
Abstract
Gastrin-releasing peptide receptors (GRPRs) are promising targets in oligometastatic prostate cancer. We have recently used 55Co (T1/2 = 17.5 h) as a label for next day PET imaging of GRPR expression obtaining high imaging contrast. The radionuclide-chelator combination can significantly influence the biodistribution of radiopeptides. Therefore, in this study, we hypothesized that the properties of 55Co-labeled PEG2-RM26 can be improved by identifying the optimal macrocyclic chelator. All analogues (X-PEG2-RM26, X = NOTA,NODAGA,DOTA,DOTAGA) were successfully labeled with radiocobalt with high yields and demonstrated high stability. The radiopeptides bound specifically and with picomolar affinity to GRPR and their cellular processing was characterized by low internalization. The best binding capacity was found for DOTA-PEG2-RM26. Ex vivo biodistribution in PC-3 xenografted mice was characterized by rapid blood clearance via renal excretion. Tumor uptake was similar for all conjugates at 3 h pi, exceeding the uptake in all other organs. Higher kidney uptake and longer retention were associated with N-terminal negative charge (DOTAGA-containing conjugate). Tumor-to-organ ratios increased over time for all constructs, although significant chelator-dependent differences were observed. Concordant with affinity measurements, DOTA-analog had the best retention of activity in tumors, resulting in the highest tumor-to-blood ratio 24 h pi, which translated into high contrast PET/CT imaging (using 55Co).
Similar content being viewed by others


Introduction
Prostate cancer is a highly heterogeneous, multifactorial disease with a complex underlying biology that evolves from initial tumorigenesis to metastatic spread and castration-resistance. During prostate cancer progression, tumors become more invasive, acquiring properties for increased metastatic potential, becoming more difficult to treat. There are no curative therapies for late stage prostate cancer and treatments are focused primarily on disease control and improving the quality of life.
In recent years, the concept of oligometastatic prostate cancer has been receiving increasing attention. Oligometastatic disease is defined as an intermediate state in prostate cancer progression characterized by limited metastatic spread1. It is hypothesized that this intermediate state has a different biology compared to widespread metastatic disease and is susceptible to treatments designed to influence the rate of progression, and potentially even cure the patients2. Therefore, diagnostic tools that can identify oligometastatic prostate cancer and stratify the patients for personalized treatment strategies are highly required.
Currently, prostate cancer diagnostic relies mainly on PSA screening and biopsies based methods, which have significant limitations. PSA screening has limited diagnostic specificity and cannot distinguish between prostate cancer and normal conditions that are associated with high PSA level3. Biopsies are invasive, have a high sampling error of up to 40%, and can lead to severe complications4. Moreover, these diagnostic modalities cannot differentiate between local, regional, and distant disease, which is a prerequisite for oligometastatic prostate cancer diagnosis.
These major limitations of the main prostate cancer diagnostic tools place molecular imaging in a unique position where it can significantly contribute not only in the initial diagnosis and patient stratification, but also in monitoring the response to therapy. However, despite its significant potential, molecular imaging of oligometastatic prostate cancer is not without challenges, and the highest sensitivity and specificity are required for imaging of small metastases5.
Imaging of oligometastatic prostate cancer requires a molecular target that is predominantly expressed in earlier stages of disease such as gastrin releasing peptide receptor (GRPR). GRPR overexpression was documented in primary tumors (63–100%), lymph node metastases (86%) and bone metastases (53%)6,7. Moreover, normal prostate tissue as well as benign prostate tumors are mostly GRPR-negative8.
Bombesin (BN), a small linear peptide, binds to GRPRs with high affinity and selectivity. The use of peptides such as bombesin-based ligands for tumor targeting offers the advantages of excellent vascular permeability and rapid access to tumors. Consequently, several bombesin derivatives have been developed in the past decades. Encouraged by the success reported for somatostatin antagonists9, the initial consensus favoring bombesin agonists has also shifted towards the use of potent bombesin antagonists10.
We have previously reported on an antagonistic analogue of bombesin NOTA-PEG2-RM26 labeled with 111In and 68Ga that had a favorable biodistribution profile with a high and specific GRPR-mediated tumor uptake11. Interestingly, the fast clearance of radioactivity from blood and normal tissues in combination with long retention in tumors resulted in a pronounced increase in tumor-to-background ratios over time up to 24 h for 111In-NOTA-PEG2-RM26. This data indicates that a long lived radionuclide would be desirable for high contrast imaging.
The use of positron emission tomography (PET) would additionally improve imaging of low abdominal lymph node involvement which requires the highest possible sensitivity. In addition to higher sensitivity, PET also provides better spatial resolution and more accurate quantification compared to single photon emission computed tomography (SPECT). Among the possible long lived positron-emitting radiometals, 55Co (T1/2 = 17.5 h) has a high positron abundance (76% β+) and good ratio between annihilation photons and co-emitted gammas. Cobalt-55 can be produced on low-energy biomedical cyclotrons at most PET centers and/or be distributed to distant sites. Production costs for cobalt-55 are comparable with copper-64. Interestingly, the use of radiocobalt for labeling of NOTA-PEG2-RM26 has led to a remarkable increase in tumor-to-organ ratios than the previously evaluated 68Ga and 111In-labeled NOTA-PEG2-RM2612. Tumor-to-blood ratio for 55/57Co-NOTA-PEG2-RM26 at the 3 h time point was twofold higher than 111In- and 4-fold higher compared to 68Ga-labeled analogues. Moreover, exceptionally high tumor-to blood, tumor-to-pancreas, tumor-to-stomach, and tumor-to kidney ratios were achieved at the later time point of 24 h post-injection (pi)12.
The remarkable potential of 55Co-NOTA-PEG2-RM26 for high contrast PET imaging of GRPR-expressing tumors, raise the question of whether the imaging properties of this conjugate can be further improved. Changes of chelating moieties are likely to provide this opportunity since it is well known that the structure and physical properties of radionuclide-chelator complex can have a large impact on the biodistribution and targeting properties of radiopeptides11,13,14,15,16.
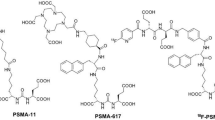
The goal of this study was to further improve the biodistribution of radiocobalt-labeled RM26. We have coupled tri- and tetra-aza macrocyclic chelators to the N-terminus of PEG2-RM26 (X-PEG2-RM26, X = NOTA, NODAGA, DOTA, DOTAGA, Fig. 1) and investigated the respective conjugates for their efficacy in imaging of GRPR-expressing tumors. Cobalt-57 (T1/2 = 271.6 d, E = 122 keV (86%), 136 keV (10%)) was used for in vitro and ex vivo experiments for convenience as proposed earlier12.

Schematic overview of the structure of different chelators: NOTA, NODAGA, DOTA and DOTAGA, coupled to RM26 peptide via a PEG2 spacer.
Results
Labeling and stability
All conjugates were successfully labeled with 57Co and 55Co with yields exceeding 98% (Table 1). Stability was evaluated for all labeled constructs after incubation in the presence of 1000-fold molar excess of EDTA for 1 h at RT. The radionuclide release was lower than 0.4% for all analogues, as shown by ITLC analysis (Table 1).
In vitro studies
The results of the in vitro binding specificity test are presented in Fig. 2. Pre-saturation of receptors with excess amount of non-labeled conjugates caused a significant reduction of 57Co-X-PEG2-RM26 uptake. This demonstrated that the uptake was receptor mediated.

Binding specificity of 57Co-X-PEG2-RM26 (X = NOTA, NODAGA, DOTA and DOTAGA) to GRPR-expressing PC-3 cells. Two groups of culture dishes containing PC-3 cells were incubated with radiolabeled conjugates (1 nM). One set of dishes was pre-saturated with excess amount of non-labeled conjugates (blocked). Data are presented as average ± standard deviation. Data were analyzed by an unpaired, two-tailed t-test.
Data concerning cellular processing and internalization of 57Co-X-PEG2-RM26 are presented in Fig. 3. The internalization pattern was similar for all analogues reaching only 10–12% of cell-associated activity after 24 h incubation. The binding pattern was characterized by a rapid increase of total cell-associated radioactivity within the first hour of incubation, followed by a gradual increase up to 24 h for all conjugates.

Binding and cellular processing of 57Co-X-PEG2-RM26 by GRPR-expressing PC-3 cells. Radiolabeled conjugates were incubated up to 24 h at 37 °C. Cell-bound activity was normalized to the maximum uptake. Data are presented as mean value ± standard deviation. Error bars may not be visible because they are smaller than point symbols.
The half-maximal inhibitory concentration (IC50) was evaluated using 125I-Tyr4-BBN as displacement radioligand (Fig. 4, Table 2). All IC50 values were in the low nanomolar range. The best binding affinity was observed for DOTA and NOTA-containing analogues. Higher IC50 values, corresponding to lower affinity were obtained for NODAGA and DOTAGA-containing constructs.

Inhibition of 125I-Tyr4-BBN binding to PC-3 cells with natCo-X-PEG2-RM26 [X = NOTA (●), NODAGA (■), DOTA (▼) and DOTAGA (◆)]. Data are presented as mean value ± standard deviation.
The binding affinity of 57Co-X-PEG2-RM26 to PC-3 cells was also assessed in real-time using LigandTracer Yellow instruments (Table 2). Similar to IC50 results, 57Co-DOTA-PEG2-RM26 had the best affinity, with a rapid binding and a slow dissociation, followed by 57Co-NOTA-PEG2-RM26. Worse KD values were obtained for NODAGA and DOTAGA-containing analogues. It must be noted that all KD values were in the low picomolar range. By measuring the time to equilibrium using real-time kinetic binding studies, the risk of premature interruption of incubation is eliminated. This is very important in the characterization of high affinity binders that require extremely long time to reach equilibrium. Therefore, the binding affinity can be severely underestimated in end-point assays (IC50 values differ in three orders of magnitude compared to KD values for the studied conjugates).
In vivo studies
Biodistribution of free cobalt was studied 3 and 24 h pi (Supplementary Fig. 1). Uniform activity distribution was observed at both time points with somewhat elevated uptake in liver that was in good agreement with previously published data17.
Biodistribution of radiocobalt-labeled conjugates and comparison of their in vivo tumor targeting was evaluated in BALB/c nu/nu mice bearing PC-3 xenografts 3 and 24 h pi (Fig. 5). The common feature of all conjugates was the rapid clearance from blood and normal tissues. Tumor uptake was similar for all analogues 3 h pi, exceeding the uptake in all normal organs, including excretory organs. Despite the similar initial uptake, radioactivity retention in tumors differed and 24 h pi tumor radioactivity was twofold higher for the DOTA-containing conjugate (4.4 ± 1.3%IA/g for DOTA, 2.1 ± 0.2%IA/g for NOTA, 2.3 ± 0.9%IA/g for NODAGA, and 2.3 ± 0.7%IA/g for DOTAGA). The accumulated activity in GRPR-expressing organs was also significantly higher for 57Co-DOTA-PEG2-RM26, with more than 6-fold higher pancreatic uptake compared to all other constructs. The main excretion pathway was through the kidneys for all analogs, although significant differences in renal uptake and retention were observed. 57Co-NOTA-PEG2-RM26 had a significantly lower kidney uptake both 3 h and 24 h pi, followed by NODAGA and DOTA-containing constructs. The highest renal activity uptake and the longest activity retention were observed for the DOTAGA-construct. The differences in kidney uptake became more pronounced at the later time point (24 h pi) with DOTAGA-conjugate having a 20-fold higher uptake compared to NOTA- and a twofold higher uptake compared to NODAGA- and DOTA- containing analogues. Interestingly, a high uptake was detected for NODAGA and DOTA-carrying radioligands in the GI tract with contents (~5%ID for NODAGA and DOTA vs ≤ 1%ID/g for NOTA and DOTAGA, 3 h pi).

Biodistribution of 57Co-X-PEG2-RM26 (X = NOTA, NODAGA, DOTA and DOTAGA) in PC-3 xenografted BALB/C nu/nu mice (A) 3 h and (C) 24 h pi. Tumor-to-normal-tissue ratios (B) 3 h and (D) 24 h pi. Data were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Significant difference (p < 0.05) at the same time point. a. Significantly different from NOTA b. Significantly different from NODAGA c. Significantly different from DOTA d. Significantly different from DOTAGA.
Tumor-to-organ ratios are presented in Fig. 5. NOTA-coupled analogue provided significantly higher tumor-to-blood and tumor-to-kidney ratios 3 h pi. However, at the later time point, the better retention in tumor of 57Co-DOTA-PEG2-RM26 resulted in a twofold higher tumor-to-blood ratio compared to NOTA-coupled analogue.
In vitro binding specificity to GRPR for 57Co-DOTA-PEG2-RM26 was confirmed in NMRI mice (Supplementary Fig. 2). Significant decrease in activity uptake was found in pancreas and tissues of GI tract (organs with GRPR expression) when higher peptide dose was injected.
Imaging
GRPR expression in PC-3 xenografted mice was successfully visualized using PET/CT after the intravenous injection of 55Co-DOTA-PEG2-RM26 (X = NOTA, NODAGA, DOTA, DOTAGA) (Fig. 6). The activity uptake in the GI tract for NODAGA and DOTA-carrying radioligands and the high kidney uptake of the DOTAGA-conjugate can be clearly visualized on the coronal MIP images acquired 3 h pi. At the later time-point of 24 h, the faster clearance of radioactivity from normal organs compared to GRPR-expressing tumors resulted in high contrast imaging. The best contrast 24 h pi was obtained for DOTA and NOTA-containing analogues.

Coronal MIP PET/CT images showing tracer distribution in PC-3 xenografted NOD-SCID mice. The animals were injected with 0.3 nmol (range: 0.24–0.32 nmol, approx. 2.5 MBq) 55Co-X-PEG2-RM26 (X = NOTA, NODAGA, DOTA and DOTAGA).
Discussion
Receptors for small regulatory peptides are often overexpressed in human tumors. One example of such receptors are GRPRs, which are overexpressed in early prostate cancer and can be targeted for diagnostic imaging and therapy of oligometastatic prostate cancer. Peptides such as the bombesin-based ligands targeting GRPRs are attractive for radionuclide molecular imaging due to their relatively small size which translates into fast blood clearance, fast tumor penetration and inexpensive production.
In this study, we have attempted to optimize the GRPR antagonist PEG2-RM26 by identifying the most suitable macrocyclic chelator for labeling with radiocobalt (55/57Co-X-PEG2-RM26, X = NOTA, NODAGA, DOTA, DOTAGA). Earlier data have shown that chelators had a profound influence on the biodistribution profile of the 68Ga and 111In-labeled X-PEG2-RM26 significantly altering the blood clearance, uptake in tumors and GRPR-expressing organs and clearance of activity from kidneys15,16. The four macrocyclic chelators differ in size, with NOTA and NODAGA containing three nitrogen donor atoms compared to the four nitrogens of DOTA and DOTAGA (Fig. 1). In addition, NODAGA is a derivative of NOTA and DOTAGA is a DOTA-derivative each containing an additional carboxylic acid group.
Two characteristics of the radiometal-chelator complex have been previously identified to have major impact on the behavior of radiopeptides: complex charge and geometry. The charge of the complex depends on the valency of the metal and the number of available carboxylic acid arms of the chelator. Labeling of analogues with radiocobalt is performed in acidic conditions that prevents oxidation of Co2+ by atmospheric oxygen to Co3+. Heppeler et al. have previously shown CoII to be stable against oxidation during labeling and complexation reactions of DOTA-containing somatostatin analogs13. Consequently, the overall charge of the Co2+-chelator complex is different compared to the previously evaluated 68Ga3+ and 111In3+ labeled analogues and will be 0 for NOTA, −1 for NODAGA and DOTA, and −2 for DOTAGA. In terms of size, cobalt ionic radius is similar to the one of gallium (0.65 Å) and smaller than indium (0.92 Å). Similarly to gallium, Co2+ is hexacoordinated to NOTA and DOTA and forms DOTA complexes with distorted octahedral coordination geometry13,18. Structures of Co2+ complexes with NODAGA and DOTAGA can be deduced from the aforementioned parent NOTA and DOTA chelators.
All tested analogues were successfully labeled with radiocobalt with yields exceeding 99% for 57Co and 98% for 55Co. Labeling did not affect the binding specificity of the conjugates to GRPR despite harsh labeling conditions. In principle, different chelator-radionuclide combinations could influence the internalization rate of radiometal-labeled peptides. However, data from cellular processing assay showed a similar internalization pattern for all analogues, with a very low fraction of internalized radioactivity. Although a low internalization is common for antagonists, the internalized fraction for the radiocobalt-labeled conjugates was three fold lower compared to the same analogues labeled with other radiometals such as 111In and 68Ga15,16. These findings were in agreement with data published for proteins labeled with cobalt12,19,20 and might indicate lower residualizing properties of the radiocatabolites for the cobalt labeled conjugates.
The binding properties of natCo-X-PEG2-RM26 were evaluated in a competitive binding assay showing IC50 values in the low nanomolar range for all conjugates. Surprisingly, IC50 values did not follow the same pattern as previously observed for indium and gallium labeled X-PEG2-RM26 or for somatostatin analogues which showed that a negative N-terminal charge will decrease the binding affinity21,22. Although the presence of a -2 charge for DOTAGA-cobalt complex resulted in the worst binding affinity, the best binding affinity was observed for negatively charged DOTA-containing analogue, followed by neutral NOTA-construct.
Equilibrium dissociation constant (KD) values obtained by measuring the binding affinity in real-time were in the picomolar range and had the same pattern as the IC50 values with the best binding affinity observed for 57Co-DOTA-PEG2-RM26, followed by NOTA-, NODAGA- and DOTAGA-conjugates indicating that N-terminus charge is not the only factor that affects the binding affinity of radiocobalt-labeled conjugates.
Biodistribution pattern of 57Co-X-PEG2-RM26 in tumor-bearing mice was characterized by rapid clearance from blood which is important for high-contrast molecular imaging. The clearance was predominantly via kidney ultrafiltration and a low activity uptake was observed for all analogues in liver and unspecific compartments. Comparison of distribution of 57Co-X-PEG2-RM26 with distribution of free cobalt corroborated the high stability of all studied cobalt complexes. Concentration of free cobalt in blood and normal organs was one-two orders of magnitude higher than for tested conjugates. Tumor uptake was on the same level for all conjugates 3 h pi, exceeding the uptake in all normal organs, including excretory organs. Interestingly, despite similar tumor uptake, the activity accumulation in GRPR-expressing organs differed significantly between conjugates. Notably, the pancreatic uptake (the organ with the most abundant GRPR expression) was more than 6-fold higher for DOTA-containing analogue compared to all other constructs. This discrepancy between the uptake in tumor and GRPR-expressing organs was previously reported for the same conjugates labeled with 111In16 and for other radiolabeled bombesin analogues23,24,25 and is most likely due to differences in affinity towards human and murine GRPR.
A clear influence of the N-terminal charge was observed on kidney activity uptake and retention with the lowest uptake and retention for neutral NOTA- and the highest uptake and the longest retention for the -2 charged DOTAGA analogue. A surprising finding was the high uptake in GI tract (with contents) for NODAGA- and DOTA-containing analogues 3 h pi that did not correlate with conjugate lipophilicity nor complex charge and geometry. Comparison of biodistribution patterns of 57Co-DOTA-PEG2-RM26 injected in low and high peptide dose and free cobalt in NMRI mice (Fig. 5A and Supplementary Fig. 1) lead us to the conclusion that the high activity uptake in GI tract observed for these conjugates partially consists of GRPR mediated uptake in GI tract tissue (specific uptake in pancreas, stomach and intestine) and partially due to GI tract content. Because we did not observe a biodistribution pattern characteristic for free cobalt (elevated activity concentration in blood, uniform high activity uptake in normal organs, and slow whole body clearance) for the cobalt labeled peptides we concluded that the elevated activity uptake in GI tract was not the result of complex instability in vivo. With high probability the GI tract content includes products of hepatic degradation of labeled peptide, either cobalt-chelate complexes or released cobalt, indicating that some excretion occurs through the bile for these conjugates. However, liver uptake was below 0.1%ID/g for all constructs 3 h pi and activity was rapidly excreted from GI tract for DOTA conjugate.
Perhaps the most remarkable finding of this study was the two fold higher retention of activity in tumors for DOTA-containing analogue that correlates with the better binding affinity found for this analogue. Due to the faster clearance of activity from blood and normal organs compared to tumors, tumor-to-organ ratios generally improved between 3 and 24 h pi, with 57Co-DOTA-PEG2-RM26 having the highest tumor-to-blood ratio 24 h pi. PET/CT imaging using 55Co-X-PEG2-RM26 were in accordance with the biodistribution results with NOTA-containing analogue demonstrating superior imaging properties 3 h pi. However, at the later time-point of 24 h pi, the best imaging contrast was obtained for 55Co-DOTA-PEG2-RM26.
Comparing indium and cobalt labeled X-PEG2-RM26 conjugates 24 h pi we have found that while for indium the best contrast was achieved for neutrally charged In-NODAGA complex16, negatively charged Co-DOTA conjugate had the best performance for cobalt labeled conjugates. Indium and cobalt labeled NODAGA-conjugates similarly had high uptake in GRPR-expressing organs and tissue in gastrointestinal tract at early time point. This activity uptake rapidly decreased for 111In-NODAGA-PEG2-RM26, but not for the conjugate labeled with cobalt.
High contrast imaging is essential for the evaluation of intraprostatic and peri-prostatic tissues and for local staging of suspect lymph nodes in prostate cancer. This is due to the fact that almost 80% of metastatic lymph nodes have a diameter of less than 8 mm, smaller than what could be detected in CT and MRI (10 mm)26.
We have compared published data for tracers suitable for next day imaging of GRPR expression using PET (labeled with copper-64 (T½ 17.5 h, β+ 18%) and cobalt-55) and SPECT (labeled with indium-111 (T½ 2.6 d)) (Supplementary Table 3). Cobalt labeled DOTA-PEG2-RM26 conjugate has appreciably higher tumor-to-blood and tumor-to-liver ratios than conjugates labeled with copper22,27. Conjugates suitable for SPECT imaging generally demonstrated high tumor-to-organ ratios16,28,29 and together with the new generation of SPECT cameras, indium labeled GRPR antagonists should provide images with high contrast. GRPR agonists demonstrated the lowest tumor-to-organ ratios as already mentioned above29,30.
In conclusion, the charge and structure of chelating moiety had a profound influence on tumor targeting and biodistribution profile of radiocobalt-labeled GRPR antagonist RM26. The net charge of the radionuclide-chelator complex influenced the uptake and retention of activity in the kidneys. The highest binding affinity observed for DOTA-containing analogue resulted in better retention of radioactivity in tumors indicating that 55Co-DOTA-PEG2-RM26 is a promising candidate for next day high contrast PET imaging of GRPR-expressing tumors.
Methods
57Co cobalt chloride was purchased from PerkinElmer Sweden (Upplands Vasby, Sweden). 55Co was produced and purified in house as previously described31,32. Buffers used for labeling were prepared in-house using high-quality Milli-Q water and chemicals supplied by Merck (Darmstadt, Germany). After preparation, buffers were pretreated with Chelex 100 resin (Bio-Rad Laboratories, Richmond, USA) to remove metal contamination.
The synthesis and characterization of X-PEG2-RM26 (RM26 = [D-Phe6, Sta13, Leu14]Bombesin[6–14], X = NOTA, NODAGA, DOTA and DOTAGA) (Fig. 1) were performed as previously described11.
The GRPR-expressing prostate cancer cell line PC-3 used in the cell studies was obtained from ATCC, LGC Promochem. Cells were cultured in RPMI-1640 media supplemented with 10% fetal calf serum (Sigma), PEST (penicillin 100 IU/mL, streptomycin 100 g/mL), and 2 mM L-glutamine (all from Biochrom AG, Berlin, Germany). This medium is referred to as complete medium in the text. The cells were detached using a trypsin-EDTA solution (0.05% trypsin, 0.02% EDTA in buffer; Biochrom AG).
An automated γ-spectrometer (3-inch NaI(Tl) detector, 2480 WIZARD2, PerkinElmer) was used to measure radioactivity.
Statistics
Data were analyzed using GraphPad Prism (version 7.03, GraphPad Software Inc.) to determine significant statistical differences (p < 0.05) by an unpaired, two-tailed t-test and by one-way ANOVA with Bonferroni correction for multiple comparisons.
Labeling and stability
Labeling of X-PEG2-RM26 with 57Co was performed using 10 nmol (aq. 1 nmol/µL) of peptide, buffered with 80 µL ammonium acetate (0.2 M, pH 5.5). For labeling with 55Co, X-PEG2-RM26 were buffered with 80 µL acetate buffer (0.2 M, pH 5.5), followed by dynamic microwave heating to 90 °C for up to 3 min in a sealed vial using a PETWave (CEM Corp.). After labeling with 55Co, radiochemical yield was evaluated by radio HPLC as previously described32. Stability was evaluated by instant thin-layer chromatography (ITLC). In this experiment, 57Co-labeled peptides were incubated in the presence of 1000-fold molar excess of EDTA disodium salt (Sigma) for 1 h at RT. The ITLC strips (150–771 Dark Green, Tec-Control Chromatography strips from Biodex Medical Systems) were eluted with citric acid (0.2 M, pH 2.0). In this system, free 57/55Co or radiocobalt chelated by EDTA migrate with the eluent front (Rf = 1.0) while the radiolabeled constructs remain on the application point (Rf = 0.0). The system was previously validated by radio-HPLC and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
In vitro studies
GRPR-binding specificity of the X-PEG2-RM26 variants after 57Co-labeling was evaluated on PC-3 cells as previously described16. Briefly, PC-3 cells (800,000 per dish) were incubated with 1 nM 57Co-X-PEG2-RM26 for 1 h at 37 °C under 5% CO2. One group of Petri dishes was pre-saturated with 200-fold excess of unlabeled peptide added 10 min prior to the addition of radiolabeled compounds. After incubation, cells were washed with serum-free media and were detached using 0.5 mL trypsin-EDTA solution. Cell-associated radioactivity was measured in an automated gamma-counter and presented as percentage from added radioactivity.
For internalization studies, PC-3 cells (800,000 per dish) were incubated with 57Co-X-PEG2-RM26 (2 nM) at 37 °C under 5% CO2. At predetermined time points (1, 2, 4, 8, and 24 h after incubation start), the incubation medium was removed and the cells were treated using the previously described acid wash method in order to collect the membrane-bound and internalized radioactivity11.
The half-inhibitory concentration (IC50) was estimated for all the natCo-labeled analogues by an in vitro competitive binding assay using 125I-Tyr4-BBN as a displacement radioligand. Briefly, PC-3 cells were incubated with 125I-Tyr4-BBN for 5 h at 4 °C in the presence of 12 increasing concentrations of natCo-X-PEG2-RM26 ranging from 0 to 1000 nM. After incubation, the cells were collected and measured in an automated gamma-counter. IC50 values were calculated by fitting the data by nonlinear regression using GraphPad Prism software.
Kinetics of 57Co-X-PEG2-RM26 binding to and dissociation from living PC-3 cells were measured in real time using LigandTracer Yellow instruments (Ridgeview Instruments AB) at RT as previously described33. The uptake curves were recorded at 0.6 and 1.8 nM. Thereafter, the 57Co-X-PEG2-RM26- containing medium was replaced by fresh medium and the dissociation curve was monitored for 1000 min. The TraceDrawer Software (Ridgeview Instruments) was used to calculate the equilibrium dissociation constant (KD) based on the association and dissociation rates.
In vivo studies
The animal experiments were planned and performed in accordance with the national regulations on laboratory animals’ protection and were approved by the Ethics Committee for Animal Research in Uppsala and Denmark, respectively. Euthanasia was performed by intraperitoneal injection of Ketalar-Rompun solution (10 mg/mL Ketalar and 1 mg/mL Rompun; 20 µL/g body weight) and all efforts were made to minimize suffering. Groups of 4 mice per data point were used.
The biodistribution of cobalt-57 chloride (pre-incubated with ammonium acetate buffer (0.2 M, pH 5.5)) was studied in NMRI mice 3 and 24 h pi (25 kBq in 100 µL PBS per mouse). Blood samples were collected by heart puncture. Lung, liver, spleen, pancreas, stomach, small intestines, kidneys, muscle, bone, and the rest of gastrointestinal tract with its content were collected, weighed and measured in a gamma-counter. The organ uptake values were expressed as a percentage of injected dose per gram of tissue weight (%ID/g) with the exception of gastrointestinal tract and the remaining carcass, which were calculated as %ID per whole sample.
The biodistribution and targeting properties of 57Co-X-PEG2-RM26 were evaluated in female BALB/c nu/nu mice (weight: 18 ± 2 g). GRPR-expressing xenografts were established by subcutaneous injection of 8 × 106 PC-3 cells/mouse, 2 weeks prior to the experiment. At the time of experiment, the mice were intravenously injected with 57Co-X-PEG2-RM26 (25 kBq, 45 pmol in 100 µL PBS per mouse) and were euthanized 3 and 24 h pi.
Specificity of 57Co-DOTA-PEG2-RM26 to GRPR was tested in NMRI mice 1 h pi of 45 or 20 nmol of labeled conjugate (25 kBq, in 100 µL PBS per mouse).
Whole body PET/CT scans were performed using a Siemens Inveon preclinical PET/SPECT/CT-scanner on PC-3 xenografted male NOD-SCID mice (in-bread). The mice were anesthetized with a mixture of 1.5–2% isoflurane and 100% oxygen and intravenously injected with 0.3 nmol (range: 0.24–0.32 nmol) 55Co-X-PEG2-RM26 (approx. 2.5 MBq). At 3 h and 24 h pi, the mice were anesthetized again using isoflurane and PET/CT scanned with PET acquisition times of 15 min. and 30 min., respectively. CT parameters were 2 bed positions, 360 projections in 360 degrees’ rotation, and bin 4. CT and PET images were co-registered and the CT-based attenuation corrected PET data was reconstructed using an OSEM3D/MAP algorithm (4 OSEM3D iterations, 16 MAP subsets, and 18 MAP iterations, target resolution 0.8 mm). PET and CT data were analyzed using Inveon Research Workplace (Siemens Healthcare) and presented as MIPs adjusted to display a color scale from 0 to the maximum tumor uptake value.
References
Ost, P. et al. Metastasis-directed therapy of regional and distant recurrences after curative treatment of prostate cancer: a systematic review of the literature. Eur. Urol. 67, 852–63 (2015).
Reyes, D. K. & Pienta, K. J. The biology and treatment of oligometastatic cancer. Oncotarget. 6, 8491–524 (2015).
Thompson, I. M. et al. Prevalence of prostate cancer among men with a prostate-specific antigen level <or =4.0 ng per milliliter. N. Engl. J. Med. 350, 2239–46 (2004).
Presti, J. Jr. Does the yield of prostate cancer biopsy and repeat biopsy justify the frequency of their use? Nat. Clin. Pract. Urol. 5, 246–7 (2008).
Zhang, H. et al. Evolution of bombesin conjugates for targeted PET imaging of tumors. PLoS One. 7, e44046 (2012).
Markwalder, R. & Reubi, J. C. Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res. 59, 1152–9 (1999).
Ananias, H. J., van den Heuvel, M. C., Helfrich, W. & de Jong, I. J. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate. 69, 1101–8 (2009).
Beer, M. et al. Profiling gastrin-releasing peptide receptor in prostate tissues: clinical implications and molecular correlates. Prostate. 72, 318–25 (2012).
Ginj, M. et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 103, 16436–41 (2006).
Cescato, R. et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 49, 318–26 (2008).
Varasteh, Z. et al. Synthesis and characterization of a high-affinity NOTA-conjugated bombesin antagonist for GRPR-targeted tumor imaging. Bioconjug. Chem. 24, 1144–53 (2013).
Mitran, B. et al. High Contrast PET Imaging of GRPR Expression in Prostate Cancer Using Cobalt-Labeled Bombesin Antagonist RM26. Contrast Media Mol. Imaging. 2017, 6873684 (2017).
Heppeler, A. et al. Metal-ion-dependent biological properties of a chelator-derived somatostatin analogue for tumour targeting. Chemistry. 14, 3026–34 (2008).
Decristoforo, C. et al. 68Ga- and 111In-labelled DOTA-RGD peptides for imaging of alphavbeta3 integrin expression. Eur. J. Nucl. Med. Mol. Imaging. 35, 1507–1515 (2008).
Varasteh, Z. et al. The effect of macrocyclic chelators on the targeting properties of the 68Ga-labeled gastrin releasing peptide receptor antagonist PEG2-RM26. Nucl. Med. Biol. 42, 446–454 (2015).
Mitran, B. et al. Selection of optimal chelator improves the contrast of GRPR imaging using bombesin analogue RM26. Int. J. Oncol. 48, 2124–34 (2016).
Bento, J. et al. Efficiency correction factors of an ACCUSCAN whole-body counter due to the biodistribution of 134Cs, 137Cs and 60Co. Radiat Prot Dosimetry. 155, 16–24 (2012).
Broan, C. J. et al. Structure and solution stability of indium and gallium complexes of 1,4,7-triazacyclononanetriacetate and of yttrium complexes of 1,4,7,10-tetraazacyclododecanetetraacetate and related ligands: kinetically stable complexes for use in imaging and radioimmunotherapy. X-Ray molecular structure of the indium and gallium complexes of 1,4,7-triazacyclononane-1,4,7-triacetic acid. J. Chem. Soc. Perkin. Trans. 2. 0, 87–99 (1991).
Rosestedt, M. et al. Evaluation of a radiocobalt-labelled affibody molecule for imaging of human epidermal growth factor receptor 3 expression. Int. J. Oncol. 51, 1765–1774 (2017).
Garousi, J. et al. The use of radiocobalt as a label improves imaging of EGFR using DOTA-conjugated Affibody molecule. Sci. Rep. 7, 5961 (2017).
Abiraj, K. et al. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 52, 1970–8 (2011).
Gourni, E. et al. N-terminal modifications improve the receptor affinity and pharmacokinetics of radiolabeled peptidic gastrin-releasing peptide receptor antagonists: examples of 68Ga- and 64Cu-labeled peptides for PET imaging. J. Nucl. Med. 55, 1719–25 (2014).
Maina, T. et al. Species differences of bombesin analog interactions with GRP-R define the choice of animal models in the development of GRP-R-targeting drugs. J. Nucl. Med. 46, 823–30 (2005).
Fournier, P. et al. Novel radiolabeled peptides for breast and prostate tumor PET imaging: (64)Cu/and (68)Ga/NOTA-PEG-[D-Tyr(6),βAla(11),Thi(13),Nle(14)]BBN(6-14). Bioconjug. Chem. 23, 1687–93 (2012).
Lane, S. R. et al. Optimization, biological evaluation and microPET imaging of copper-64-labeled bombesin agonists, [64Cu-NO2A-(X)-BBN(7-14)NH2], in a prostate tumor xenografted mouse model. Nucl. Med. Biol. 37, 751–761 (2010).
von Eyben, F. E., Picchio, M., von Eyben, R., Rhee, H. & Bauman, G. 68Ga-Labeled Prostate-specific Membrane Antigen Ligand Positron Emission Tomography/Computed Tomography for Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. Focus. 4, 686–693 (2018).
Gourni, E. et al. Copper-64 Labeled Macrobicyclic Sarcophagine Coupled to a GRP Receptor Antagonist Shows Great Promise for PET Imaging of Prostate Cancer. Mol Pharm. 12, 2781–90 (2015).
Mansi, R. et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging 38, 97–107 (2011).
Mansi, R. et al. Evaluation of a 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid–Conjugated Bombesin-Based Radioantagonist for the Labeling with Single-Photon Emission Computed Tomography, Positron Emission Tomography, and Therapeutic Radionuclides. Clin Cancer Res 15, 5240–49 (2009).
Dam, J. H., Olsen, B. B., Baun, C., Høilund-Carlsen, P. F. & Thisgaard, H. In Vivo Evaluation of a Bombesin Analogue Labeled with Ga-68 and Co-55/57. Mol. Imaging Biol. 18, 368–76 (2016).
Thisgaard, H., Olesen, M. L. & Dam, J. H. Radiosynthesis of 55Co‐ and 58mCo‐labelled DOTATOC for positron emission tomography imaging and targeted radionuclide therapy. J. Labelled Comp. Radiopharm. 54, 758–762 (2011).
Thisgaard, H. et al. Evaluation of cobalt-labeled octreotide analogs for molecular imaging and auger electron-based radionuclide therapy. J. Nucl. Med. 55, 1311–6 (2014).
Xu, B. et al. Detecting ligand interactions with G protein-coupled receptors in real-time on living cells. Biochem. Biophys. Res. Commun. 441, 820–4 (2013).
Acknowledgements
This work was supported by the Swedish Cancer Society (grants CAN 2017/425 (AO), CAN 2018/436 (VT)), the Swedish Research Council (grants 2015–02509 (AO) and 2015–02353 (VT)). The bioimaging experiments reported in this paper were performed at DaMBIC, a bioimaging research core facility, at the University of Southern Denmark. DaMBIC was established by an equipment grant from the Danish Agency for Science Technology and Innovation and by internal funding from the University of Southern Denmark. Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
B.M., H.T., V.T., A.O. and U.R. conceived and designed the experiments; B.M., H.T., S.R., J.H.D., F.A., V.T., A.O. and U.R. performed the experiments; B.M., H.T., V.T., A.O. and U.R. analyzed the data; B.M., H.T., J.H.D., A.O. and U.R. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mitran, B., Thisgaard, H., Rinne, S. et al. Selection of an optimal macrocyclic chelator improves the imaging of prostate cancer using cobalt-labeled GRPR antagonist RM26. Sci Rep 9, 17086 (2019). https://doi.org/10.1038/s41598-019-52914-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52914-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
