
Abstract
Echocardiography has become an indispensable tool for the study of heart performance, improving the monitoring of individuals with cardiac diseases. Diverse genetic factors associated with echocardiographic measures have been previously reported. The impact of several apoptotic genes in heart development identified in experimental models prompted us to assess their potential association with human cardiac function. This study aimed at investigating the possible association of variants of apoptotic genes with echocardiographic traits and to identify new genetic markers associated with cardiac function. Genome wide data from different studies were obtained from public repositories. After quality control and imputation, a meta-analysis of individual association study results was performed. Our results confirmed the role of caspases and other apoptosis related genes with cardiac phenotypes. Moreover, enrichment analysis showed an over-representation of genes, including some apoptotic regulators, associated with Alzheimer’s disease. We further explored this unexpected observation which was confirmed by genetic correlation analyses. Our findings show the association of apoptotic gene variants with echocardiographic indicators of heart function and reveal a novel potential genetic link between echocardiographic measures in healthy populations and cognitive decline later on in life. These findings may have important implications for preventative strategies combating Alzheimer’s disease.
Similar content being viewed by others

Introduction
Echocardiographic assessment of cardiac structure offers prognostic information about cardiac conditions such as heart failure (HF)1,2. Pathological processes including cardiomyocyte cell death, inflammatory cell response and changes in interstitial tissue of the heart are factors leading to adverse remodelling and HF3.
A number of apoptotic genes have been investigated as potential targets to prevent cardiomyocyte death, but it is now increasingly evident that caspase-dependent cell death plays a minor if any role in adult myocyte loss4, which involves Cyclophilin D5 and calpains6. By contrast, caspase proteins are now recognized as important factors for initial differentiation of stem cells to cardiomyocytes7, and its deficiency in vivo was shown to induce abnormal heart development8,9. In rodent cardiomyocytes, caspase-3 is involved in WNT signalling and myocyte growth10,11, contributing also to muscle-specific gene splicing by cleaving PTB12. In addition, apoptotic DNA nucleases were shown to play a role in the developmental process of C.elegans including the C.elegans Caspase-associated DNase (CAD), ENDOG and TATD orthologues13. Furthermore, ENDOG also contributes to the signalling pathways determining myocyte size through the control of reactive oxygen radicals (ROS)14,15. These facts lead us to hypothesize that caspases and the nucleases ENDOG and TATD play relevant functions in cardiomyocyte proliferation and maturation during development.
Recent genome wide association studies have been performed for evaluating comprehensive sets of echocardiographic traits in well characterized individuals included in large cohort studies16,17. Increased left ventricular mass (LVM) is a well-established predictor of adverse cardiovascular events and premature death18,19. End-Diastolic LV Internal Dimension (LVID) and LV Wall Thickness (LVWT) are other measures of left ventricular hypertrophy (LVH), usually a pathological compensatory mechanism of the LV overload which evolves towards progressive LV dysfunction and HF. The principal role of the left atrium (LA) is to modulate LV filling and cardiovascular performance, being LA enlargement and dysfunction also predictors of cardiovascular events, especially in patients with atrial fibrillation (AF)20. The risk of aortic aneurysm is strictly related to the diameter of the ascending aorta, and recent studies have shown high prevalence of aortic root (AROT) enlargement in the hypertensive population21. Using data from publicly available repositories, we aimed to explore the association between a selected group of candidate apoptosis-related genes and these echocardiographic phenotypes by means of meta-GWAS. Furthermore, we aimed to assess previously reported signals in our study datasets and performed an agnostic analysis to investigate relevant pathways revealed for each trait. Following the leading results of this analysis, we further explored the unsuspected genetic relationship between Alzheimer’s disease (AD) and these echocardiographic traits, by estimating their genetic correlation, and identifying common genetic determinants of these conditions.
Materials and Methods
Experimental design
This study is aimed at identified genetic variants associated with echocardiographic traits, using both a candidate gene approach and an agnostic approach in retrospective datasets available in public repositories.
Study cohorts
The four cardiovascular datasets analysed in this study were downloaded from dbGAP (https://www.ncbi.nlm.nih.gov/gap) after requesting the appropriate permissions. In the case of a multi-ethnic study, only Caucasian samples after principal component analysis (PCA) were retained for analysis. A summary of the clinical characteristics of these populations is shown in Table 1. A full description of each of them is provided in the Supplementary Info.
A total of seven AD datasets were used to further explore the observed enrichment of top genes derived from the analysis of echocardiographic traits on genes involved in AD pathways. As for previously described datasets, only Caucasian samples after principal component analysis (PCA) were retained for analysis. Demographic characteristics of these datasets are summarized in the Supplementary Table 1.
Phenotypes
Data from the most recent available echocardiographic examinations of each cohort were included in this study. The following five phenotypes were analysed: LVM (g), AROT (cm), LVID (cm), LA size LAS, cm), and LVWT (cm). The latter was defined as the sum of the End-Diastolic Thicknesses of the Posterior Wall (TPW) and End-Diastolic Thicknesses of the Interventricular Septum (TIS). LVM was calculated using the formula 0.8 [1.04{(LVID + TIS + TPW)3 −(LVID)3}] + 0.622.
Genotyping and imputation
The cardiovascular datasets included in this study were genotyped using different platforms: CARDIA and MESA were genotyped using the Affymetrix Genome-Wide Human 6.0 array, whereas the FHS was genotyped using the Affymetrix Human 500k array and the CHS cohort with the Illumina HumanCNV370-Duo v1.0.
AD datasets were genotyped using the Illumina arrays Human 610‐Quad BeadChip (ADNI1, AddNeuroMed batch 1), the HumanOmniExpress BeadChip (ADNI2/GO, AddNeuroMed batch 2, ADGC dataset 3), the Human660W-Quad (ADGC datasets 1 and 2) and the HumanHap300-Duo BeadChip (The Mayo study) or the Affymetrix 250k NspI (the Neocodex-Murcia study), 500k (the TGEN and GenADA studies) or 6.0 (ROSMAP study) arrays.
Prior to imputation, we first performed an extensive quality control excluding individuals with more than 3% missing genotypes, with excess autosomal heterozygosity (>0.35), those showing a discrepancy between genotypic and reported sex, as well as individuals of non-European ancestry based on PCA using SMARTPCA23. At the genotype level, we removed single nucleotide polymorphisms (SNPs) with missing genotype rate > 5%, not in Hardy-Weinberg equilibrium (p < 10−6) and SNPs with minor allele frequency (MAF) < 1%. Duplicated and related individuals were identified and removed by means of Identity By State (IBS) estimates within and across studies.
Genotype imputation is aimed at estimating unobserved genotypes using as reference known haplotypes from a well characterized population. Imputation was performed using the minimac3 algorithm and the SHAPEIT tool for haplotype phasing at the University of Michigan server using the HRC reference panel24. After imputation, SNPs with an R2 quality estimate lower than 0.3 were excluded from further analyses in accordance with software instructions.
Statistical analysis
All analyses were performed in Caucasian populations only. Individuals with prevalent myocardial infarction (MI) or congestive heart failure (CHF) were excluded from the study. Linear regression models available from PLINK software25 were fitted to investigate the association between genotypes and quantitative phenotypes, with age, sex, body mass index and the four principal components as covariates. For each phenotype, we obtained summary estimates of association across studies by using a fixed-effects model meta-analysis procedure implemented also in PLINK. For the genome wide SNP analysis, the conventional GWAS significance threshold was used (p = 5 × 10−8)26, whereas signals with p value < 10−5 were considered as suggestive of association and reported in the Supplementary Tables 2–6.
Gene-wise statistics were computed using MAGMA software, which takes into account physical distance and linkage disequilibrium (LD) between markers to estimate a summary gene p-value using a known approximation of the sampling distribution27. All SNPs with MAF above 1% were used in these analyses. At each trait, genes were ranked according to the global p mean value.
Top results from MAGMA genome wide analyses were tested for over-representation of genes involved in known pathways, functions and diseases. For these enrichment analyses, we applied a moderated threshold of p < 10−3, which has been showed to have a good power for detecting relevant pathways and functions while maintaining type 1 error controlled27. For these analyses, we used the R packages Webgestalt (GO terms, KEGG, Wikipathways and Reactome) and enrichR28,29 (Aging perturbations from GEO, Biocarta, Panther Disease perturbations from GEO Disease signatures from GEO, dbGaP and OMIM) with default parameters.
In order to explore genetic correlation between different traits we used a bivariate GREML analysis with GTCA software30. This method allows to identify pleiotropic gene effects associated with different diseases or disorders, providing a single measure of the proportion of shared genetic determinants. Furthermore, we obtained summary estimates of association across phenotypes performing unweighted meta-analysis of Fisher p-values.
Results
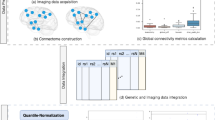
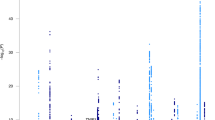
Overall, our study included data from 11,559 individuals with echocardiographic phenotypes from four different datasets (Table 1). Figure 1 illustrates the data analysis roadmap as explained in Materials and Methods. After imputation and quality control, we obtained about 7 million SNPs with MAF > 0.01 that were tested for association with echocardiographic traits at each study. We then performed a meta-GWAS to obtain summary estimates of association for each SNP. Genomic inflation factor (λ) ranged from 0.994 to 1.022 in these analyses, indicating absence of population stratification due to hidden population structure (Fig. 2). MAGMA software was used for summarizing the meta-GWAS SNP results in order to obtain a gene-wise statistic of the association between 18,480 genes and the five phenotypes.

Data analysis workflow.

Manhattan plots of the meta-analyses for the different echocardiographic traits. The threshold for genome-wide significance (P < 5 × 10−8) is indicated by the red line, while the blue line represents the suggestive threshold (P < 1 × 10−5). Loci previously associated with echocardiographic traits are shown in blue, and newly associated loci are shown in red.
Association of apoptosis-related genes with cardiac phenotypes
Because there is experimental evidence supporting the role of some apoptosis-related genes with cardiac development and disease, we first analysed the potential association of polymorphisms in a series of apoptosis-related genes with the cardiac phenotypes. Table 2 shows the association results of the 20 apoptosis-related candidate genes. Study-wide statistically significant results were observed for the association of a genetic locus on 2q33.1 involving two initiator caspases (CASP8 and CASP10) and the apoptosis regulator protein CFLAR (CASP8 And FADD Like Apoptosis Regulator, c-FLIP) with LVM. The same three genes were also linked to LVID with p < 0.05, along with the Fas receptor-associated adaptor FADD (Fas-associated protein with death domain) and BCL2 (B-cell lymphoma-2) genes. BCL2, FADD and TATDN1 (TatD DNase domain containing-1) showed a trend for association with AROT. We did not find evidence of association of any calpain family member with any of the analysed traits (data not shown).
Agnostic GWAS of genetic variants associated with cardiac phenotypes and enrichment analysis of top genes on echocardiographic traits
Although we did not find any GWAS significant signal at SNP-level (p < 5 × 10−8) related with the analysed phenotypes, we observed several suggestive signals at the p < 10−5 level (Supplementary Tables 2–6), most of them intragenic (Fig. 2). For each genotype, we additionally performed a gene-level, ranking genes according to the MAGMA computed SNP-wise p-value, which uses the full spectrum of SNP-level meta-analysis results (Table 3 and Supplementary Tables 7–11). Overall, these top genes from MAGMA gene level analyses (p < 10−3) showed little overlap across phenotypes (Fig. 3a), with the exception of TECTB/ACSL5 locus for LVID and LVWT, genes associated to cholesterol and fatty acid metabolism respectively, and ZNF678, a zinc finger protein involved in immune response, for LVM and LVWT genes. Enrichment analysis (Fig. 3b, Supplementary Tables 12–16) suggested some processes underlying or related to one or more echocardiographic traits such as lymphocyte activity, cardiomyopathy, left ventricular hypertrophy or oxidative stress response Interestingly, enrichment analysis also suggested an over-representation of genes differentially expressed in AD samples versus control as well as genes involved in tau, presenilin and amyloid biology (APH1B, KLC3, KLC4, MAPK4, MAPK13;MAPK14, PPP2R5D, RBPJ, TCF7L1).

Results from gene-wise analysis of echocardiographic traits. (a) Venn diagram showing the overlap of top genes of the different echocardiographic traits analysed. (b) Plot summarising main results of the enrichment analysis of top genes from gene-wise statistics. RF (rich factor): number of genes from the input list included in each category divided by the total number of genes in the category.
Genetic correlation analysis between echocardiographic traits and AD
The association of gene variants related to echocardiographic measures with mental illnesses, the increasing interest in the vascular aspects of AD and availability of diverse AD datasets, prompted us to explore in more depth the relationship between the echocardiographic phenotypes and AD. We performed a genetic correlation analysis using the study dataset (comprised by 11,559 individuals with echocardiographic phenotypes) along with 12,730 AD cases and controls both from internal and publicly available databases. First, we estimated the proportion of variance, as a proxy of trait heritability, explained by all SNPs in each one of these traits31, which was higher for AD (0.38) than for the echocardiographic phenotypes (range: 0.17-0.36). Then, we looked for shared genetic loci across the genome between echocardiographic traits and AD using GREML analyses as suggested by the enrichment analyses. Specifically, we observed a positive correlation between AD and LAS (rG = 0.167, p = 0.0334), and negative correlations between AD and LVID (rG = −0.196, p = 0.0056), and AD and LVM (rG = −0.198, p = 0.0165); of note, LVM and LVID are the most correlated echocardiographic traits (rG = 0.988, p < 0.00001) (Fig. 4a). The sign of the rG estimates determines whether a direct or inverse relationship between the two phenotype traits is observed. Therefore, our results suggest that SNPs that increase the risk of AD tend to be associated with increasing LAS values. On the contrary, we found that SNPs associated with increased risk of AD tend to be associated with decreasing ventricular measures (or vice versa), in particular LVID and LVM.

Results from the combined analysis of echocardiographic traits and AD. (a) Genetic correlations (rG) between echocardiographic traits and AD. (b) Venn diagram showing the overlap of top genes derived from gene-wise association analysis. (c) Enrichment analysis of top genes from meta-analysis of echocardiographic traits and Alzheimer’s disease. RF (rich factor): number of genes from the input list included in each category divided by the total number of genes in the category.
Based on these findings we performed a SNP-wise meta-analysis by pooling Fisher association p-values of two or more phenotypes aimed at identifying genes contributing most to both heart measures and AD. Thus, we combined in these meta-analyses p-values for LAS&AD, LVID&AD, LVM&AD, LAS&LVID&LVM&AD and LVID&LVM&AD and calculated gene-wise statistics (Table 4, Supplementary Tables 17–21). We observed a large overlap of genes falling under the p < 10−3 threshold in the different meta-analyses performed, with a group of 30 genes consistently associated in all of them, including the CASP8/CASP10/CFLAR locus and the GABRR1 GABA receptor (Fig. 4b). Consequently, apoptosis related pathways driven by the CASP8, CASP10 and CFLAR locus were persistently observed in the subsequent enrichment analyses (Fig. 4c, Supplementary Tables 22–25). We also observed an enrichment on genes present at the neuronal synapse such as glutamate (GRM5), cholinergic (CHRNA2) and GABA receptors (GRIN2C, GABRR1, GABRR2 and GABBR2), teneurin (TENM2), calsyntenin (CLSTN3), or the cytoskeletal alpha-actin protein (ACTN2) for the diverse meta-analyses involving ventricular measures and AD.
Discussion
Our study, based on data from 11,559 individuals free of cardiovascular disease, shows that variants affecting diverse genes involved in apoptosis regulation associate with echocardiographic phenotypes in humans. We have obtained these results by both hypothesis-driven and agnostic approaches. In addition, novel findings from the analysis of our GWAS also include previously unnoticed associations of variants in genes involved in cell proliferation, DNA replication and mRNA splicing with left ventricular morphology. Our data also suggest the existence of a set of genes, mainly related to apoptosis/inflammation signalling, whose variants are associated with both cardiac phenotype and AD.
Our hypothesis-driven analysis showed that caspases 8 and 10 and the regulatory CFLAR (cFLIP) gene are strong predictors of LVM. Furthermore, our hypothesis-free approach found that caspase dependent pathways were overrepresented among top genes associated with the LVM phenotype. These results support our a priori hypothesis that the apoptotic signalling influences heart morphology with potential impact on heart performance. Our hypothesis was based in previous experimental work, including our own, showing that deficiency9,11 or overexpression32 of key apoptotic genes altered normal cardiomyocyte differentiation and heart development independently of cell death. Indeed, although these genes are best known for their role in regulating apoptotic cell death, experimental evidences show that the same genes also regulate myocyte proliferation, inflammation and hypertrophy in the heart9,11,33,34,35. Because cell death is not a major event during heart development, and based on the above experimental work, we suggest that the relationship between the apoptotic genes and the cardiac phenotype might involve non-apoptotic functions.
In order to estimate the robustness of our GWAS analysis, we asked whether we had reproduced some signals already observed in previous genetic studies. Indeed, we found the already published link between SMG6, TSR1 and SRR genes and AROT16,17, but failed to detect statistically significant associations for other gene variants, possibly due to the limited power of this study. Given the strong influence of age in echocardiographic parameters, the diverse population structure of the study cohorts could also affect our power in the meta-analysis, with signals not replicating in populations largely differing in their median age. However, our analysis demonstrated genetic association between variants of genes previously shown to influence heart function and cardiac hypertrophy in experimental models, such as MLF136, which associates with LVM phenotype in our study, and KCNIP2 (KChIP2)37,38,39 and TRAF3IP240,41, which have also been associated with LVWT and LVM phenotypes respectively in humans and in this study as well. Also, from the top list of genes whose variants are associated to LIVD, ANKS6 has been associated with heart malformations42.
The GWAS analysis also showed association of variants of a group of odorant receptors genes located in the 11p15.4 chromosomic region coding (subfamilies 51, 52 and 56) or in chromosome 16 (OR2C1) with LVWT and LVID. Expression of these genes in non-neural tissues is related to the control of different processes, including glucose and oxygen homeostasis or cell cycle control43, and has been shown to be involved in the regulation of cardiac function in rodent experimental models through interaction with fatty acids44. Therefore, our genetic results open the possibility that odorant receptors’ activity can influence cardiac function in humans. Interestingly, low OR expression has been found in the cortex of neuro-psychiatric patients45 and a genetic microduplication in the 11p15.4 region has been associated with familial intellectual disability and autism46.Unexpectedly, we found enrichment on AD related pathways for LAS, LVID and LVM that led us to explore comprehensively a possible link between AD and all these echocardiographic traits. Intriguingly, our results revealed for the first time a potential genetic link between AD and LAS, paired with a negative correlation between AD and either LVM or LVID. Interestingly, in line with our observation, recent reports found that LAS and LVID were independently associated to cognitive function in older adults, being predictors of cognitive decline after 14 years of follow-up47,48. The opposite direction of the correlation coefficients for atrial and ventricular measures could be related to the different development patterns of these chambers during embryogenesis49 and with the described strong association of left atrial size with long-term exposure to vascular risk factors, particularly high blood pressure and obesity. In fact, the CARDIA Brain MRI Substudy found association of higher left atrial volume in early adulthood with impairment of white matter integrity in midlife, but not for ventricular measures50.
A recent GWAS by our group on AD clinical endophenotypes revealed that vascular processes were the main causal mechanisms in pure AD51. But our findings are compatible to a broader definition of- dementia which includes other diseases such vascular dementia, fronto-temporal dementia or Lewy Body diseases among others. In fact, the rule is the presence of multiple pathologies in the brain of people with dementia including pervasive vascular lesions52. The link between dementia and cardiac conditions is not well understood. The old concept of cardiogenic dementia was based on the high incidence of cardiac dysrhythmias observed in patients with dementia due to vascular causes53. However, the relation between coronary heart disease (CHD) or HF and AD in epidemiological studies remains controversial, with some studies showing an association with cognitive impairment and dementia54,55,56 whereas others found no association57,58. The fact that both conditions are competing risks complicates the study of their relationship. Lower cardiac index levels are related to lower cerebral blood flow in older adults free of CVD59, but individuals with cardiac conditions that did not result in premature death might include many individuals chronically exposed to brain hypoperfusion due to reduced cardiac output that adaptively decreased cerebrovascular resistance through arteriolar dilatation. This kind of antagonistic pleiotropy between these phenotypes has been previously suggested by Beeri et al. after observing that better cognitive performance was associated with worse cardiac functioning in very elderly subjects60.
Moreover, whereas enlarged ventricular volume (LV hypertrophy) is a marker of diastolic dysfunction, LVM is also a marker of cardiovascular health, positively correlated with physical activity and cardiorespiratory fitness61,62. Population based studies have shown an inverted U-shaped association for LVM values and age, since they rise in adolescence and decline with increased age63,64. Furthermore, a U-shaped association between left ventricular ejection fraction (LVEF), a marker of systolic dysfunction, and abnormal cognitive decline has been reported, with increased dementia risk at the lowest and highest LVEF quintiles65.
To our knowledge, this is the first report analysing shared genetic factors between echocardiographic measures and AD. This method for estimating genome-wide pleiotropy has the advantage of being free of potential confounders determined by shared epidemiological risk factors such as high blood pressure or atherosclerosis. Our results show a negative genetic correlation for the ventricular measures LVID and LVM and AD, pointing to antagonist pleiotropic effects of shared genes by AD and LV cardiac measures, the main known functions of which are related to apoptosis (CASP8, CASP10, CFLAR, PHIP) and neurotransmission (GABRR1). Interestingly, some CASP8 variants have been previously associated with AD66, and the protein has been shown to be involved in amyloid related pathways and activated in both blood and brain cells from AD patients1,67,68,69. In our study, the GABA receptor GABRR1 was consistently associated in all meta-analyses involving heart traits and AD. Neurotransmission affects both cardiac and neuronal performance, and a few studies have examined synapse and neuron loss in AD brains and suggested that synaptic changes precede neuron loss70,71. In fact, GABA signalling is reduced in AD and other mental disorders such as dementia with Lewy bodies or frontotemporal lobar degeneration72,73,74. By contrast, augmented inhibitory GABAergic neurotransmission has been reported in animals models of HF and LVH through the involvement of the paraventricular nucleus of the hypothalamus75,76.
In summary, our GWAS data suggest the influence of gene variants affecting the apoptotic/inflammation signalling pathway on left ventricular morphology and cardiac function, uncover novel candidate gene variants regulating echocardiographic phenotypes and establish a genetic link between cardiac morphology alterations, mental illness and AD involving key genes in the regulation of apoptotic signalling that deserve functional assessment due to their diagnostic and therapeutic potential. These results should be replicated in larger datasets in order to confirm the observed pleiotropic effects for genes associated with echocardiographic traits.
Data availability
Full meta-analysis results for echocardiographic traits are available at Mendeley data (https://data.mendeley.com/, https://doi.org/10.17632/22jdjghnsp.1).
References
Folin, M. et al. Caspase-8 activation and oxidative stress are involved in the cytotoxic effect of β-amyloid on rat brain microvascular endothelial cells. Int. J. Mol. Med. (2006).
Paulus, W. J. et al. How to diagnose diastolic heart failure: A consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. in. European Heart Journal 28, 2539–2550 (2007).
Piek, A., de Boer, R. A. & Silljé, H. H. W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 21, 199–211 (2016).
Inserte, J. et al. Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling. Basic Res. Cardiol. 111, 18 (2016).
Baines, C. P. et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 (2005).
Inserte, J., Hernando, V. & Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovascular Research 96, 23–31 (2012).
Fujita, J. et al. Caspase Activity Mediates the Differentiation of Embryonic Stem Cells. Cell Stem Cell 2, 595–601 (2008).
Lakhani, S. A. et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–51 (2006).
Cardona, M. et al. Executioner Caspase-3 and 7 Deficiency Reduces Myocyte Number in the Developing Mouse Heart. PLoS One 10, e0131411 (2015).
Abdul-Ghani, M. et al. Wnt11 promotes cardiomyocyte development by caspase-mediated suppression of canonical Wnt signals. Mol. Cell. Biol. 31, 163–78 (2011).
Putinski, C. et al. Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. 110, E4079–E4087 (2013).
Ye, J. et al. A pathway involving HDAC5, cFLIP and caspases regulates expression of the splicing regulator polypyrimidine tract binding protein in the heart. J. Cell Sci. 126, 1682–91 (2013).
Parrish, J. Z. & Xue, D. Functional genomic analysis of apoptotic DNA degradation in C. elegans. Mol. Cell 11, 987–96 (2003).
McDermott-Roe, C. et al. Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function. Nature 478, 114–8 (2011).
Blasco, N. et al. Cardiomyocyte hypertrophy induced by Endonuclease G deficiency requires reactive oxygen radicals accumulation and is inhibitable by the micropeptide humanin. Redox Biol. 16, 146–156 (2018).
Vasan, R. S. et al. Genetic Variants Associated With Cardiac Structure and Function. JAMA 302, 168 (2009).
Wild, P. S. et al. Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J. Clin. Invest. 127, 1798–1812 (2017).
Koren, M. J., Devereux, R. B., Casale, P. N., Savage, D. D. & Laragh, J. H. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann. Intern. Med., https://doi.org/10.7326/0003-4819-114-5-345 (1991).
Drazner, M. H. et al. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: The Cardiovascular Health Study. J. Am. Coll. Cardiol., https://doi.org/10.1016/j.jacc.2003.11.064 (2004).
Hoit, B. D. Left atrial size and function: Role in prognosis. Journal of the American College of Cardiology, https://doi.org/10.1016/j.jacc.2013.10.055 (2014).
Covella, M. et al. Echocardiographic aortic root dilatation in hypertensive patients: A systematic reviewand meta-analysis. Journal of Hypertension, https://doi.org/10.1097/HJH.0000000000000286 (2014).
Devereux, R. B. et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am. J. Cardiol. 57, 450–8 (1986).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet., https://doi.org/10.1038/ng.3643 (2016).
Purcell, S. et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
de Bakker, P. I. W. et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 17, 122–128 (2008).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 11 (2015).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Wang, J., Duncan, D., Shi, Z. & Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41 (2013).
Lee, S. H., Yang, J., Goddard, M. E., Visscher, P. M. & Wray, N. R. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012).
Walters, R., Churchhouse, C. & Hosking, R. Heritability 201: types of heritability and how we estimate it. Available at: http://www.nealelab.is/blog/2017/9/13/heritability-201-types-of-heritability-and-how-we-estimate-it.
Nelson, D. P. et al. Proinflammatory consequences of transgenic fas ligand expression in the heart. J. Clin. Invest. 105, 1199–208 (2000).
Yokoyama, T. et al. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation 95, 1247–52 (1997).
Wollert, K. C. et al. The cardiac Fas (APO-1/CD95) Receptor/Fas ligand system: relation to diastolic wall stress in volume-overload hypertrophy in vivo and activation of the transcription factor AP-1 in cardiac myocytes. Circulation 101, 1172–8 (2000).
Badorff, C. et al. Fas receptor signaling inhibits glycogen synthase kinase 3 beta and induces cardiac hypertrophy following pressure overload. J. Clin. Invest. 109, 373–81 (2002).
Rangrez, A. Y. et al. Myeloid leukemia factor-1 is a novel modulator of neonatal rat cardiomyocyte proliferation. Biochim. Biophys. Acta 1864, 634–644 (2017).
Sanguinetti, M. C. Reduced transient outward K+ current and cardiac hypertrophy: causal relationship or epiphenomenon? Circ. Res. 90, 497–9 (2002).
Thomsen, M. B. et al. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ. Res. 104, 1382–9 (2009).
Cheng, C.-F., Kuo, H.-C. & Chien, K. R. Genetic modifiers of cardiac arrhythmias. Trends Mol. Med. 9, 59–66 (2003).
Valente, A. J., Clark, R. A., Siddesha, J. M., Siebenlist, U. & Chandrasekar, B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 53, 113–124 (2012).
Yariswamy, M. et al. Cardiac-restricted Overexpression of TRAF3 Interacting Protein 2 (TRAF3IP2) Results in Spontaneous Development of Myocardial Hypertrophy, Fibrosis, and Dysfunction. J. Biol. Chem. 291, 19425–36 (2016).
Hoff, S. et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 45, 951–956 (2013).
Chen, Z., Zhao, H., Fu, N. & Chen, L. The diversified function and potential therapy of ectopic olfactory receptors in non-olfactory tissues. J. Cell. Physiol. 233, 2104–2115 (2018).
Jovancevic, N. et al. Medium-chain fatty acids modulate myocardial function via a cardiac odorant receptor. Basic Res. Cardiol. 112, 13 (2017).
Ansoleaga, B. et al. Decrease in olfactory and taste receptor expression in the dorsolateral prefrontal cortex in chronic schizophrenia. J. Psychiatr. Res. 60, 109–116 (2015).
Sofos, E. et al. A novel familial 11p15.4 microduplication associated with intellectual disability, dysmorphic features, and obesity with involvement of the ZNF214 gene. Am. J. Med. Genet. Part A 158A, 50–58 (2012).
Alosco, M. L. et al. Left atrial size is independently associated with cognitive function. Int. J. Neurosci. 123, 544–552 (2013).
Furuäng, L., Wollmer, P., Siennicki-Lantz, A. & Elmståhl, S. Cardiac ventricular dimensions predict cognitive decline and cerebral blood flow abnormalities in aging men. BMC Geriatr., https://doi.org/10.1186/1471-2318-13-45 (2013).
Lamers, W. H. & Moorman, A. F. M. Cardiac septation: A late contribution of the embryonic primary myocardium to heart morphogenesis. Circulation Research 91, 93–103 (2002).
Cermakova, P. et al. Subclinical cardiac dysfunction and brain health in midlife: CARDIA (Coronary Artery Risk Development in Young Adults) brain magnetic resonance imaging substudy. J. Am. Heart Assoc. 6 (2017).
Moreno-Grau, S. et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer disease and three causality networks of AD: the GR@ACE project. bioRxiv, https://doi.org/10.1101/528901 (2019).
Schneider, J. A., Arvanitakis, Z., Bang, W. & Bennett, D. A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology, https://doi.org/10.1212/01.wnl.0000271090.28148.24 (2007).
Lane, R. J. ‘Cardiogenic dementia’ revisited. J. R. Soc. Med. 84, 577–9 (1991).
Roberts, R. O. et al. Coronary heart disease is associated with non-amnestic mild cognitive impairment. Neurobiol. Aging 31, 1894–1902 (2010).
Newman, A. B. et al. Dementia and Alzheimer’s Disease Incidence in Relationship to Cardiovascular Disease in the Cardiovascular Health Study Cohort. J. Am. Geriatr. Soc. 53, 1101–1107 (2005).
Qiu, C. et al. Heart failure and risk of dementia and Alzheimer disease: A population-based cohort study. Arch. Intern. Med., https://doi.org/10.1001/archinte.166.9.1003 (2006).
Knopman, D. S., Petersen, R. C., Cha, R. H., Edland, S. D. & Rocca, W. A. Coronary artery bypass grafting is not a risk factor for dementia or Alzheimer disease. Neurology 65, 986–90 (2005).
Petrovitch, H. et al. Influence of myocardial infarction, coronary artery bypass surgery, and stroke on cognitive impairment in late life. Am. J. Cardiol. 81, 1017–21 (1998).
Jefferson, A. L. et al. Lower cardiac index levels relate to lower cerebral blood flow in older adults. Neurology 89, 2327–2334 (2017).
Beeri, M. S. et al. Better Cognitive Performance Associated With Worse Cardiac Functioning Suggests Antagonistic Pleiotropy in Very Elderly Subjects. Am. J. Geriatr. Psychiatry 17, 911–912 (2009).
Gidding, S. S. et al. Low cardiovascular risk is associated with favorable left ventricular mass, left ventricular relative wall thickness, and left atrial size: the CARDIA study. J. Am. Soc. Echocardiogr. 23, 816–22 (2010).
Whalley, G. A. et al. Association of fat-free mass and training status with left ventricular size and mass in endurance-trained athletes. J. Am. Coll. Cardiol. 44, 892–896 (2004).
Cain, P. A. et al. Age and gender specific normal values of left ventricular mass, volume and function for gradient echo magnetic resonance imaging: A cross sectional study. BMC Med. Imaging 9 (2009).
Cain, P. A. et al. Physiological determinants of the variation in left ventricular mass from early adolescence to late adulthood in healthy subjects. Clin. Physiol. Funct. Imaging 27, 254–262 (2007).
Jefferson, A. L. et al. Relation of left ventricular ejection fraction to cognitive aging (from the Framingham Heart Study). Am. J. Cardiol. 108, 1346–1351 (2011).
Rehker, J. et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS One, https://doi.org/10.1371/journal.pone.0185777 (2017).
Vaisid, T., Barnoy, S. & Kosower, N. S. Calpain activates caspase-8 in neuron-like differentiated PC12 cells via the amyloid-β-peptide and CD95 pathways. Int. J. Biochem. Cell Biol., https://doi.org/10.1016/j.biocel.2009.07.010 (2009).
Tacconi, S. et al. Increased caspase activation in peripheral blood mononuclear cells of patients with Alzheimer’s disease. Exp. Neurol., https://doi.org/10.1016/j.expneurol.2004.07.009 (2004).
Yew, D. T., Ping L, W. & Liu, W. K. Fas and activated caspase 8 in normal, Alzheimer and multiple infarct brains. Neurosci. Lett., https://doi.org/10.1016/j.neulet.2004.05.091 (2004).
Selkoe, D. J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 399, A23–A31 (1999).
Congdon, E. E. & Sigurdsson, E. M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol., https://doi.org/10.1038/s41582-018-0013-z (2018).
Luchetti, S. et al. Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol. Aging, https://doi.org/10.1016/j.neurobiolaging.2009.12.014 (2011).
Jiang, S. et al. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl. Psychiatry, https://doi.org/10.1038/s41398-018-0319-z (2018).
Khundakar, A. A. et al. Analysis of primary visual cortex in dementia with Lewy bodies indicates GABAergic involvement associated with recurrent complex visual hallucinations. Acta Neuropathol. Commun., https://doi.org/10.1186/s40478-016-0334-3 (2016).
Cauley, E. et al. Neurotransmission to parasympathetic cardiac vagal neurons in the brain stem is altered with left ventricular hypertrophy-induced heart failure. Am. J. Physiol. - Hear. Circ. Physiol., https://doi.org/10.1152/ajpheart.00445.2015(2015).
Li, Y. F. & Patel, K. P. Paraventricular nucleus of the hypothalamus and elevated sympathetic activity in heart failure: The altered inhibitory mechanisms. In Acta Physiologica Scandinavica, https://doi.org/10.1046/j.1365-201X.2003.01043.x(2003).
Acknowledgements
General. Data collection and sharing for this project was partially funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. For the Alzheimer’s Disease Neuroimaging Initiative: Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp19content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. The AddNeuroMed data are from a public-private partnership supported by EFPIA companies and SMEs as part of InnoMed (Innovative Medicines in Europe), an Integrated Project funded by the European Union of the Sixth Framework program priority FP6-2004-LIFESCIHEALTH-5. Clinical leads responsible for data collection are Iwona Kłoszewska (Lodz), Simon Lovestone (London), Patrizia Mecocci (Perugia), Hilkka Soininen (Kuopio), Magda Tsolaki (Thessaloniki), and Bruno Vellas (Toulouse), imaging leads are Andy Simmons (London), Lars-Olad Wahlund (Stockholm) and Christian Spenger (Zurich) and bioinformatics leads are Richard Dobson (London) and Stephen Newhouse (London). This dataset was downloaded from Synapse (https://doi.org/10.7303/syn2790911). Funding support for the Alzheimer’s Disease Genetics Consortium (ADGC) was provided through the NIA Division of Neuroscience (U01-AG032984). This study was downloaded from NIH dbGaP repository (phs000372.v1). The Coronary Artery Risk Development in Young Adults Study (CARDIA) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the University of Alabama at Birmingham (N01-HC95095 & N01-HC48047), University of Minnesota (N01-HC48048), Northwestern University (N01-HC48049), and Kaiser Foundation Research Institute (N01-HC48050). This manuscript was not approved by CARDIA. The opinions and conclusions contained in this publication are solely those of the authors, and are not endorsed by CARDIA or the NHLBI and should not be assumed to reflect the opinions or conclusions of either. Genotyping for the CARDIA GENEVA cohort was supported by grant U01 HG004729 from the National Human Genome Research Institute. This study was downloaded from NIH dbGaP repository (phs000285.v3.p2). The Cardiovascular Heart Study (CHS) was supported by contracts HHSN268201200036C, HHSN268200800007C, N01-HC85079, N01-HC-85080, N01-HC-85081, N01-HC-85082, N01-HC-85083, N01-HC-85084, N01-HC-85085, N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, and N01-HC-85239; grant numbers U01 HL080295 and U01 HL130014 from the National Heart, Lung, and Blood Institute, and R01 AG-023629 from the National Institute on Aging, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at https://chs-nhlbi.org/pi. This manuscript was not prepared in collaboration with CHS investigators and does not necessarily reflect the opinions or views of CHS or the NHLBI. Support for the genotyping through the CARe Study was provided by NHLBI Contract N01-HC-65226. This study was downloaded from NIH dbGaP repository (phs000287.v5.p1). The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195 and HHSN268201500001I). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI. “Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02-HL64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding for Affymetrix genotyping of the FHS Omni cohorts was provided by Intramural NHLBI funds from Andrew D. Johnson and Christopher J. O’Donnell. This dataset was obtained from the NIH dbGaP repository (phs000007.v29.p10). The genotypic and associated phenotypic data used in the study, “Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer’s Disease (GenADA)” were provided by the GlaxoSmithKline, R&D Limited. The datasets used for analyses described in this manuscript were obtained from NIH dbGaP repository (phs000219.v1.p1). The Mayo Clinic Alzheimer’s Disease Genetic Studies, led by Dr. Nilüfer Ertekin-Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer’s Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. This dataset was downloaded from Synapse (https://doi.org/10.7303/syn5550404). The MESA study was supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 from the National Heart, Lung, and Blood Institute, and by grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 from NCATS. The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. This dataset was obtained from the NIH dbGaP repository (phs000209.v6.p2). The Neocodex-Murcia study was funded by the Fundación Alzheimur (Murcia), the Ministerio de Educación y Ciencia (Gobierno de España), Corporación Tecnológica de Andalucía and Agencia IDEA (Consejería de Innovación, Junta de Andalucía). The Diabetes Research Laboratory, Biomedical Research Foundation. University Hospital Clínico San Carlos has been supported by CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM); CIBERDEM is an ISCIII Project. The ROS/MAP study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute. This dataset was downloaded from Synapse (https://doi.org/10.7303/syn3219045). The TGEN study was supported by Kronos Life Sciences Laboratories, the National Institute on Aging (Arizona Alzheimer’s Disease Center P30 AG19610, RO1 AG023193, Mayo Clinic Alzheimer’s Disease Center P50 AG16574, and Intramural Research Program), the National Alzheimer’s Coordinating Center (U01 AG016976), and the state of Arizona. TGEN investigators provided free access to genotype data to other researchers via Coriell Biorepositories (http://www.coriell.org). The results published here are in part based on data obtained from the AMP-AD Knowledge Portal accessed at https://doi.org/10.7303/syn2580853. D.S. research is supported by Grant 20153810 from Fundació La Marató de TV3 and Grant SAF2013-44942-R from the Ministerio de Economía y Competitividad (MINECO) and, with J.X.C., Grant 2009SGR-346 from the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) from the Government of Catalonia. A.B. has a predoctoral contract from Fundació La Marató de TV3. A.R. research is also supported by grants PI13/02434 and PI16/01861. Acción Estratégica en Salud, integrated in the Spanish National R&D&I Plan and financed by ISCIII (Instituto de Salud Carlos III)-Subdirección General de Evaluación and the European Regional Development Fund (ERDF – “A way to make Europe”), by Fundación banca “La Caixa” and Grifols SA (GR@ACE project). This work was also partly supported by the ADAPTED consortium, which has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115975. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and the European Federation of Pharmaceutical Industries and Associations.
Author information
Authors and Affiliations
Contributions
A.R. and D.S. contributed to the design of the study, acquisition of data and interpretation of the results; M.E.S. contributed to the design, acquisition of data, statistical analysis, interpretation of the results and redaction of the manuscript; A.G. contributed to the design of the study and interpretation of the results and redaction of the manuscript; B.H.-O., S.M.G., I.R. and A.B. contributed to the statistical analysis and critical review of the manuscript; G.M., A.O. and S.V. contributed to the acquisition of data and critical review of the manuscript; J.X.C. critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sáez, M.E., González-Pérez, A., Hernández-Olasagarre, B. et al. Genome Wide Meta-Analysis identifies common genetic signatures shared by heart function and Alzheimer’s disease. Sci Rep 9, 16665 (2019). https://doi.org/10.1038/s41598-019-52724-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52724-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
