d), indicating that the physical properties depend only marginally on the diimine substituents. For the benzoannulated complexes, the contribution from the annulated benzene moieties to the LUMO is larger than that to the HOMO (Fig. 8e,f). In the case of Pc, the introduction of electron-donating groups destabilizes the HOMO46, while electron-donating substituents stabilize the LUMO, in agreement with the experimental absorption spectra of 2a and 2b.
The blue-shift of the intense near-IR band upon benzoannulation was also reproduced by the calculations. The LUMO of 2a’ was destabilized, supporting a cathodic shift of the reduction potential of 2a. The changes in MO energy can be explained by an MO diagram based on a simple theory50,51. Benzoannulation can be interpreted as an MO interaction between parent 1a’ and 1,3-butadiene (Fig. 8g). The symmetry of the interacting orbitals determines whether the orbitals are able to interact or not. The HOMO of 1,3-butadiene favors cooperative interactions with the appropriate position of the LUMO of 1a’, leading to an occupied bonding MO (not shown) and an unoccupied antibonding orbital (LUMO of 2a’), while the HOMO of 1a’ cannot interact with the MO of 1,3-butadiene due to a mismatch of symmetry. The net effect of these interactions is an unaltered HOMO and a destabilized LUMO, resulting in a blue-shifted MLCT absorption upon benzoannulation of 1a’.
Discussion
The one-step condensation of cyanoaryls and diimines in the presence of a metal (Ru or Fe) afforded novel ball-shaped (Temari) complexes that absorb near-IR light beyond 700 nm. Various azaporphyrin precursors could be used in this synthesis and a series of compounds with various substituent groups/benzoannulated moieties/central metals were successfully prepared and characterized. MCD spectra and theoretical calculations revealed that MLCT transitions significantly contribute to this near-IR absorption band. As summarized in Fig. 9, the effects of the peripheral substituents and central metal on the optical and electrochemical properties were rationalized based on a combination of theoretical calculations and experimental results. These features highlight the advantages of azaporphyrins (Fig. 1) for industrial applications, which are currently under investigation. However, the concept underlying the synthetic strategy in this work should open the door to a variety of applications of metal-based near-IR materials.

Summary of the substituent effects on the physical properties.
Methods
Typical procedure for the synthesis of Temari-shaped ruthenium complexes such as 1a
Under an Ar atmosphere, diimine 5a (89.3 mg, 0.25 mmol) and ruthenium trichloride (31.5 mg, 0.15 mmol) were dissolved in benzonitrile (1.0 mL, 9.7 mmol) and dimethylaminoethanol (0.5 mL), followed by the addition of DBU (0.1 mL, 0.67 mmol). The mixture was stirred under reflux for 12 h, before it was concentrated. The residue was dissolved and extracted with CHCl3. The organic layer was washed with brine and dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by column chromatography on silica gel (eluent: CHCl3/Hexane = 1/1 v/v) to provide 1a as a moss-green solid (23.6 mg, 18.8 μmol, 15%) after recrystallization from methanol. 1H NMR (500 MHz, CDCl3, 1 mM): δ 9.53 (s, 4H, imine-NH), 7.98 (d, 8H, J = 8.5 Hz, pyrrole-tBuPh-PhH), 7.80–7.78 (m, 8H, ArCN-PhH), 7.50 (d, 8H, J = 8.5 Hz, pyrrole-tBuPh-PhH), 7.29–7.26 (m, 12H, ArCN-PhH), 1.41 (s, 36H, pyrrole-tBuPh-tBuH); UV-Vis-NIR (CHCl3): λmax = 767 nm (ε = 35000 M−1 cm−1) and 345 nm (ε = 63000 M−1 cm−1); HR-MS (MALDI-MS): m/z = 1230.5290, calcd. for (C76H76N10Ru)+ = 1230.5313 [(M)+].
Theoretical calculations
DFT geometry optimizations were carried out using the B3LYP functional of the Gaussian 09 software package52. The Ru atoms were described using an SDD53. The 6–31G* basis set was used for all the other atoms (denoted as 631SDD). After geometry optimization, TD-DFT calculations were performed using ωB97XD54 and the same basis set. All calculations used a relatively simple self-consistent reaction field (SCRF) method based on the polarizable continuum model (PCM)55 that mimicked the solvation effect of chloroform (ε = 4.7113). For further details, calculated coordinates, and TD-DFT output, see the Supplementary Information.
Data availability
Crystallographic data (CIF files) for RuTAP, 1d, and 3 have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publications under reference numbers CCDC 1892288 (RuTAP), CCDC 1892286 (1d), and CCDC 1892287 (3). These data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the article and its Supplementary Information.
References
Kojima, T., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
O’Regan, B. & Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353, 737–740 (1991).
Blankenship, R. E. et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 332, 805–809 (2011).
Okamura, M. et al. A pentanuclear iron catalyst designed for water oxidation. Nature 530, 465–468 (2016).
Vikesland, P. J. & Wigginton, K. R. Nanomaterial enabled biosensors for pathogen monitoring - A review. Environ. Sci. Technol. 44, 3656–3669 (2010).
Chen, G., Roy, I., Yang, C. & Prasad, P. N. Nanochemistry and nanomedicine for nanoparticle-based diagnostics and therapy. Chem. Rev. 116, 2826–2885 (2016).
Agostinis, P. et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 61, 250–281 (2011).
Kinoshita, T. et al. Spectral splitting photovoltaics using perovskite and wideband dye-sensitized solar cells. Nat. Commun. 6, 8834 (2015).
Kimura, M., Nomoto, H., Masaki, N. & Mori, S. Dye molecules for simple co-sensitization process: Fabrication of mixed-dye-sensitized solar cells. Angew. Chem., Int. Ed. 51, 4371–4374 (2012).
Yao, Z. et al. Dithienopicenocarbazole-based acceptors for efficient organic solar cells with optoelectronic response over 1000 nm and an extremely low energy loss. J. Am. Chem. Soc. 140, 2054–2057 (2018).
Zitzler-Kunkel, A. et al. NIR-absorbing merocyanine dyes for BHJ solar cells. Chem. Mater. 26, 4856–4866 (2014).
Zhu, M. et al. Metal-free photocatalyst for H2 evolution in visible to near-infrared region: Black phosphorus/graphitic carbon nitride. J. Am. Chem. Soc. 139, 13234–13242 (2017).
Whittemore, T. J. et al. New Rh2(II, II) complexes for solar energy applications: Panchromatic absorption and excited-state reactivity. J. Am. Chem. Soc. 139, 14724–14732 (2017).
Ravertz, B. D. et al. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 565, 343–346 (2019).
Anderson, E. D., Gorka, A. P. & Schnermann, M. J. Near-infrared uncaging or photosensitizing dictated by oxygen tension. Nat. Commun. 7, 13378 (2016).
Amemori, S., Sasaki, Y., Yanai, N. & Kimizuka, N. Near-infrared-to-visible photon upconversion sensitized by a metal complex with spin-forbidden yet strong S0-T1 absorption. J. Am. Chem. Soc. 138, 8702–8705 (2016).
Xie, J. et al. A NIR dye with high-performance n-type semiconducting properties. Chem. Sci. 7, 499–504 (2016).
Yamaguchi, Y., Ogawa, K., Nakayama, K.-I., Ohba, Y. & Katagiri, H. Terazulene: A high-performance n-type organic field-effect transistor based on molecular orbital distribution control. J. Am. Chem. Soc. 135, 19095–19098 (2013).
Zhou, Y. et al. A phosphorus phthalocyanine formulation with intense absorbance at 1000 nm for deep optical imaging. Theranostics 6, 688–697 (2016).
Myochin, T. et al. Development of a series of near-infrared dark quenchers based on Si-rhodamines and their application to fluorescent probes. J. Am. Chem. Soc. 137, 4759–4765 (2015).
Welsher, K. et al. A route to brightly fluorescent carbon nanotubes for near-infrared imaging in mice. Nat. Nanotech. 4, 773–780 (2009).
Li, B., Lu, L., Zhao, M., Lei, Z. & Zhang, F. An efficient 1064 nm NIR-II excitation fluorescent molecular dye for deep-tissue high-resolution dynamic bioimaging. Angew. Chem., Int. Ed. 57, 7483–7487 (2018).
Kawauchi, K. et al. An anionic phthalocyanine decreases NRAS expression by breaking down its RNA G-quadruplex. Nat. Commun. 9, 2271 (2018).
Mitsunaga, M. et al. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 17, 1685–1691 (2011).
Gorman, A. et al. In vitro demonstration of the heavy-atom effect for photodynamic therapy. J. Am. Chem. Soc. 126, 10619–10631 (2004).
Fukuda, T. & Kobayashi, N. In Handbook of Porphyrin Science (eds Kadish, K. M., Smith, K. M. & Guilard, R.); Vol. 9, p. 1–650 (World Scientific, Singapore, 2010).
Claessens, C. G., González-Rodríguez, D., Rodríguez-Morgade, M. S., Medina, A. & Torres, T. Subphthalocyanines, subporphyrazines, and subporphyrins: singular nonplanar aromatic systems. Chem. Rev. 114, 2192–2277 (2014).
Mack, J. & Kobayashi, N. Low symmetry phthalocyanines and their analogues. Chem. Rev. 111, 281–321 (2011).
Juris, A. et al. Ru(II) polypyridine complexes: Photophysics, photochemistry, electrochemistry, and chemiluminescence. Coord. Chem. Rev. 84, 85–277 (1988).
Whittemore, T. J., White, T. A. & Turro, C. New ligand design provides delocalization and promotes strong absorption throughout the visible region in a Ru(II) complex. J. Am. Chem. Soc. 140, 229–234 (2018).
Al-Afyouni, M. H., Rohrabaugh, T. N. Jr., Al-Afyouni, K. F. & Turro, C. New Ru(II) photocages operative with near-IR light: new platform for drug delivery in the PDT window. Chem. Sci. 9, 6711–6720 (2018).
Jiang, T., Polizzi, N. F., Rawson, J. & Therien, M. J. Engineering high-potential photo-oxidants with panchromatic absorption. J. Am. Chem. Soc. 139, 8412–8415 (2017).
Granda, J. M., Donina, L., Dragone, V., Long, D.-L. & Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 559, 377–381 (2018).
Klucznik, T. et al. Efficient synthesis of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 4, 522–532 (2018).
Rawling, T. & McDonagh, A. Ruthenium phthalocyanine and naphthalocyanine complexes: Synthesis, properties and applications. Coord. Chem. Rev. 251, 1128–1157 (2007).
Yoshida, T., Zhou, W., Furuyama, T., Leznoff, D. B. & Kobayashi, N. An extremely air-stable 19π porphyrinoid. J. Am. Chem. Soc. 137, 9258–9261 (2015).
Furuyama, T., Yoshida, T., Hashizume, D. & Kobayashi, N. Phosphorus(V) tetraazaporphyrins: porphyrinoids showing an exceptionally strong CT band between the Soret and Q bands. Chem. Sci. 5, 2466–2474 (2014).
Kimura, T. et al. Preparation, optical and electrochemical properties, and molecular orbital calculations of tetraazaporphyrinato ruthenium(II) bis(4-methylpyridine) fused with one to four diphenylthiophene units. J. Inorg. Biochem. 158, 35–44 (2016).
Cammidge, A. N., Berber, G., Chambrier, I., Hough, P. W. & Cook, M. J. Octaalkylphthalocyaninato ruthenium(II) complexes with mixed axial ligands and supramolecular porphyrin: phthalocyanine structures derived from them. Tetrahedron 61, 4067–4074 (2005).
Awada, A. et al. New acridine-based tridentate ligand for ruthenium(II): Coordination with a twist. Inorg. Chem. 57, 15430–15437 (2018).
Büldt, L. A., Prescimone, A., Neuburger, M. & Wenger, O. S. Photoredox properties of homoleptic d6 metal complexes with the electron-rich 4,4′,5,5′-tetramethoxy-2,2′-bipyridine ligand. Eur. J. Inorg. Chem. 4666–4677 (2015).
Kobayashi, N., Muranaka, A. & Mack, J. Circular Dichroism and Magnetic Circular Dichroism Spectroscopy for Organic Chemists. (Royal Society of Chemistry, London, 2011).
Gonca, E., Baklaci, Ü. G. & Dinçer, H. A. Synthesis and spectral properties of novel seco-porphyrazines with eight 4-biphenyl groups. Polyhedron 27, 2431–2435 (2008).
Gan, Q. et al. Synthesis and photophysical properties of a series of octaphenyl-porphyrazine-magnesium. Inorg. Chem. Commun. 8, 285–288 (2005).
Kobayashi, N., Nakajima, S.-I., Ogata, H. & Fukuda, T. Synthesis, spectroscopy, and electrochemistry of tetra-tert-butylated tetraazaporphyrins, phthalocyanines, naphthalocyanines, and anthracocyanines, together with molecular orbital calculations. Chem.-Eur. J. 10, 6294–6312 (2004).
Furuyama, T., Satoh, K., Kushiya, T. & Kobayashi, N. Design, synthesis and properties of phthalocyanine complexes with main-group elements showing main absorption and fluorescence beyond 1000 nm. J. Am. Chem. Soc. 136, 765–776 (2014).
Kobayashi, N., Ogata, H., Nonaka, N. & Luk’yanets, E. A. Effect of peripheral substitution on the electronic absorption and fluorescence spectra of metal-free and zinc phthalocyanines. Chem.-Eur. J. 9, 5123–5134 (2003).
Lever, A. B. P. In Phthalocyanines: Properties and Applications. (eds Leznoff, C. C. & Lever, A. B. P.) Vol. 3, p. 1 (VCH, Weinheim, Germany, 1993).
Gouterman, M. In The Porphyrins. (ed. Dolphin, D.) Vol. 3, Part A (Academic Press, New York, 1978).
Hanson, K. et al. A paradigm for blue- or red-shifted absorption of small molecules depending on the site of π-extension. J. Am. Chem. Soc. 132, 16247–16255 (2010).
Ortí, E., Piqueras, M. C., Crespo, R. & Brédas, J. L. Influence of annelation on the electronic properties of phthalocyanine macrocycles. Chem. Mater. 2, 110–116 (1990).
Frisch, M. J. et al. Gaussian 09 (Gaussian, Inc., 2009, http://www.gaussian.com/).
Andrae, D., Haeussermann, U., Dolg, M., Stoll, H. & Preuss, H. Energy-adjusted ab initio pseudopotentials for the 2nd and 3rd row transition-elements. Theor. Chem. Acc. 77, 123–141 (1990).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094 (2005).
Acknowledgements
This work was partly supported by a JST PRESTO grant (JPMJPR18TB), a JSPS KAKENHI grant (18K19071), the Sumitomo Foundation, the Japan Prize Foundation, the JGC-S Scholarship Foundation, the Uehara Memorial Foundation, the Kanazawa University frontier science and innovation cooperation young investigator grant, and the Kanazawa University SAKIGAKE Project 2018. The authors thank Prof. Shigehisa Akine and Prof. Yoko Sakata (Kanazawa University) for X-ray measurements, and the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan for the mass spectrometry measurements (Dr. Akio Miyazato at JAIST).
Author information
Authors and Affiliations
Contributions
T.F. directed the project and performed the single-crystal X-ray diffraction analysis as well as theoretical calculations. F.S. and N.S. carried out the synthesis and characterization of all compounds, and performed the optical and electrochemical measurements. H.M. and M.S. participated in helpful discussions. T.F. and F.S. co-wrote the paper with feedback from H.M. and M.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Furuyama, T., Shimasaki, F., Saikawa, N. et al. One-step synthesis of ball-shaped metal complexes with a main absorption band in the near-IR region. Sci Rep 9, 16528 (2019). https://doi.org/10.1038/s41598-019-53014-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53014-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
