
Abstract
U.S. public health agencies have employed next-generation sequencing (NGS) as a tool to quickly identify foodborne pathogens during outbreaks. Although established short-read NGS technologies are known to provide highly accurate data, long-read sequencing is still needed to resolve highly-repetitive genomic regions and genomic arrangement, and to close the sequences of bacterial chromosomes and plasmids. Here, we report the use of long-read nanopore sequencing to simultaneously sequence the entire chromosome and plasmid of Salmonella enterica subsp. enterica serovar Bareilly and Escherichia coli O157:H7. We developed a rapid and random sequencing approach coupled with de novo genome assembly within a customized data analysis workflow that uses publicly-available tools. In sequencing runs as short as four hours, using the MinION instrument, we obtained full-length genomes with an average identity of 99.87% for Salmonella Bareilly and 99.89% for E. coli in comparison to the respective MiSeq references. These nanopore-only assemblies provided readily available information on serotype, virulence factors, and antimicrobial resistance genes. We also demonstrate the potential of nanopore sequencing assemblies for rapid preliminary phylogenetic inference. Nanopore sequencing provides additional advantages as very low capital investment and footprint, and shorter (10 hours library preparation and sequencing) turnaround time compared to other NGS technologies.
Similar content being viewed by others
Introduction
U.S. public health agencies routinely perform surveillance on microbial foodborne pathogens, and in the U.S. alone each year, approximately 1 in 6 individuals are sickened by foodborne illnesses, resulting in approximately 3,000 deaths1. During outbreak responses, identification of the source is instrumental to inform surveillance and public health strategies. However, specific characterization of foodborne pathogens during these surveillance programs in food production and distribution is important, as it allows for early warnings and fast removal of the contaminated food product(s) from public circulation before the development of an outbreak1. To that end, U.S. public health agencies have employed next-generation sequencing (NGS) using short-read sequencing technology in surveillance activities and outbreak response2. In addition to utilizing whole genome sequencing (WGS) for pathogen identification, more detailed information on the pathogen such as virulence, antimicrobial resistance, serotype, and inference of possible links between the sources of contamination is obtained3. WGS has provided faster identification of contaminated sources of outbreaks, reduced the number of illnesses and deaths due to the foodborne infections, and decreased the number of isolates needed to link the illness to the source of contamination4,5.
Although WGS is now a routine procedure in epidemiologic investigation and surveillance of foodborne pathogens, short-read sequencing technology faces challenges such as resolving repetitive regions, which introduce ambiguities that lead to inaccurate sequence reconstruction and incomplete and fragmented de novo assemblies6,7,8,9. These gaps can lead to the inability to determine accurate genome organization or architecture, which can be important in determining if genes are co-regulated or co-transmissible in the case of genes associated with mobile elements10. Even though the short-reads are accurate, closed whole genome assemblies are now commonly accomplished using a combination of both short-read (for base accuracy) and long-read sequencing technologies (for structural accuracy)9,11,12.
Long-read sequencing, enabled by single-molecule real-time (SMRT) sequencing technology that has been utilized since 2004, can produce reads averaging 11 kb in length, which facilitates the completion of bacterial genome assemblies that are either lacking in sequencing depth at certain repetitive areas of the genome or have areas that are missing reads completely13. The long-reads span across these large repetitive regions14,15,16 and can provide unbiased coverage of regions sequenced poorly with other technologies due to G/C content or other characteristics13,17. However, there is a need for an approach that generates inexpensive, long-read data in a short turnaround time. Such approach will offer benefits for rapid detection of an organism, complete sequencing of bacterial chromosomes and plasmids, and complementation to other sequencing technologies used in both outbreak investigations and foodborne pathogen surveillance.
The MinION (Oxford Nanopore), which is pocket-size (10 cm × 2 cm × 3.3 cm) and powered directly by a USB port from a laptop computer, is a nanopore-technology sequencer that produces long, single-molecule reads18 and can address these trade-offs. It is portable, field-deployable, inexpensive, and provides sequencing of both DNA and RNA in real time. Since the release of the MinION platform, bioinformatics tools have been steadily evolving, with the goal of using nanopore data to assemble accurate, whole, bacterial genomes independent of any other sequencing technology19. However, the relatively high error rate of the obtained raw reads is a recognized concern in nanopore sequencing data20.
In this study, utilizing only nanopore technology, we aimed to simultaneously sequence and assemble complete genomes of two pathogenic bacterial strains that can cause human illness worldwide, Salmonella enterica subsp. enterica serovar Bareilly and Escherichia coli O157:H7. In addition, we aimed to develop an improved bioinformatics workflow that provides accurate assemblies and to determine whether shorter sequencing time would still provide reliable results. Utilizing publicly-available tools, we report a reproducible bioinformatics workflow which assembled the circularized bacterial genomes and associated plasmids with the lowest error rate reported to date. We also demonstrate that utilizing the proposed sequencing and bioinformatics approach, sequencing of the entire chromosome and plasmid can be achieved with significantly shortened run time. This study shows that long-read nanopore sequencing can be used as a low-cost method to sequence the whole microbial genomes of foodborne pathogens. These closed assemblies provide information on genome organization and can complement existing characterization data from other technologies such as short-read sequencing.
Materials and Methods
Bacterial cultures and DNA extraction
The Salmonella Bareilly isolate (CFSAN000189) was isolated from raw shrimp in India (Biosample SAMN04364135), and the E. coli O157:H7 isolate (FSIS11705876) was isolated from domestic, raw, ground beef collected by the U.S. Department of Agriculture Food Safety and Inspection Services (USDA-FSIS) as part of routine sampling of a U.S. establishment (Biosample SAMN08167607). Both bacterial isolates were grown on sheep blood agar (SBA) for 24 hours at 35 °C. Total DNA from each isolate was extracted using the DNeasy Blood and Tissue Kit (Qiagen, USA) following manufacturer’s instructions. DNA concentrations throughout the experiment were determined by using the Qubit® dsDNA HS Assay Kit on a Qubit® fluorometer 3.0 (Thermo Fisher Scientific, USA).
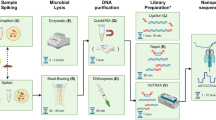
Library preparation and MinION sequencing
The 1D gDNA long read selection protocol was used with the SQK-LSK108 kit (Oxford Nanopore Technologies, UK) to prepare MinION-compatible libraries. The DNA shearing step was eliminated from the protocol with the aim of selecting for very long reads. Approximately, 2 µg of E. coli DNA and 2 µg of Salmonella DNA in a total of 100 µL each were added to the NEBNext® Ultra™ II End Repair/dA-Tailing module (New England Biolabs, USA) for end repair and dA-Tailing, following manufacturer’s instructions, and purified using Agencourt AMPure XP beads (Beckman Coulter, USA). Each purified, end-prepped DNA product was barcoded using a separate barcode from the 1D Native barcoding kit (EXP-NBD103, ONT) and following the 1D Native barcoding genomic DNA protocol. The samples were then bead-purified (Beckman Coulter), and equimolar amounts of each barcoded sample were pooled together for a final quantity of 700 ng. Adapters were ligated to the pooled sample using Blunt/TA ligase (New England Biolabs) following the 1D gDNA long read selection protocol. The MinION device was used to sequence the created library on a new FLO-MIN106 R9.4 flow cell21,22. The standard 48 hr 1D sequencing protocol was initiated using the MinKNOW software (ONT, UK). Average quality and coverage of the raw sequencing data were determined using CG-pipeline23.
MiSeq sequencing and quality control
To verify the newly developed approach used in this study, libraries for short-read WGS of the Salmonella Bareilly and E. coli isolates were prepared using the Nextera XT kit (Illumina, USA) according to the manufacturer’s protocol. The libraries were loaded separately into a single flow cell of the 300 and 500 cycle MiSeq Reagent Kits v2 for Salmonella Bareilly and E. coli, respectively, and paired-end sequencing (2 × 150 bp for Salmonella Bareilly and 2 × 250 bp for E. coli) was performed on the MiSeq instrument (Illumina, USA). The produced raw data were analyzed using SPAdes version 3.7124. Average quality and coverage of the raw sequencing data were determined using CG-pipeline23.
MinION bacterial bioinformatics workflow for whole genome assembly
To analyze the MinION sequencing data, a customized workflow was developed. For subsequent time analysis, the data was also analyzed at intervals from the start of the sequencing – at 15, 30, 60, 120, 240, 480, 960 and 1500 minutes (mins). Reads were basecalled using Albacore (v 2.0.2b, Oxford Nanopore Technologies) and subsampled for assembly using Filtlong (v.0.2.0)25 to a target depth of 75X with read quality weighted more heavily than length (‘mean_q_weight 5’). The filtered reads were assembled using the Unicycler pipeline (v.0.4.7)26. This pipeline utilizes a minimap/miniasm/racon iterative approach to assemble long-read-only data. Since Unicycler sometimes fails to detect valid end overlaps, assemblies were circularized using a custom script based on minimus227 (available in the workflow source repository). Circular contigs were rotated to start at a fixed position based on the reference. The consensus sequences were subjected to two rounds of polishing using Nanopolish (v.0.10.2)28, for which the full run (subject to time-based sub-setting but prior to Filtlong subsampling) was used, and Benchmarking Universal Single-Copy Orthologs (BUSCO v.3.0.2)29 was used to evaluate the completeness of coding sequences and degree of gene fragmentation in the polished assemblies. To evaluate assembly accuracy, two procedures were used for the Salmonella Bareilly isolate, which has previously been sequenced and published30. DNAdiff (MUMMER v.3.23)31 was used to evaluate both base-level and structural accuracy in the MinION assembly compared to the published reference. For the E. coli isolate, lacking a published reference, Illumina MiSeq reads were mapped to the assembly using BWA (v0.7.17), and LoFreq (v.2.1.3.1)32 was used to call single nucleotide polymorphisms (SNPs) and small indels, from which the assembly accuracy was calculated. Utilizing the short-read data, Pilon (v1.2.2)33 was used to error-correct small errors (‘--fix bases’) in the assemblies using existing short-read data from the same isolates (SRA accession SRR498276 for Salmonella Bareilly; SRA accession SRR6373397 for E. coli O157:H7).
MinION annotation
The polished-MinION assemblies after 4 hours of sequencing were initially annotated using the “Annotate From” tool within Geneious 11.1.5 and the published Salmonella Bareilly strain CFSAN000189 (GenBank Accession NC_021844) and E. coli O157:H7 strain 9234 (GenBank Accession CP017446) sequences as references. ResFinder v.3.1 was used to locate any antimicrobial resistance genes and any point mutations that would result in antimicrobial resistance34. Additionally, to confirm the 4-hour assembly annotation, the pilon-corrected, final genome sequences were submitted to GenBank to be processed through the NCBI Prokaryotic Genomic Annotation Pipeline (PGAP) before being released.
Phylogenetic analysis
Twenty-three Salmonella reference datasets (Supplementary Table S1) used in tracing a foodborne outbreak in the U.S that were previously published30,35 were downloaded. For the MinION-only data to be comparable, the eight sub-sampled (15 mins to 1500 mins) unpolished S. Bareilly assemblies obtained in this experiment were used to generate simulated Illumina datasets using ART (150 × 2, 50X coverage, MiSeq platform, 300 bp mean fragment length, 50 bp standard deviation)36. All datasets were analyzed with a SNP-calling pipeline using strain CFSAN000212 as a reference. Briefly, reads were optionally trimmed using Trim Galore (Illumina datasets), aligned to the reference using BWA-MEM37, SNPs were called using LoFreq32, and filtered using local scripts according to specific criteria. For Illumina datasets, the VCF files were filtered by removing indels as well as any SNPs with an alternate allele frequency of <90%. Sites meeting one or more of the following criteria were flagged as suspect, and these loci were ignored during matrix generation: (i) sites within 3 bp of a homopolymeric stretch of 4 bp or more; (ii) sites occurring in a variant cluster (multiple variants within 2 bp of each other; (iii) sites within 10 bp of a dam or dcm methylation motif; and (iv) sites with observed A- > G or T- > C transition mutations. The remaining SNPs were used to create a matrix of variable sites for phylogenetic reconstruction. MEGA6 (v.6.06) was used to generate a Neighbor-joining SNP trees utilizing the Maximum Composite Likelihood model with 1000 bootstrap iterations38. Three separate trees were constructed. The first tree was built using the SNP matrix obtained from the 23 Salmonella reference datasets35 (Supplemental Table S1). The second tree was constructed by replacing the reference Illumina data of the CFSAN000189 strain with the MinION-only data obtained by sequencing the same strain in this study (240 and 1500 mins time points were used). A third tree that contained both the Illumina and the MinION-only data of the CFSAN000189 strain was also built for comparison.
Availability of workflows, tools and code
The full NextFlow workflow, Conda environment configuration, and other associated code used in the analyses are publicly-available on GitHub (https://github.com/jvolkening/minion_bacterial).
Results
Analysis of MinION and MiSeq raw data
Before subsampling of the reads, the raw MinION sequencing data was used to estimate the mean depth for Salmonella Bareilly and E. coli, respectively. A total of 2.8 billion bases from 333,298 Salmonella Bareilly reads, with an average read length of 8638 nucleotides (nt), yielded a mean depth of 599X. For E. coli, a total of 3.8 billion bases from 429,909 reads with an average read length of 8979 nt were sequenced, and the mean depth was calculated to be 692X (Table 1). The shortest MinION read was 85 nt, which was from the E. coli isolate, while the longest read was from Salmonella Bareilly and was 129,119 nt. Both sets of MinION data had a mean read quality score above the standard (Q ≥ 10).
Illumina MiSeq data was also analyzed using the same bioinformatics tool. The MiSeq raw data had a depth of 57X for Salmonella Bareilly and 111X for E. coli. This sequencing technology produced 288 million bases from 1,930,511 Salmonella Bareilly reads, with an average read length of 150 nt. For E. coli, a total of 556 million bases from 2,291,825 reads were sequenced that had an average read length of 243 nt (Table 1). The minimum read length from both sets of bacterial sequences was 35 nt, while the longest was 151 nt for Salmonella Bareilly and 251 nt for E. coli; the MiSeq mean read quality was above the Q30 benchmark.
Assembly of MinION sequencing data
The raw MinION data for both isolates were subsampled on the basis of cumulative run time in order to simulate the effect of run time on final assembly quality. Subsets of reads generated in the first 15, 30, 60, 120, 240, 480, and 960 mins, in addition to the full run length of 1500 mins, were analyzed (Table 2). Four hours (240 mins) was determined as the shortest run time sufficient to assemble circular sequences from all chromosomes and plasmids from both isolates and represented a point after which longer run times resulted in significantly diminishing gains in final accuracy (Supplemental Fig. S1). Detailed data at each of the other run time subsets is available in Tables 2–4; however, the following analyses herein refer to the data collected in the first four hours of sequencing.
The MinION sequencing data was assembled using a custom Nextflow39 workflow that utilized publicly-available tools. Filtlong quality- and length-based subsampling resulted in 28,492 reads for the Salmonella Bareilly isolate, which were assembled into two circular contigs, the chromosome (4,724,389 bp) and plasmid (81,761 bp), with an average nucleotide identity of 99.87% and coverage of 100% compared to the reference genome (Table 2). For the E. coli isolate, 19,589 subsampled reads produced two circular contigs, the chromosome (5,482,542 bp) and plasmid (94,503 bp), with an average nucleotide identity of 99.89% compared to the available MiSeq data of the same bacterium (Table 2).
The final genome assemblies utilized two rounds of polishing using Nanopolish, which represented, by far, the most time-consuming and resource-intensive portion of the analysis workflow. However, it also increased the overall accuracy (Fig. 1a) due to a decrease in both SNPs (Fig. 1b) and chromosomal insertions or deletions (Fig. 1c). The largest gains in accuracy were achieved from the first round of polishing, while much less but still noticeable improvement was achieved with the second round, particularly when examining completeness of genome annotation as measured by BUSCO. However, further rounds (>2) of polishing did not significantly impact the overall assembly (Fig. 1). The central processing units (CPU) time and memory consumption for the assembling and polishing steps of the workflow can be found in Supplemental Table S2.

Polishing Results of the MinION-only Assemblies Using Multiple Rounds of Nanopolish. Due to the errors remaining in the MinION-only assemblies, a signal-level consensus software, Nanopolish, was used to increase the assembly accuracy. The overall accuracy, the Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness, BUSCO Fragmented, BUSCO Missing, number of indels per kb, and number of SNPs per kb are shown after 0, 1, 2, 3 and 4 rounds of Nanopolish. After two rounds of polishing, the overall accuracy and the number of Indels and SNPs per kb did not considerably change.
Detailed statistics of the assemblies’ accuracy are provided in Tables 3 and 4. For the Salmonella Bareilly assembly after 4 hours of sequencing, the rate of single nucleotide polymorphisms (SNPs) per kilobase (kb) decreased from 2.41 to 0.42 after one round of polishing and to 0.26 after two rounds of polishing. At the same time point, the insertions or deletions (indels) per kb decreased from 3.91 to 1.14 and 1.03 after one and two rounds of polishing, respectively (Table 3). For the E. coli assembly at the same time point, the SNPs per kb decreased from 2.20 to 0.37 after only one round of polishing and to 0.2 after two rounds of polishing. The indels per kb also decreased from 3.86 to 1 to 0.89 (Table 4). Additionally, the BUSCO tool was used to further analyze the polished data to determine the completeness of the gene content based on quality and length of alignment. The “BUSCO completeness” (fraction of expected gene complement with full-length reading frames) value of both bacterial assemblies and the rounds of polishing were directly related, increasing from 21 and 23% for the Salmonella and E. coli assemblies, respectively, with no polishing to 65 and 69% after two rounds of polishing; the BUSCO fragmented (decreased length alignment of genes) and BUSCO missing (no significant matches) values decreased correspondingly (Tables 3 and 4).
MinION assembly annotation
Both 4-hour MinION assemblies, after two rounds of polishing with Nanopolish, were annotated using Geneious and the most closely related, published, annotated genomes for each bacterial species. Since the Salmonella Bareilly genome was already completed and closed by a hybrid Illumina/PacBio approach and published, we confirmed that the Geneious genome annotation of the sequence of the same bacterium produced by MinION was accurately reconstructed (loci of protein-coding genes), by using the PGAP annotations tool on the final, corrected assembly; for example, but not limited to, the two major serotyping antigens located on the chromosome: the flagellin FliC CDS and the O-antigen polymerase.
The presence of major virulence factors in the E. coli MinION-only assembly were identified, as well as genes that would cause possible antimicrobial resistance, using Geneious (Fig. 2a,b). The locus of enterocyte effacement (LEE), one of the major virulence factors of enterohemorrhagic E. coli40,41 that includes the gene intimin for adhesion and the type III secretion system, was annotated between positions 4,603,699 and 4,636,299 in this MinION-only assembly (Fig. 2a). Additionally, the genes expressing the Shiga toxins (Stx), responsible for causing host cell damage40,42, were annotated from position 3,181,004 to 3,181,963 for Stx subunit A and from position 3,180,723 to 3,180,992 for Stx2 subunit B (Fig. 2a). The multidrug resistance gene Mdf(A), which encodes a membrane protein that confers resistance to a multitude of clinically important drugs, including macrolides, lincosamides, and streptogramin B43, was also identified at position 1,012,477 to 1,013,709. No other genes or point mutations that would confer antimicrobial resistance were detected. Not only was the full-length chromosome of this E.coli O157:H7 isolate sequenced using MinION, but also the full-length pO157 (Fig. 2b). Genes that encode E. coli O157-specific virulence factors40, such as hemolysin (ehx), catalase-peroxidase (katP), and the type II secretion system (T2SS) were identified in the sequenced plasmid.

Annotation of the MinION assembly of Escherichia coli. (a) The E. coli O157:H7 chromosome was sequenced and assembled into a final consensus of 5,482,542 nucleotides. The annotation of the genome provided the location of 5,748 coding sequences (CDS), 106 tRNAs, 29 rRNAs, 6 regulatory regions, and 1 repeat regions. For imaging purposes, only the 6 regulatory regions (green), the one repeat region (brown) and the CDS of two virulence factors (yellow) are shown magnified. The LEE (locus of enterocyte effacement) is highlighted at position 4,603,699 to 4,636,299, and the Shiga Toxin subunits are shown at position 3,181,004 to 3,180,992 for demonstration purposes. (b) The E. coli pO157 plasmid was sequenced and assembled into a final consensus of 94,503 nucleotides. The annotation shows all 124 coding sequences (CDS) in yellow. The CDS of three well-known virulence factors are highlighted: hemolysin (ehx) at position 16,584 to 19,578, catalase-peroxidase (katP) at position 76,704 to 78,356, and the type II secretion system (T2SS) at position 64,056 to 85,694 for demonstration purposes.
Additional polishing of the MinION assemblies with MiSeq Data
One of the main objectives of the presented work is to determine if MinION alone can be utilized to obtain fully closed genomes and plasmids from important foodborne pathogens. However, for submission of final sequences to GenBank, the most accurate assemblies attainable were used. To this end, for both samples, assemblies produced using the full run length, were utilized and further error-corrected using Pilon, together with available MiSeq data. Pilon utilizes the low error rate of Illumina reads mapped to the draft assembly to drastically improve the local accuracy of the final sequence. The error rate for both samples after Pilon polishing decreased, with accuracy rates of 99.99% and 100%, and BUSCO completeness rates of 99.7% and 99.99% for Salmonella and E.coli, respectively. There were also a reduction in SNPs per kb to 0.002 and 0.001 and indels per kb to 0.008 and 0.002 for Salmonella and E. coli, respectively. The assembled, polished, and short-read error-corrected data from the full 25-hour run were the final assemblies annotated and submitted to GenBank (Accession numbers CP034177- CP034178 and CP035545-CP035546, Bioproject PRJNA498670).
Phylogenetic inference (SNP tree)
The constructed SNPs trees are presented in Fig. 3. The tree built with the reference Salmonella datasets used for phylogenetic pipeline validation for foodborne pathogen surveillance35 is depicted in Fig. 3a. To demonstrate the potential of the MinION-only sequencing for rapid preliminary phylogenetic inference, the SNPs data for strain CFSAN000189 sequenced in this study, was replaced with the data from our assemblies, and the resulting tree is depicted in Fig. 3b. For simplicity, 240 mins and 1500 mins timepoints were used for the reconstruction. The comparison between the trees built with the reference datasets and the tree utilizing the MinION-only data for the CSAFN000189 strain demonstrates topological congruence between the trees. The results using all eight time points showed identical topology (data not shown). An additional tree using both the Illumina and the MinION data of strain CFSAN000189 was constructed (Fig. 3c). The results showed clustering in a monophyletic branch (98% branch support) of all CFSAN000189 data. The constructed trees were also congruent to the standard tree provided by Timme et al.35.

SNPs trees of Salmonella reference datasets and data obtained with MinION. (a) Constructed with SNPs of twenty-three Salmonella reference datasets which were used for phylogenetic pipeline validation for foodborne pathogen surveillance35; (b) The CFSAN000189 data is replaced with SNPs from the 240 mins and 1500 mins MinION-only assemblies obtained in this study; (c) The tree includes both the reference dataset and the MinION-only data for the CFSAN000189 strain along with the SNPs of the remaining 22 Salmonella reference datasets.
Discussion
In this study, we demonstrate that long-read, nanopore sequencing technology can be used as a single tool to sequence full length bacterial chromosomes and plasmids. Utilizing a customized workflow, optimized and tailored for bacterial sequencing results, and MinION-only data, whole genome sequences with as little as 0.1% error rate, were produced. These assemblies are 0.4% and 3.1% more accurate compared to previous reports10,19. The tools used in our customized bioinformatics workflow are publicly-available25,26 and the Conda environment configuration, along with other associated code used in the analyses, are also provided for public use.
Using MinION sequencing alone, two completely closed contigs, one chromosome and one plasmid for each pathogen, were assembled. This capability and the low cost make the MinION highly accessible as both a primary sequencing platform, as well as a secondary platform to complement laboratories’ existing sequencing infrastructure. The initial investment required for the MinION is drastically lower (starter pack costs $1000) than other sequencing technologies, each flow cell can be used for multiple runs, and samples can be multiplexed together per run to further reduce the cost21,44. Based on the results of barcoding and simultaneous sequencing of two whole bacterial genomes and plasmids shown here, we estimate that six bacterial samples could be multiplexed together to further decrease cost and sequenced in approximately 16 hours to obtain complete genomic data with high accuracy.
The effects of increased sequencing run lengths, different criteria and weights to subsample data for assembly, and increased rounds of polishing, were examined for their effect on the final assembly completeness and accuracy. Filtlong subsampling is not random but keeps the longest and highest quality reads from the input, which targets maximum sequencing depth (total bases). It was observed that the nanopore reads were long enough on average that over-aggressive length-based filtering resulted in reduced representation. Such extensive subsampling would result in less complete assembly of small plasmids, which can contain virulence factors of great interest for diagnostic and food safety purposes. It therefore proved critical to evaluate filtering and subsampling criteria to take full advantage of the technology. Read quality was weighted more heavily than length, as testing showed this was necessary to retain sufficient coverage of small plasmids.
Our results suggest that at least one round of polishing with Nanopolish is needed to achieve acceptable accuracy, and a second round provides additional improvement if the near-doubling of the analysis time is warranted. The data in Supplemental Table S2 are provided when only one core is utilized, but due to the wide availability of high-performance computers, the analysis time for two rounds of polishing can decrease to 6 hours using 124 cores, for example. In MinION-only assemblies, it is known that putative pseudogenes caused by systematic indel errors (often near homopolymeric tracts19,45), leading to reading frame shifts can be an issue, as evident from the “BUSCO fragmented” column in Tables 3 and 4. Even after polishing, this value was observed to be greater than 20% of expected coding genes, which must be taken into consideration during annotation. However, the polished assemblies, with only 0.1% error are accurately reconstructed and reveal serotype and important genes responsible for the virulence, metabolism, defense, and pathogenesis of the bacterium.
In outbreak situations, a rapid turn-around time is necessary. Therefore, polymerase chain reaction (PCR), real-time PCR assays, and other rapid diagnostic assays are still deployed. However, WGS has become routine in use and coupled with proper bioinformatics analysis can provide complete genome sequences in a couple of days2. With the MinION platform and sufficient computational resources (which can be cloud-based and thus widely available), basecalled sequence data can be analyzed in near-real-time as it comes off of the machine46. Therefore, the MinION can be used for rapid diagnostics as initial sequencing data from pure cultures can be provided in approximately 9 to 10 hours47. Furthermore, the MinION-only results have potential for rapid preliminary phylogenetic inference as demonstrated by the congruent topology between trees (and to the standard tree provided by Timme et al.35) built with the Illumina and the MinION-only data (only after four hours of sequencing). Of note, due to the higher MinION sequencing error rate, the distances between the MinION-only results and references were higher compared to the reference tree. However, the nanopore and bioinformatics are constantly improving, the quality and accuracy of the sequences steadily increase, and the MinION-only results would likely be epidemiologically informative in the near future. The complete MinION data can be further analyzed and polished after the entire sequencing run to obtain more accurate whole genomes that provide detailed data on subtyping, virulence genes, antimicrobial resistance genes, and other genetic characteristics. Same-day detection of antimicrobial resistance genes with 99.75% accuracy (with polishing) after enriching for plasmid DNA and MinION sequencing has been recently demonstrated48.
In conclusion, this low-cost, rapid, random-priming nanopore sequencing approach, coupled with our customized workflow, provides sufficient data where complete genomes, including plasmids, can be assembled into a single contiguous sequence with 99.89% accuracy (highest reported-to-date). These data allowed accurate gene identification and genomic organization without the need for additional sequencing tools to close gaps that are required by other sequencing methods. As the nanopore chemistry and bioinformatics continue to evolve, this method is promising in providing a sufficient amount of accurate data to complement the current sequencing methods by resolving repetitive regions of the genome, which will be instrumental in increasing the number of available complete genome assemblies.
Data availability
The final assemblies generated during the current study are available in GenBank (Accession CP034177- CP034178 and CP035545-CP035546). The raw data generated during the current study are available under BioProject number PRJNA498670, BioSamples numbers SAMN04364135 and SAMN08167607, and SRA Accession numbers SRR9603470 and SRR9603471.
References
Scallan, E., Griffin, P. M., Angulo, F. J., Tauxe, R. V. & Hoekstra, R. M. Foodborne illness acquired in the United States–unspecified agents. Emerg Infect Dis 17, 16–22, https://doi.org/10.3201/eid1701.091101p2 (2011).
Sekse, C. et al. High Throughput Sequencing for Detection of Foodborne Pathogens. Front Microbiol 8, 2029, https://doi.org/10.3389/fmicb.2017.02029 (2017).
Forbes, J. D., Knox, N. C., Ronholm, J., Pagotto, F. & Reimer, A. Metagenomics: The Next Culture-Independent Game Changer. Front Microbiol 8, 1069, https://doi.org/10.3389/fmicb.2017.01069 (2017).
Struelens, M. J., Palm, D. & Takkinen, J. Enteroaggregative, Shiga toxin-producing Escherichia coli O104:H4 outbreak: new microbiological findings boost coordinated investigations by European public health laboratories. Euro Surveill 16 (2011).
Dallman, T. J. et al. The utility and public health implications of PCR and whole genome sequencing for the detection and investigation of an outbreak of Shiga toxin-producing Escherichia coli serogroup O26:H11. Epidemiology and infection 143, 1672–1680, https://doi.org/10.1017/S0950268814002696 (2015).
van Dijk, E. L., Jaszczyszyn, Y. & Thermes, C. Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res 322, 12–20, https://doi.org/10.1016/j.yexcr.2014.01.008 (2014).
Chain, P. S. et al. Genomics. Genome project standards in a new era of sequencing. Science 326, 236–237, https://doi.org/10.1126/science.1180614 (2009).
Nagarajan, N. & Pop, M. Sequence assembly demystified. Nat Rev Genet 14, 157–167, https://doi.org/10.1038/nrg3367 (2013).
Orlek, A. et al. Plasmid Classification in an Era of Whole-Genome Sequencing: Application in Studies of Antibiotic Resistance Epidemiology. Front Microbiol 8, 182, https://doi.org/10.3389/fmicb.2017.00182 (2017).
Greig, D. R., Dallman, T. J., Hopkins, K. L. & Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microb Genom, https://doi.org/10.1099/mgen.0.000213 (2018).
Margos, G. et al. Lost in plasmids: next generation sequencing and the complex genome of the tick-borne pathogen Borrelia burgdorferi. BMC Genomics 18, 422, https://doi.org/10.1186/s12864-017-3804-5 (2017).
Gonzalez-Escalona, N., Yao, K. & Hoffmann, M. Closed Genome Sequence of Salmonella enterica Serovar Richmond Strain CFSAN000191, Obtained with Nanopore Sequencing. Microbiol Resour Announc 7, https://doi.org/10.1128/MRA.01472-18 (2018).
Koren, S. et al. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome biology 14, R101, https://doi.org/10.1186/gb-2013-14-9-r101 (2013).
Utturkar, S. M. et al. Evaluation and validation of de novo and hybrid assembly techniques to derive high-quality genome sequences. Bioinformatics 30, 2709–2716, https://doi.org/10.1093/bioinformatics/btu391 (2014).
Koren, S. & Phillippy, A. M. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol 23, 110–120, https://doi.org/10.1016/j.mib.2014.11.014 (2015).
Brown, S. D. et al. Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of Clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant Clostridia. Biotechnol Biofuels 7, 40, https://doi.org/10.1186/1754-6834-7-40 (2014).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10, 563–569, https://doi.org/10.1038/nmeth.2474 (2013).
Feng, Y., Zhang, Y., Ying, C., Wang, D. & Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genomics Proteomics Bioinformatics 13, 4–16, https://doi.org/10.1016/j.gpb.2015.01.009 (2015).
Loman, N. J., Quick, J. & Simpson, J. T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12, 733–735, https://doi.org/10.1038/nmeth.3444 (2015).
Lewandowski, K. et al. Metagenomic Nanopore sequencing of influenza virus direct from clinical respiratory samples. bioRxiv, 676155, https://doi.org/10.1101/676155 (2019).
Butt, S. L. et al. Rapid virulence prediction and identification of Newcastle disease virus genotypes using third-generation sequencing. Virology journal, https://doi.org/10.1101/349159 (2018).
Phan, H. T. T. et al. Illumina short-read and MinION long-read WGS to characterize the molecular epidemiology of an NDM-1 Serratia marcescens outbreak in Romania. J Antimicrob Chemother, https://doi.org/10.1093/jac/dkx456 (2017).
Kislyuk, A. O. et al. A computational genomics pipeline for prokaryotic sequencing projects. Bioinformatics 26, 1819–1826, https://doi.org/10.1093/bioinformatics/btq284 (2010).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Wick, R. Fitlong: quality filtering tool for long reads, https://github.com/rrwick/Filtlong (2007).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13, e1005595, https://doi.org/10.1371/journal.pcbi.1005595 (2017).
Sommer, D. D., Delcher, A. L., Salzberg, S. L. & Pop, M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8, 64, https://doi.org/10.1186/1471-2105-8-64 (2007).
Simpson, J. Nanopolish: Signal-level algorithms for MinION data, https://github.com/jts/nanopolish (2018).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Hoffmann, M. et al. Tracing Origins of the Salmonella Bareilly Strain Causing a Food-borne Outbreak in the United States. J Infect Dis 213, 502–508, https://doi.org/10.1093/infdis/jiv297 (2016).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome biology 5, R12, https://doi.org/10.1186/gb-2004-5-2-r12 (2004).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic acids research 40, 11189–11201, https://doi.org/10.1093/nar/gks918 (2012).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67, 2640–2644, https://doi.org/10.1093/jac/dks261 (2012).
Timme, R. E. et al. Benchmark datasets for phylogenomic pipeline validation, applications for foodborne pathogen surveillance. PeerJ 5, e3893, https://doi.org/10.7717/peerj.3893 (2017).
Huang, W., Li, L., Myers, J. R. & Marth, G. T. ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594, https://doi.org/10.1093/bioinformatics/btr708 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint, arXiv:13033997 (2013).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729, https://doi.org/10.1093/molbev/mst197 (2013).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat Biotechnol 35, 316–319, https://doi.org/10.1038/nbt.3820 (2017).
Lim, J. Y., Yoon, J. & Hovde, C. J. A brief overview of Escherichia coli O157:H7 and its plasmid O157. J Microbiol Biotechnol 20, 5–14 (2010).
Franzin, F. M. & Sircili, M. P. Locus of enterocyte effacement: a pathogenicity island involved in the virulence of enteropathogenic and enterohemorragic Escherichia coli subjected to a complex network of gene regulation. BioMed research international 2015, 534738, https://doi.org/10.1155/2015/534738 (2015).
Baranzoni, G. M. et al. Characterization of Shiga Toxin Subtypes and Virulence Genes in Porcine Shiga Toxin-Producing Escherichia coli. Front Microbiol 7, 574, https://doi.org/10.3389/fmicb.2016.00574 (2016).
Edgar, R. & Bibi, E. MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J Bacteriol 179, 2274–2280 (1997).
Ring, N. et al. Resolving the complex Bordetella pertussis genome using barcoded nanopore sequencing. Microb Genom 4, https://doi.org/10.1099/mgen.0.000234 (2018).
Tyson, J. R. et al. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res 28, 266–274, https://doi.org/10.1101/gr.221184.117 (2018).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome biology 17, 132, https://doi.org/10.1186/s13059-016-0997-x (2016).
Quick, J. et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome biology 16, 114, https://doi.org/10.1186/s13059-015-0677-2 (2015).
Lemon, J. K., Khil, P. P., Frank, K. M. & Dekker, J. P. Rapid Nanopore Sequencing of Plasmids and Resistance Gene Detection in Clinical Isolates. Journal of clinical microbiology 55, 3530–3543, https://doi.org/10.1128/JCM.01069-17 (2017).
Acknowledgements
The authors would like to thank Drs. Ruth Timme and James Pettengill with the FDA Center for Food Safety and Applied Nutrition for the Salmonella Bareilly isolate used in this manuscript. This work was supported by U.S. Department of Agriculture, ARS CRIS Project 6612-32000-072. The mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
Conceptualization – T. Taylor, E. DeJesus, G. Tillman and C. Afonso.; Methodology - T. Taylor and E. DeJesus.; Software - J. Volkening and M. Simmons.; Formal Analysis - T. Taylor, J. Volkening, M. Simmons and K. Dimitrov; Resources – G. Tillman, D. Suarez and C. Afonso.; Original Draft Preparation - T. Taylor; Review & Editing - T. Taylor, J. Volkening, E. DeJesus, M. Simmons, K. Dimitrov, G. Tillman, D. Suarez and C. Afonso; Supervision, Project Administration, Funding Acquisition - D. Suarez and C. Afonso.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, T.L., Volkening, J.D., DeJesus, E. et al. Rapid, multiplexed, whole genome and plasmid sequencing of foodborne pathogens using long-read nanopore technology. Sci Rep 9, 16350 (2019). https://doi.org/10.1038/s41598-019-52424-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52424-x
This article is cited by
-
Novel Organism Verification and Analysis (NOVA) study: identification of 35 clinical isolates representing potentially novel bacterial taxa using a pipeline based on whole genome sequencing
BMC Microbiology (2024)
-
A preliminary study of the use of MinION sequencing to specifically detect Shiga toxin-producing Escherichia coli in culture swipes containing multiple serovars of this species
Scientific Reports (2023)
-
Nanopore metatranscriptomics reveals cryptic catfish species as potential Shigella flexneri vectors in Kenya
Scientific Reports (2022)
-
MicroPIPE: validating an end-to-end workflow for high-quality complete bacterial genome construction
BMC Genomics (2021)
-
Trycycler: consensus long-read assemblies for bacterial genomes
Genome Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
