
Abstract
Pseudoxanthoma Elasticum (PXE) is a rare disorder characterized by fragmentation and progressive calcification of elastic fibres in connective tissues. Although arterial hypertension (AHT) has been reported in PXE patients, its impact on pathological manifestations has as yet been unexplored. We investigated the consequences of experimental AHT on Abcc6−/− PXE mouse models. Experimental AHT was induced by deoxycorticosterone acetate (DOCA-salt) in uni-nephrectomised mice. Blood pressure (BP) and vascular reactivity were monitored using tail-cuff plethysmography and myography respectively. Calcium content and fibrosis were assessed using colorimetry, Von Kossa and Sirius red staining respectively. The gene expression implicated in vascular biology was measured using quantitative polymerase chain reaction. DOCA-salt induced a matching rise in BP in Abcc6−/− and WT mice. Aortic ring contraction and relaxation in vitro were comparable. Calcium accumulated in the hearts of hypertensive Abcc6−/− mice along with significant fibrosis in the myocardium and aorta by contrast with the WT mice. In hypertensive Abcc6−/− mouse aortas, these results were corroborated by gene expression patterns favouring calcification, fibrosis and extracellular matrix remodelling. Abcc6 loss-of-function is associated with greater cardiovascular calcification and fibrosis in mice subjected to DOCA-Salt hypertension. These results suggest likely cardiovascular deterioration in PXE patients with AHT, necessitating diligent BP monitoring.
Similar content being viewed by others


Introduction
Pseudoxanthoma Elasticum (PXE, OMIM 264800) is a genetic disease characterized by fragmentation and progressive calcification of elastic fibers in the skin, eyes and blood vessels1. The most common vascular manifestations are peripheral arterial disease (PAD) and increased risk of cerebral infarction1. PXE is caused by mutations in the ABCC6 gene which encodes a transmembrane adenosine triphosphate (ATP)-binding cassette (ABC) transporter primarily expressed in the liver and kidney2,3. Several studies have demonstrated that normal tissues exposed to PXE patient serum/plasma tend to calcify, indicating a metabolic etiology underlying this disease consistent with inexistent or low-level ABCC6 transporter expression in affected tissues4. No ABCC6 endogenous substrates have as yet been emphatically identified, cause of ectopic calcification5,6.
Typical PXE manifestations include yellowish papules on the neck and large skin folds, retinal angioid streaks and cardiovascular (CV) complications7 such as: diminished or absent peripheral vascular pulsations; hypertension of unknown origin; echographic opacities due to arterial calcification (especially in the kidneys, spleen and pancreas); AHT; angina pectoris; intermittent claudication (often regarded as the first sign of accelerated atherosclerosis); gastrointestinal hemorrhage; arteriosclerosis; and increased risk of myocardial and cerebral infarction7. There is a higher risk of vascular events in PXE patients than in the general population, presumably resulting from arterial medial calcification along with arterial stiffening and thickening8. Adverse arterial remodelling and increased aortic stiffness are associated with resistant AHT in the general population9. Around 20–25% of PXE patients present CV complications related to high morbidity and mortality such as higher prevalence of accelerated atherosclerosis involving greater risk of complete vascular occlusion10,11.
Since Abcc6−/− mouse models mimic the majority of human PXE manifestations, their use for research into PXE is warranted12. In the present study, Abcc6−/− and WT mice were submitted to deoxycorticosterone acetate (DOCA)-salt-induced AHT, representing an experimental model characterized by an initial rise in AHT over the first few days followed by sustained elevated BP for weeks13. Our aim was to investigate the consequences of DOCA-salt-induced AHT on CV reactivity and remodelling in Abcc6−/− mice by comparison with WT mice.
Results
Comparable DOCA-salt induced arterial hypertension and changes in vascular reactivity observed in Abcc6−/− and WT mice
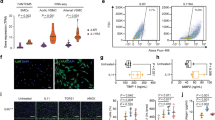
No difference in resting hemodynamic parameters could be found between Abcc6−/− and WT mice (Table 1). DOCA-salt treatment caused a progressive rise in systolic blood pressure (SBP) over a period of 19 days in Abcc6−/− and WT mice (Fig. 1). At end of treatment, SBP was 141.4 ± 5.5 mmHg in Abcc6−/− DOCA vs 109.7 ± 5.1 mmHg in Abcc6−/− mice (p < 0.01) and 153.4 ± 8,8 mmHg in WT DOCA vs 117 ± 3.7 mmHg in WT mice (p < 0.01). There was no difference in SBP between Abcc6−/− and WT mice. Vascular contraction and endothelium-dependent and -independent relaxation were measured in the aortic rings using myography. Contractile response to KCl was unaffected by DOCA-salt and was comparable in both mouse strains (Fig. 2A). Contractile response to 5-hydroxytryptamine (5-HT) increased in both Abcc6−/− and WT DOCA specimens with no evidence of significant genotype effect (Fig. 2B). Similarly, the pre contractions in response to phenylephrine (1 µM prior Ach) were equivalent according to the genotype (tension in mN: 8.1 ± 4.6 vs 5.4 ± 3.4 for untreated WT and Abcc6−/−; 11.6 ± 1.9 vs 11.1 ± 4.3 for DOCA WT vs Abcc6−/−), while a significant gain of contraction associated to the DOCA treatment was found (p = 0.007, data not shown). Endothelium-dependent (acetylcholine, ACh) and -independent (sodium nitroprusside, SNP) relaxation was unaffected by DOCA-salt and was comparable in both mouse strains (Fig. 2C,D).

Comparable DOCA-salt-induced hemodynamic parameters changes in Abcc6−/− and WT mice. Systolic blood pressure kinetic characteristics as well as mean values over ultimate five days of DOCA-salt treatment. Data represent mean ± SEM of six mice from each group *p < 0.05, **p < 0.01 WT untreated versus DOCA; #p < 0.05, ##p < 0.01 Abcc6−/− untreated versus DOCA.

Comparable DOCA-salt-induced vascular reactivity changes in Abcc6−/− and WT mice. (A) Contraction of aortic rings in response to KCl, (B) Dose-response contraction curve in response to 5-HT and (C,D) endothelium-dependent and -independent relaxation (ACh and SNP, respectively) evaluated using wire myography. Data represent mean ± SEM of six mice from each group *p < 0.05, **p < 0.01 WT untreated versus DOCA; #p < 0.05, ##p < 0.01 Abcc6−/− untreated versus DOCA.
DOCA-salt induced calcium accumulation and fibrosis in Abcc6−/− murine hearts
Heart weight to tibia length (HW/TL) ratio was measured to evaluate cardiac hypertrophy, revealing no difference between untreated WT and Abcc6−/− mice. DOCA-salt treatment induced a significant increase in HW/TL ratio in WT (88 ± 8 to 127 ± 8; p < 0.01) but not in Abcc6−/− mice (Fig. 3A).

DOCA-salt-induced cardiac remodelling in Abcc6−/− mice: hypertrophy, calcium accumulation and fibrosis. (A) Heart weight normalized to tibia length (HW/TL) (B) Colorimetric determination of total tissue calcium level in left ventricles and (C) representative images and (D) quantification of Sirius red staining. Data represent mean ± SEM of six mice from each group *p < 0.05, **p < 0.01, ***p < 0.001.
Colorimetric quantification of total tissue calcium showed more significant accumulation in the left ventricles (LV) of Abcc6−/− DOCA than in Abcc6−/− mice (p < 0.05) (Fig. 3B), indicative of dystrophic cardiac calcification (DCC). Comparable tissue calcium increase was not detected in WT mice. All of the Von Kossa staining slides analyzed were negative in the heart LV specimens from the four series of mouse models (data not shown). Heart fibrosis, evidenced by Sirius red staining, occurred in Abcc6−/− DOCA but not Abcc6−/− mice. This was not observed in WT DOCA as opposed to WT mice (Fig. 3C,D). No significant correlation between heart hypertrophy (HW/TL) and the fibrotic score could be evidenced in both WT DOCA mice and Abcc6−/− DOCA mice (data not shown).
DOCA-salt induced vascular fibrosis as distinct from hypertrophy in Abcc6−/− mice
Aortic remodelling arose from hypertension and wall thickness were measured in these animals. Significant vascular hypertrophy was found in WT but not in Abcc6−/− DOCA-treated mice (82.7 ± 2.4 µm vs 75.2 ± 5.6 µm; p < 0.05) by comparison with untreated WT and Abcc6−/− mice (60.7 ± 2 µm vs 67.5 ± 3.1 µm; p > 0.05) (Fig. 4A). Von Kossa staining was negative in the entire TA slides analyzed (Fig. 4B). Fibrosis was significantly more extensive in Abcc6−/− DOCA mice than in Abcc6−/− mice. No positive Sirius red staining was recorded in WT DOCA (Fig. 4C,D). No significant correlation between thoracic aorta (TA) hypertrophy and the fibrotic score could be evidenced neither in WT DOCA nor in Abcc6−/− DOCA mice (data not shown).

DOCA-salt-induced aortic fibrosis excepting hypertrophy in Abcc6−/− mice. (A) Wall thickness measured in TA sections. (B) Representative images of Von Kossa staining performed on TA sections. (C) Representative images and (D) quantification of Sirius red staining performed on TA sections. Data represent mean ± SEM of six mice from each group **p < 0.01, ***p < 0.001.
Molecular approach to vascular remodelling in hypertensive Abcc6−/− and WT mice
We evaluated expression of gene coding in the proteins involved in fibrosis (Fig. 5A), ectopic calcification (Fig. 5B) and matrix remodelling (Fig. 5C) in the TA. No statistical differences were found in gene expression levels between untreated Abcc6−/− and WT mice. Of the genes involved in the fibrotic process, significant increase in collagen 1 and 3 (Col1a1 and Col3a1) and Tgfβ2 expression was found exclusively in Abcc6−/− DOCA mice while a significant increase in connective tissue growth factor (Ctgf) expression was found exclusively in WT DOCA mice (Fig. 5A). Among the genes involved in ectopic mineralization, transcription factor Sp7 expression decreased while osteopontin (Spp1) and matrix Gla protein (Mgp) expression increased in both Abcc6−/− DOCA and WT DOCA mice by comparison with untreated Abcc6−/− and WT mice. Interestingly, pro-mineralizing bone morphogenetic protein 4 (Bmp4) and periostin (Postn) gene expression significantly increased in Abcc6−/− DOCA compared to untreated Abcc6−/− but not in WT DOCA mice, reaching statistical significance subject to genotype (Fig. 5B). Finally, in the genes involved in matrix remodelling, expression of tissue inhibitor of metalloproteinase 1 (Timp1) was higher while matrix metalloproteinase 9 (Mmp9) was lower in both Abcc6−/− DOCA and WT DOCA mouse aortas than in untreated Abcc6−/− and WT mice. Matrix metalloproteinase 2 (Mmp2), plasminogen activator urokinase (Plau) and plasminogen activator inhibitor-1 (Serpine1) expression increased significantly in Abcc6−/− DOCA mice alone. Excepting Timp2 and Plat, significant differences were recorded subject to genotype (Fig. 5C).

Molecular pattern of vascular remodelling in hypertensive Abcc6−/− and WT mice. Gene coding for proteins involved in (A) fibrosis, (B) ectopic calcification and (C) matrix remodelling evaluated by RT-qPCR in the TA. Data represent fold change versus untreated ± SEM from four to six mice per group (Student’s t-test). *p < 0.05, **p < 0.01 untreated versus DOCA. #p < 0.05, ##p < 0.01 WT DOCA versus Abcc6−/− DOCA.
Discussion
In the present study, we evidence that DOCA-salt-induced AHT gives rise to profound CV remodelling in Abcc6−/− mice which was associated with (i) calcium deposits in the heart characteristic of DCC; (ii) myocardial fibrosis; (iii) extensive vascular fibrosis and (iv) vascular gene expression involved in calcification and fibrosis.
This CV remodelling cannot be explained solely by a higher blood pressure since following DOCA-salt treatment, Abcc6−/− mice developed the same kinetic and amplitude of blood pressure rise (and the other hemodynamic parameters) as WT mice. This suggests that PXE patients may develop AHT as the general population and especially in salt-rich diet condition14.
Despite a similar gain in BP, only hypertensive Abcc6−/− mice developed myocardial calcifications. Progressive ectopic calcification is the hallmark of human and mouse PXE2,15. In mouse, such phenotype is evidenced by chronic age-related vibrissae calcifications16 or calcium deposits following acute cardiac damage, corresponding to DCC phenotype. DCC results from local tissue damage and cellular necrosis that occurs in infarcted tissue during myocardial healing17. To our knowledge, there has been no evidence of myocardial calcification following sustained hemodynamic changes in Abcc6−/− mice or PXE patients. These findings suggest that pressure overload imposed on the heart by increased peripheral arterial resistance could constitute a stress that contributes to induce dystrophic calcifications. Although DCC has not been reported in human PXE18, this phenotype may potentially predict the propensity of soft tissues to calcify when submitted to mechanical strain19. The fact that calcium deposits were not found by Von Kossa staining, a method routinely used on patient skin biopsies for diagnosis of PXE, may be explained by the patchy mineral deposition pattern in the LV of Abcc6−/− DOCA mice, as previously reported in the TA of old Abcc6−/− mice20.
In parallel to calcification, hypertensive Abcc6−/− mice developed a marked cardiac fibrosis.
There are very few data available on the incidence of CV fibrosis in PXE. Rau et al. proposed Abcc6 as a candidate gene responsible of stress-induced cardiac fibrosis21. Moreover, Götting et al. demonstrated greater xylosyltransferase 1 (a marker of fibrosis) activity in the serum of PXE patients22. DOCA-salt model engenders CV remodelling that is representative of human volume-overload-induced AHT, involving vascular inflammation, hypertrophy, endothelial dysfunction and fibrosis13 Despite DOCA-salt model was reported to cause heart dysfunction witnessed by diastolic blood pressure (DBP) impairment23, we observed no differential effect of Abcc6−/− genotype on DBP or heart rate (HR). The absence of cardiac functional repercussion may be due to the lack of sensitivity of the method (tail-cuff plethysmography). Also, it is not excluded that investigation at later stage of hypertension would have given different results leading to myocardial dysfunction in the long term as previously reported in this model23. Of note, Abcc6 deficiency mitigates cardiac hypertrophy that commonly characterizes elevated BP. This contrasts with our previous study evidencing that old Abcc6−/− mice developed spontaneous heart hypertrophy but no fibrosis24. These data suggest that the pathological remodelling of the heart in the context of Abcc6 deficiency is different during ageing and hypertension.
Of note, we found increased fibrosis in the LV and TA of Abcc6−/− mice treated with DOCA-salt. Vascular fibrosis was associated with an increase of fibrosis-related genes regarding Col3a1, Col1a1, Mmp2, Tgfβ2, Serpine1, Plau and Postn in the aortic wall25,26. In human PXE fibroblasts, an upregulation of TGFβ2 was reported27 although the exact nature of this dysregulation remains unclear26. MMP2 and MMP9 are thought to play a role in PXE, due to evidence of an increase in their serum levels in PXE patients28. From a cellular perspective, only differential expression of MMP2 could be demonstrated in PXE-fibroblasts29,30. In the present study, we found higher levels of Mmp2 and Timp1 expression in the TA of Abcc6−/− DOCA compared to untreated Abcc6−/− mice, likely indicative of ongoing ECM remodelling and increased collagen turnover in hypertensive Abcc6−/− mice29. Serpine1, also known as PAI-1 (Plasminogen activator inhibitor-1), is the principal inhibitor of tissue plasminogen activator (PLAT) and urokinase plasminogen activator (PLAU), and acts as a fibrinolysis inhibitor31. PLAU activity, converting plasminogen to plasmin, is directly controlled by Serpine1 itself modulated by TGFβ family members and contribute to PLAU dysregulation in various age-related diseases, demonstrating impaired tissue regeneration, inflammation, and fibrosis32. The findings from our study, revealed higher levels of Serpine1 and Plau expression in the TA of hypertensive Abcc6−/− suggesting an exacerbated fibrotic process in Abcc6−/− DOCA mice. Periostin (Postn) is an ECM protein expressed predominantly by fibroblasts and plays an key role in cardiac hypertrophy and fibrosis following myocardial infarction33. We measured a greater expression of Postn in the TA of Abcc6−/− DOCA compared to untreated mice, evoking the link previously described between Postn and fibrotic response34. An increase in collagen type I synthesis and in the collagen type I/III ratio in vascular smooth muscle cells leads to matrix stiffness35. In the present study, we found higher levels of Col1a1, Col3a1 expression in the TA of Abcc6−/− DOCA mice suggesting some remodelling associated with fibrosis and potentially underlying arterial ECM stiffening.
Another consequence of Abcc6 deficiency on vascular remodelling can be evidenced by a pro-calcifying genes expression pattern in the TA of hypertensive Abcc6−/− mice. There is a growing evidence that the involvement of TGFβ superfamily signalling pathway (mainly TGFβ and BMPs) and inorganic pyrophosphate (PPi) in PXE26, that may synergistically contribute to ectopic calcification. It has been shown that BMP signalling pathway is activated in hearts and aortas of Abcc6−/− mice36. Bmp4, a protein involved in vascular smooth muscle cell calcification36, was significantly higher in Abcc6−/− hypertensive mice than in WT mice. Interestingly, Bmp4 could be potentially instrumental in DCC following ischemia-reperfusion injury in Abcc6−/− mice36. We also evidenced a decrease in alkaline phosphatase (Alp) and an increase in ectonucleotidase (Enpp1) expression in WT DOCA mice, both favouring a reduction in Pi/PPi ratio preventing vascular calcification37,38. These changes were abrogated in Abcc6−/− mice, suggesting a lack of an anti-mineralization mechanism in these animals. Altogether, these results highlight a propensity for vascular calcification in Abcc6−/− mice treated with DOCA-salt. Medial arterial calcification is a hallmark of vascular aging39. Our results show that vascular calcification in hypertensive Abcc6−/− mice mimics what observed in older Abcc6−/− mice20. Hypertension accelerates age-related vascular remodelling and dysfunction40. Aging may impact the severity of vascular damage in hypertension40. Thus, close interactions between biological aging and the effect of high BP could exist40. The relevance of such specific remodelling pathways has yet to be validated in human PXE patients.
We can assume that such remodelling could lead in the long-term to the deterioration of cardiac and vascular function in Abcc6−/− mice and likely in PXE patients. Such hypothesis may be corroborated both in mouse and in human. Cardiac function of Abcc6−/− mice was previously assessed by our group. With the exception of a mild heart hypertrophy in the oldest pre-senescent animals, no major abnormalities could be evidenced in Abcc6−/− mice24. However, the situation may be different in pathological conditions. Unintuitively, the DOCA-salt-induced hypertrophic remodelling was limited both in LV and aorta of Abcc6−/− mice. No significant correlation between heart or TA hypertrophy and the fibrotic score could be evidenced neither in Abcc6−/− nor in WT mice DOCA (not shown). Although speculative, the conclusion may be that early fibrosis (or calcification) would have blunt the hypertrophic process.
Vascular function investigations of medial thoracic aorta revealed that despite a gain of contractile response to serotonin (and phenylephrine), DOCA-salt treatment did not impair endothelial function neither in WT nor in Abcc6−/− mice. However, we cannot exclude that an endothelial dysfunction would have been assessed in resistance arteries of hypertensive DOCA-salt mice as it has been previously reported41. Such apparent discrepancy with our findings may result from the difference between resistance and compliance vessels in the context of a DOCA-salt regimen41.
In our previous study conducted on both resistant and conductance arteries of WT and Abcc6−/− mice, no differences were found in endothelial dependent and -independent relaxation as well as phenylephrine and thromboxane analogue-dependent contraction20. Here, in pathological conditions, the vascular impact of Abcc6 deficiency was still not revealed functionally. This may be explained by the fact that fragments used for vascular reactivity (medial thoracic aorta) were different from those used for histological analysis which was more proximal (adjoining aortic arch) and prone to fibrosis/calcification20. Also, histological quantification of fibrosis using Sirius red is semi quantitative. Although the staining was corroborated by a molecular approach it is difficult to evaluate the real extent of fibrosis and its functional impact. Absence of endothelial dysfunction in hypertensive Abcc6−/− mice may nevertheless suggest that PXE-associated PAD contrasts with other calcific metabolic diseases such as diabetes, chronic renal insufficiency and atherosclerosis, which incur early onset of endothelial dysfunction42. To our knowledge, no incidence of endothelial dysfunction or flow-mediated dilation has been reported in PXE patients. With reference to contractile response, our results are coherent with previous data showing that loss of Abcc6 had a limited impact on receptor-dependent arterial contraction, while it was associated with increased myogenic tone20.
To our knowledge, there is no data reporting CV remodelling of Abcc6−/− mice. Extensive fibrosis and worsening of calcifications were not expected and raised the question of the therapeutic potential of anti-hypertensive drugs. Different treatments targeting renin angiotensin system or endothelin receptors43,44,45 have been shown to potently reduce cardiac fibrosis in the DOCA-salt model without affecting BP rise while sympatholytic such as hydralazine reduces BP with however incomplete effect on fibrosis46, evidencing the dissociation between efficacy of pharmacological treatment to prevent fibrosis and to blunt BP. From our findings, we can assume that calcifications and myocardial fibrosis in DOCA-salt mice could be independent on elevated blood pressure. Based on the studies cited above, it would be interesting to see whether antihypertensive treatments would work efficiently to reduce fibrosis in Abcc6−/− mice and whether these treatments could also improve calcification, which constitutes a specificity of this model.
Indeed, in a model of heart failure21, Abcc6−/− mice were shown to develop heart fibrosis and calcification following myocardial infarction or necrotic heart lesions47. However, in these studies the cardiac function was not investigated. Hence, the functional repercussion of this CV remodelling remains questioned, in these pathological situations as well as during hypertension. In human PXE patients, Campens et al. reported heart and vascular dysfunction including increased pulse wave velocity and altered left ventricular diastolic function11. The first most likely reveals arterial stiffness due to excessive calcium deposit in the arterial wall as previously reported in Abcc6−/− mice20 and PXE patients8,11,20. We believe that the second may be linked to structural (fibrosis and/or calcification?) alterations of the myocardium.
Vascular stiffness has been reported in Abcc6−/−20 mice and PXE patients8,11. Cardiovascular remodelling in PXE is an important question, especially in the context of common pathological condition such as hypertension. We think that the calcific fibrotic remodelling we evidence here may originate long term CV dysfunction in agreement with the clinical feature of PXE patients. The investigation of the cardiac function of hypertensive Abcc6−/− mice as well as the potential protective effect of anti-hypertensive molecules would deserve specific attention.
Materials and Methods
Ethical standards
The data that support the findings of this study are available from the corresponding author upon reasonable request. The corresponding author had full access to all the data in the study and takes responsibility for its integrity and the data analysis. The lead author wrote the first draft of the manuscript, and all co-authors participated in and approved subsequent revisions.
Animals
The investigation was conducted in accordance with guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes (authorization of the laboratory 849007003). The protocol was approved by the Institutional Animal Care and Use Committee (IACUC): Committee on the Ethics of Animal Experiments (CEAA) of “Pays de la Loire” (permits CEEA.Pdl n°06, and APAFIS#4570-20l6031716512454 v3 from the “Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche”). Abcc6−/− deficient mice (Abcc6tm1Aabb hereafter referred to as Abcc6−/− mice) were generated as previously described12. Mice were backcrossed to a C57BL/6 J background more than 10 times, and reproduction was obtained using heterozygous breeders. Adult male mice aged 23 to 50 weeks were used to avoid bias arising from age-related arterial stiffness.
Models of hypertension
Abcc6−/− and WT mice were unilaterally nephrectomised under isoflurane inhalation anaesthesia (2.2–2.6% isoflurane in 100% O2). Hind-paw withdrawal, blink reflex and respiratory rate were monitored in these mice. A 1.5-cm incision was made through the skin and abdominal muscle caudal to the rib cage. The renal artery and vein were ligated with 4-0 silk sutures (Ethicon) and the left kidney was removed. Skin and muscle layers were closed separately with 4-0 silk sutures. In addition, a 1-cm incision was made in the back, at the base of the neck to implant DOCA pellets subcutaneously (Innovative Research of America, Sarasota, Florida, USA), to provide a dose of 1 mg/kg/day. Mice were then administered water containing 0.9% NaCl and 0.2% KCl. Upon recovery, they were monitored each day for 19 days to measure SBP, DBP and HR using tail-cuff plethysmography48.
Ex Vivo Pharmacological investigation
Mice were sacrificed at day 19 after DOCA pellet implantation. Immediately after euthanasia, TA arteries were dissected in ice-cold physiological saline solution (PSS: mmol/L 130.0, NaCl; 15.0, NaHCO3; 3.7, KCl; 1.6, CaCl2; 1.2, MgSO4 and 11.0, glucose). Pharmacological investigation was conducted on 2 mm-long arterial segments mounted on a wire-myograph (DMT, Aarhus, DK). Initial contraction to potassium chloride (KCl 80 mM), and cumulative concentration-dependent contraction in response to serotonin (5-HT) was tested in resting arteries. Endothelium-dependent and -independent relaxation were measured using ACh and nitric oxide donor (SNP), respectively. Relaxing responses, measured by cumulative concentration-response curves, were established on phenylephrine-contracted (1 μM) arteries.
Calcium quantification
Calcium quantification in the LV of the heart was determined using colorimetric analysis49. One half of each murine LV was weighed, dried and incubated at room temperature for 48 hours in 0.15 N HCl. These samples were then centrifuged at 15,000 × g, and total calcium content of the supernatant was calculated using the Calcium Liquicolor Test Kit (Stanbio, Boerne, TX). The results were normalized to tissue weight and values were expressed in mg/g dry tissue.
Pathological analysis
The remaining half of each murine LV and TA tissues were embedded in OCT (Optimal Cutting Temperature) compound. Von Kossa staining to determine calcium deposits and Sirius red staining to determine type I and III collagen fibers were undertaken on 5μm-thick frozen sections. Tissue sections were analyzed under an Olympus AX70 microscope (Olympus, Tokyo, Japan). Quantification of red-stained fibrotic areas was obtained from five different fields for each tissue sample at 400x magnification (corresponding to a field size of 0.344-mm2), using Cell-F software and a ColorView III camera (Olympus, Tokyo, Japan). Results were expressed as the mean value of the five different field findings from each tissue sample. Aorta thickness was measured in 5 μm-thick frozen sections using Image J software (National Institutes of health NIH, USA) (three to six aorta sections per animal).
RT-qPCR
Gene expression was investigated using quantitative polymerase chain reaction after reverse transcription of total RNA (RT-qPCR). Segments of dissected TA were stored at −20 °C in RNAlater Stabilization Reagent (Qiagen, Valencia, CA, USA) until use. RNA was extracted using the RNeasy® Micro Kit (Qiagen, Valencia, CA, USA) as per manufacturer instructions. RNA extracted from these aortas (200 ng) was used to synthesize cDNA using the QuantiTect® Reverse Transcription Kit (Qiagen, Valencia, CA, USA). RT-qPCR was performed with Sybr® Select Master Mix (Applied Biosystems Inc., Lincoln, CA, USA) reagent using a LightCycler 480 Real-Time PCR System (Roche, Branchburg, NJ, USA). Primer sequences are shown in Supplementary Table S1. Gapdh, Hprt and Gusb were used as housekeeping genes. Analysis was not performed when Ct values exceeded 35. Results were expressed as: 2(Ct target-Ct housekeeping gene).
Data analysis and statistics
Results were expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism software (La Jolla, CA, USA). The respective roles of DOCA-salt treatment and genotype were tested by means of a 2-way ANOVA test. Where interaction between the 2 factors was observed, the effect of DOCA-salt treatment was studied in each genotype using the Bonferroni post-hoc test. 2-way repeated measures ANOVA was used to analyse hypertension and ex vivo pharmacological findings. Vascular gene expression data were compared using the Student’s t-test. A p-value <0.05 was considered statistically significant. For genes expression, the statistical analysis was performed on raw data to evaluate (i) the effect of hypertension in both genotype separately (ii) the effect of the genotype in control (non-hypertensive) mice and (iii) the effect of the genotype in hypertensive mice. For sake of readability results were shown as fold change in gene expression levels compared to untreated mice.
Translational perspective
The present study demonstrates that Abcc6 loss-of-function is associated with CV calcification and fibrosis following chronic AHT (Fig. 6). Hypertension leads to a vicious circle of pathological vascular remodelling including thickness, stiffness, fibrosis, calcification and aging25,40. Thus CV aging may be responsible for high residual lifetime risk for developing AHT in middle-aged and elderly individuals50. Our findings suggest accelerated pathological CV remodelling in hypertensive Abcc6−/− mice leading to fibrosis and calcification in heart and aorta independent of atherosclerosis, inflammation and thrombosis as detected in PXE patients51. Finally, the current treatment approach for slowing or minimizing CV manifestations of PXE is based on reducing CV risk factors through lifestyle changes (smoking cessation, weight loss, moderate physical exercise)18. There are no specific recommendations for AHT management in PXE patients since no randomized trial has investigated this issue to date. While arterial thickness and stiffness are increased in PXE patients8 regardless of BP and given the impact of AHT on thickness and stiffness50, our results pinpoint the importance of diligent monitoring of BP in PXE patients, in accordance with the Blood Pressure Treatment Guideline Recommendations and Cardiovascular Risk52. Choice of BP medication should target central arterial pressure to prevent aortic thickness and stiffness as demonstrated in the Conduit Artery Function Evaluation (CAFE) study53.

General synthesis of the study suggesting an evidence of cardiovascular calcification and fibrosis in Pseudoxanthoma Elasticum Mouse Models Subjected to DOCA-Salt Hypertension.
Ethics approval and participant consent
All procedures described in this report have received ethics committee approval.
Conclusion
This study aimed at assessing the interplay between hypertension and PXE. While CV calcifications is a hallmark of PXE, Abcc6 deficiency allowed extensive fibrosis to occur in mice subjected to DOCA-Salt hypertension. Perturbations in the expression of genes implicated in the synthesis, organization, and degradation of ECM co-occurred with the activation of osteogenic signalling in PXE mice. Future investigations should be designed to characterize cardiac and vascular molecular signature of this abnormal remodelling in order to orientate on proper therapies to prevent hypertension-associated CV complications in PXE.
References
Leftheriotis, G. et al. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: contribution of a genetic disease to the understanding of vascular calcification. Front. Genet. 4 (2013).
Bergen, A. A. et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 25, 228–231 (2000).
Le Saux, O. et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25, 223–227 (2000).
Jiang, Q., Endo, M., Dibra, F., Wang, K. & Uitto, J. Pseudoxanthoma elasticum is a metabolic disease. J. Invest. Dermatol. 129, 348–354 (2009).
Jansen, R. S. et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 110, 20206–20211 (2013).
Jansen, R. S. et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 34, 1985–1989 (2014).
Quaglino, D., Boraldi, F., Annovi, G. & Ronchetti, I. The Multifaceted Complexity of Genetic Diseases: A Lesson from Pseudoxanthoma Elasticum. In Advances in the Study of Genetic Disorders (ed. Ikehara, K.) https://doi.org/10.5772/22161 (InTech 2011).
Kranenburg, G. et al. Arterial stiffening and thickening in patients with pseudoxanthoma elasticum. Atherosclerosis 270, 160–165 (2018).
Sharman, J. E., Boutouyrie, P. & Laurent, S. Arterial (Aortic) Stiffness in Patients with Resistant Hypertension: from Assessment to Treatment. Current Hypertension Reports 19 (2017).
Pingel, S. et al. Increased vascular occlusion in patients with pseudoxanthoma elasticum. VASA 46, 47–52 (2017).
Campens, L. et al. Characterization of cardiovascular involvement in pseudoxanthoma elasticum families. Arterioscler. Thromb. Vasc. Biol. 33, 2646–2652 (2013).
Gorgels, T. G. M. F. et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 14, 1763–1773 (2005).
Basting, T. & Lazartigues, E. DOCA-Salt Hypertension: an Update. Curr. Hypertens. Rep. 19, 32 (2017).
Garfinkle, M. A. Salt and essential hypertension: pathophysiology and implications for treatment. J Am Soc Hypertens 11, 385–391 (2017).
Klement, J. F. et al. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol. Cell. Biol. 25, 8299–8310 (2005).
Le Corre, Y. et al. Quantification of the calcification phenotype of Abcc6-deficient mice with microcomputed tomography. Am. J. Pathol. 180, 2208–2213 (2012).
Meng, H. et al. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc. Natl. Acad. Sci. USA 104, 4530–4535 (2007).
Germain, D. P. Pseudoxanthoma elasticum. Orphanet J Rare Dis 12, 85 (2017).
Bouchareb, R. et al. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J. Mol. Cell. Cardiol. 67, 49–59 (2014).
Kauffenstein, G. et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arterioscler. Thromb. Vasc. Biol. 34, 1045–1056 (2014).
Rau, C. D. et al. Mapping Genetic Contributions to Cardiac Pathology Induced by Beta-Adrenergic Stimulation in Mice. Circulation: Cardiovascular. Genetics 8, 40–49 (2015).
Götting, C. et al. Elevated xylosyltransferase I activities in pseudoxanthoma elasticum (PXE) patients as a marker of stimulated proteoglycan biosynthesis. J. Mol. Med. 83, 984–992 (2005).
Silberman, G. A. et al. Uncoupled Cardiac Nitric Oxide Synthase Mediates Diastolic Dysfunction. Circulation 121, 519–528 (2010).
Prunier, F. et al. Pseudoxanthoma elasticum: cardiac findings in patients and Abcc6-deficient mouse model. PLoS ONE 8, e68700 (2013).
van Varik, B. J. et al. Mechanisms of arterial remodeling: lessons from genetic diseases. Front Genet 3, 290 (2012).
Van Gils, M., Nollet, L., Verly, E., Deianova, N. & Vanakker, O. M. Cellular signaling in pseudoxanthoma elasticum: an update. Cell. Signal. 55, 119–129 (2019).
Hosen, M. J., Coucke, P. J., Le Saux, O., De Paepe, A. & Vanakker, O. M. Perturbation of specific pro-mineralizing signalling pathways in human and murine pseudoxanthoma elasticum. Orphanet J Rare Dis 9, 66 (2014).
Diekmann, U. et al. Elevated circulating levels of matrix metalloproteinases MMP-2 and MMP-9 in pseudoxanthoma elasticum patients. Journal of Molecular Medicine 87, 965–970 (2009).
Quaglino, D. et al. Dermal fibroblasts from pseudoxanthoma elasticum patients have raised MMP-2 degradative potential. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1741, 42–47 (2005).
Faust, I. et al. Characterization of dermal myofibroblast differentiation in pseudoxanthoma elasticum. Experimental Cell Research 360, 153–162 (2017).
Ghosh, A. K. & Vaughan, D. E. PAI-1 in tissue fibrosis. Journal of Cellular Physiology 227, 493–507 (2012).
Sudol, M. From Rous sarcoma virus to plasminogen activator, src oncogene and cancer management. Oncogene 30, 3003 (2011).
Oka, T. et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 101, 313–321 (2007).
Walker, J. T., McLeod, K., Kim, S., Conway, S. J. & Hamilton, D. W. Periostin as a multifunctional modulator of the wound healing response. Cell Tissue Res. 365, 453–465 (2016).
Pauschinger, M. et al. Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation 99, 2750–2756 (1999).
Mungrue, I. N. et al. Abcc6 Deficiency Causes Increased Infarct Size and Apoptosis in a Mouse Cardiac Ischemia-Reperfusion Model. Arteriosclerosis, Thrombosis, and Vascular Biology 31, 2806–2812 (2011).
Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch 77, 4–12 (2010).
Albright, R. A. et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun 6, 10006 (2015).
Jiang, L. et al. Calpain-1 regulation of matrix metalloproteinase 2 activity in vascular smooth muscle cells facilitates age-associated aortic wall calcification and fibrosis. Hypertension 60, 1192–1199 (2012).
Harvey, A., Montezano, A. C. & Touyz, R. M. Vascular biology of ageing—Implications in hypertension. Journal of Molecular and Cellular Cardiology 83, 112–121 (2015).
Silva, G. C., Silva, J. F., Diniz, T. F., Lemos, V. S. & Cortes, S. F. Endothelial dysfunction in DOCA-salt-hypertensive mice: role of neuronal nitric oxide synthase-derived hydrogen peroxide. Clinical Science 130, 895–906 (2016).
Davignon, J. & Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 109, III27–32 (2004).
Peng, H., Carretero, O. A., Alfie, M. E., Masura, J. A. & Rhaleb, N.-E. Effects of Angiotensin-Converting Enzyme Inhibitor and Angiotensin Type 1 Receptor Antagonist in Deoxycorticosterone Acetate–Salt Hypertensive Mice Lacking Ren-2 Gene. Hypertension 37, 974–980 (2001).
Grobe, J. L., Mecca, A. P., Mao, H. & Katovich, M. J. Chronic angiotensin-(1–7) prevents cardiac fibrosis in DOCA-salt model of hypertension. American Journal of Physiology-Heart and Circulatory Physiology 290, H2417–H2423 (2006).
Ammarguellat, F., Larouche, I. & Schiffrin, E. L. Myocardial Fibrosis in DOCA-Salt Hypertensive Rats: Effect of Endothelin ET A Receptor Antagonism. Circulation 103, 319–324 (2001).
Klanke, B. et al. Blood pressure versus direct mineralocorticoid effects on kidney inflammation and fibrosis in DOCA-salt hypertension. Nephrology Dialysis Transplantation 23, 3456–3463 (2008).
Brampton, C. et al. The level of hepatic ABCC6 expression determines the severity of calcification after cardiac injury. Am. J. Pathol. 184, 159–170 (2014).
Wilde, E. et al. Tail-Cuff Technique and Its Influence on Central Blood Pressure in the Mouse. J Am Heart Assoc 6 (2017).
Brampton, C. et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 10, 1810–1820 (2011).
Cheng, H.-M. et al. Vascular aging and hypertension: Implications for the clinical application of central blood pressure. Int. J. Cardiol. 230, 209–213 (2017).
Bartstra, J. W., de Jong, P. A. & Spiering, W. Accelerated peripheral vascular aging in pseudoxanthoma elasticum – proof of concept for arterial calcification-induced cardiovascular disease. Aging 11, 1062–1064 (2019).
Colantonio, L. D. et al. 2017 ACC/AHA Blood Pressure Treatment Guideline Recommendations and Cardiovascular Risk. J. Am. Coll. Cardiol. 72, 1187–1197 (2018).
Williams, B. et al. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 113, 1213–1225 (2006).
Acknowledgements
The authors wish to thank the Patients’ Association “PXE France” especially its president Ms Karine Unger, the “Fondation Groupama” and the “CHU de Rennes – Corect” for their support for our work. The authors wish to thank Mrs Hazel Chaouch (hctranslationservices@outlook.com) for the formatting of the article and English language correction. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
G.K.: Conception and design of the protocol. L.O., C.R., J.F. and G.K.: Data Acquisition. L.O., C.R., C.L., J.F., A.J. and G.K.: Data Analysis and writing the report. L.O., C.R., C.L., J.F., D.H., G.M., G.L., L.M., A.J. and G.K.: Data interpretation and drafted the work. Each author revised the report and approved the submitted version of the manuscript. Each author has agreed both to be personally accountable for the author’s own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Omarjee, L., Roy, C., Leboeuf, C. et al. Evidence of Cardiovascular Calcification and Fibrosis in Pseudoxanthoma Elasticum Mouse Models Subjected to DOCA-Salt Hypertension. Sci Rep 9, 16327 (2019). https://doi.org/10.1038/s41598-019-52808-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52808-z
This article is cited by
-
Vascular calcification in different arterial beds in ex vivo ring culture and in vivo rat model
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
