
Abstract
Human fetuin-B plays a key physiological role in human fertility through its inhibitory action on ovastacin, a member of the astacin family of metallopeptidases. The inhibitor consists of tandem cystatin-like domains (CY1 and CY2), which are connected by a linker containing a “CPDCP-trunk” and followed by a C-terminal region (CTR) void of regular secondary structure. Here, we solved the crystal structure of the complex of the inhibitor with archetypal astacin from crayfish, which is a useful model of human ovastacin. Two hairpins from CY2, the linker, and the tip of the “legumain-binding loop” of CY1 inhibit crayfish astacin following the “raised-elephant-trunk mechanism” recently reported for mouse fetuin-B. This inhibition is exerted by blocking active-site cleft sub-sites upstream and downstream of the catalytic zinc ion, but not those flanking the scissile bond. However, contrary to the mouse complex, which was obtained with fetuin-B nicked at a single site but otherwise intact, most of the CTR was proteolytically removed during crystallization of the human complex. Moreover, the two complexes present in the crystallographic asymmetric unit diverged in the relative arrangement of CY1 and CY2, while the two complexes found for the mouse complex crystal structure were equivalent. Biochemical studies in vitro confirmed the differential cleavage susceptibility of human and mouse fetuin-B in front of crayfish astacin and revealed that the cleaved human inhibitor blocks crayfish astacin and human meprin α and β only slightly less potently than the intact variant. Therefore, the CTR of animal fetuin-B orthologs may have a function in maintaining a particular relative orientation of CY1 and CY2 that nonetheless is dispensable for peptidase inhibition.
Similar content being viewed by others


Introduction
Metallopeptidases (MPs) are engaged in spatially and temporally regulated physiological processes including maturation, shedding and inactivation1,2,3. Owing to the irreversibility of peptide bond cleavage in vivo, MPs must be tautly restrained to prevent aberrant activity, which gives rise to dysfunction and disease. Control mechanisms include transcriptional regulation4, cellular and molecular compartmentalization5, zymogen-mediated latency3,6, and colocalization of specific or broad-spectrum protein inhibitors7. One of the many MP families described7,8 are the astacins, which were named for the eponymous digestive enzyme from the crayfish Astacus astacus9,10,11,12,13. Astacins have a common ~200-residue zinc-dependent catalytic domain (CD) whose architecture generally conforms to that of MPs of the metzincin clan8,14,15,16,17. Astacins are produced as zymogens with a pro-segment for latency upstream of the CD and a variable number of downstream domains (see Fig. 1 in11 for details), and six family members are found in humans: bone morphogenetic protein 1 and its major splice variant, mammalian tolloid, which are also known as procollagen C-proteases; tolloid-like proteins 1 and 2; meprin α and β; and ovastacin (see11,12 and http://degradome.uniovi.es/met.html 18). The first three MPs constitute the tolloid subgroup and process precursors of fibrillar procollagens, proteoglycans, laminins, and anchoring fibrils. They are thus important for extracellular-matrix assembly11,19. Tolloid astacins also cleave growth factors and their antagonists, which are crucial for dorso-ventral patterning during embryo gastrulation20. Next, meprins are membrane-bound proteins involved in tissue differentiation and pericellular signaling by processing of biologically active peptides, cytokines, chemokines, growth factors, and peptidase zymogens21,22. They also cleave components of the extracellular matrix such as the basal lamina, procollagens, and adhesion proteins21,23,24. Finally, ovastacin is a hatching enzyme expressed in the oocyte25. The enzyme cleaves the glycoprotein matrix surrounding the oocyte, dubbed the zona pellucida, after gamete fusion. This leads to rigidification of the matrix, blocks further sperm binding, and is essential for the survival of the developing embryo25,26,27.
Small astacins, but not large multidomain forms such as the meprins, are inhibited by the non-specific pan-peptidase inhibitor α2-macroglobulin28,29,30. Specific inhibition of tolloid astacins has been reported for Xenopus laevis sizzled/ogon31,32. By contrast, meprins, crayfish astacin, nephrosin from cyprinid fishes, and ovastacin are strongly inhibited by fetuin-B forms from mammals, which are strictly selective for astacins33,34,35,36, and by fish fetuin, which acts as the physiological antagonist of nephrosin37. By blocking ovastacin, fetuin-B prevents premature hardening of the zona pellucida and maintains female fertility26,33,34. Fetuin-B belongs to the I25 family of peptidase inhibitors according to the MEROPS database of peptidases and inhibitors (www.ebi.ac.uk/merops)7. The archetype of this family is chicken egg-white cystatin (ovocystatin), a 116-residue reversible inhibitor specific for cysteine peptidases38,39. Within the family, fetuins are type-3 cystatins (subfamily I25C), which include glycosylated proteins with two or three cystatin-like modules40,41. Recent crystal structures of the mouse ortholog (mFB), isolated and in complex with crayfish astacin36, have revealed that the inhibitor consists of the tandem cystatin-type modules 1 and 2 (CY1 and CY2), which are united by a linker (LNK) with a “CPDCP-trunk” and followed by a C-terminal region (CTR). The inhibitor blocks the active-site cleft of the MP following a novel “raised-elephant-trunk” mechanism36.
To complement these studies, we here report the crystal structure of the complex between the human ortholog of fetuin-B (hFB), which is the physiologically relevant species for studying human fertility42, and 202-residue mature crayfish astacin, which is a useful model for the 197-residue catalytic domain of human ovastacin (35% sequence identity; 48% similarity; see also35). These studies revealed unexpected differences with mFB in terms of proteolytic susceptibility and the spatial arrangement of the cystatin domains, which enabled us to identify dispensable structural elements for inhibition. We verified these structural findings by means of biochemical studies with crayfish astacin and human meprins in vitro.
Results and Discussion
Crystallization and proteolytic susceptibility of human fetuin-B in the presence of crayfish astacin
We have previously reported the structure of isolated mFB, which was recombinantly produced in mammalian cells and processed with endoglycosidase H36. This treatment clipped down the N-glycans attached to N40 and N139 (mFB amino-acid numbering in superscript and italics according to UniProt entry [UP] Q9QXC1) to single N-acetylglucosamine moieties. In particular, the sugar attached to N40 participates in a packing contact that is crucial for crystal formation. In contrast, no crystals have been obtained with protein heterologously produced in insect cells, which attach different glycans. However, for the astacin·mFB complex crystal structure, protein from insect cells has been successfully employed in the presence of MP excess36. In this study, hFB was recombinantly produced and purified from mammalian cells, and crystallization of the astacin·hFB complex was also only successful with peptidase excess. SDS-PAGE, Western blotting, and N-terminal Edman degradation of the complex in solution revealed that most of the CTR was removed through cleavages at positions G302-S303 (hFB residue numbering in superscript according to UP Q9UGM5), E322-A323 and somewhere upstream within the CTR to yield species migrating according to ~30–35 kDa instead of the ~50 kDa of intact hFB (Fig. 1A,C). Cleaved forms were isolated by astacin affinity chromatography for comparative inhibitory studies (see Fig. 1E and below). In contrast to hFB, mFB proved more resistant to astacin degradation under equivalent conditions (Fig. 1B). This is consistent with the aforementioned crystallization experiments36, which revealed that peptidase excess cleaves mFB at bond S296-S297 within the CTR of the otherwise intact molecule36. The structure of the mouse complex has shown that the CTR is an irregularly folded domain that is partially disordered and devoid of regular secondary structure, which explains its proteolytical susceptibility.

Digestion and inhibitory studies. Coomassie staining of hFB (A) and mFB (B) at 25 pmol/lane incubated with astacin and separated by reducing (left) and non-reducing (right) 14% SDS-PAGE. Bands subjected to N-terminal Edman degradation are pinpointed by asterisks and labeled 1, 1′, 1″ and 2, respectively. (C) Western-blot analysis of hFB (10 pmol/lane) incubated with astacin and separated by reducing and non-reducing 14% SDS-PAGE. Immunodetection was performed with anti-full-length hFB antibody (red) and anti-penta-His antibody (green). (D) Plot of fractional velocity (logarithmic scale) of peptidase inhibition by hFB. Enzyme and substrate concentrations were 1.0 nM and 170 mM for astacin, 1 nM and 25 mM for meprin α, and 0.5 nM and 25 mM for meprin β, respectively. Error bars indicate standard errors. The upper-right inset depicts the derived Ki values. (E) Coomassie staining of cleaved (3 µg/lane) and intact (1 µg/lane) hFB separated by 14% SDS-PAGE. Species pinpointed by a hashtag were treated with urea as described in the Materials and Methods section. (F) Plot of fractional velocity (logarithmic scale) of peptidase inhibition by urea-treated intact and cleaved hFB. Enzyme and substrate concentrations as in (D). Error bars indicate standard deviations. The upper-right inset indicates the derived Ki values of intact (left) and cleaved (right) hFB.
Astacin·hFB crystals diffracted to a similar resolution as those of the mouse complex (3.0 Å vs. 3.1 Å) and likewise contained two enzyme·inhibitor complexes per asymmetric unit (A·B and C·D). However, they belonged to a different space group (P61 vs. P212121; compare Table 1 and Table 1 in36), had different cell constants, and contained significantly more solvent (57% vs. 47%).
Structure of cleaved human fetuin-B
The structure spans domains CY1 (A25/N27-S143) and CY2 (S156-E258/S259), which are connected by the linking segment LNK (K144-P155), plus a small fragment of the CTR (R364/T365-A375/S376) (Fig. 2A). Characteristic of cystatin-type domains, CY1 and CY2 consist of an antiparallel five-stranded curled β-sheet of up-and-down connectivity (strands β1-β5 in CY1 and β6-β10 in CY2) and a perpendicular α-helix (α1 in CY1 and α3 in CY2) inserted between the first two strands. The second/third and fourth/fifth strands of each sheet are joined by short connections (hairpins I and II, respectively), while the third and fourth strands are connected by long “legumain-binding loops” (LBLs)36,43 of 21 and 19 residues in CY1 and CY2, respectively. In the latter domain, the LBL is disordered for C216-S228. In contrast, in CY1 it is well-defined and creates a hydrophobic pillow at its most exposed segment spanning I108-F110. Residing on this pillow, LNK comprises helix α2 and the CPDCP-trunk (C151-P155), which protrudes from the molecular surface (Fig. 2A). Finally, a 12-residue fragment of the CTR is attached to CY1 through disulfide C36-C368. Further disulfides are found in the LBLs (C93-C104 in CY1 and, probably, C216-C224 in CY2), as well as between β4 and β5 of CY1 (C117-C137) and β9 and β10 of CY2 (C237-C254). A last disulfide (C151-C154) tightly crosslinks LNK to provide rigidity to the CPDCP-trunk. Within CY1, two N-glycan chains are attached to N37 and N136, respectively (Fig. 2A).

The cleaved human fetuin-B structure. (A) Ribbon-type plot of cleaved hFB (molecule B), which consists of domains CY1 (in orange) and CY2 (turquoise), linker LNK with the CPDCP-trunk (red) and a short fragment of the CTR (purple). Inter- and intra-domain disulfides are indicated as yellow sticks (for the respective residue numbers, see Fig. 4A). N-linked glycans are attached to N37 (①) and N136 (②). The regular secondary structure elements are labeled (β1-β10, α1-α3; for the respective spanning residues see Fig. 4A). The legumain-binding loops are labelled, as well as hairpins I and II of each cystatin domain. Relevant residues from the CPDCP-trunk and the tips of CY2 hairpin I and II are shown for their side chains. Residue H245 is in double occupancy in the A·B complex. The LBL of CY2 is partially disordered (dotted line). After the last visible residue of strand β10 (magenta arrow), the polypeptide chain only becomes defined again at CTR residues R364/T365 (turquoise arrow). (B) Superposition of hFB molecules B (in plum) and D (turquoise) reveals a hinge motion about K144 and significant variations around A100 and P126. Turquoise and plum arrows pinpoint the last CY2 residues and the first CTR residues visible in the final Fourier map.
The two hFB moieties in the asymmetric unit (B and D) superpose with an rmsd of 1.1 Å. While the respective CY2 and LNK moieties fit well, the CY1 domains are rotated by ~5° around K144Cα, which leads to a displacement of maximally ~4.5 Å (at P126Cα) (Fig. 2B). In addition to this rigid-body displacement, which in general keeps the same conformation in both CY1 domains, it is remarkable that significant rearrangement is found in segment R97-M106 within the LBL, which is displaced by ~5 Å maximally (at A100Cα).
Inhibition of crayfish astacin by human fetuin-B
Crayfish astacin is a bipartite molecule of 202 residues consisting of two equally large upper and lower sub-domains (USD and LSD), which form an extended, deep active-site cleft at their interface9,10,44. The cleft harbors the catalytic zinc ion, which is bound by three histidines from a zinc-binding consensus sequence (H92-EXXHXXGXX-H102; mature residue numbering of astacin in subscript; for numbering of the preproprotein according to UP P07584, add 49), which further contains the general base/acid for catalysis (E93) and is a hallmark of the astacins11,12 and other metzincin MPs8,14,17.
In the complex, the hFB moiety inserts like a chock into the active-site cleft of astacin through contacts made by the LNK, hairpins I and II of CY2, and the tip of the LBL of CY1 (Fig. 3A,B). This causes the cleft of both MP protomers in the asymmetric unit (A and C) to slightly open, triggered by a ~7°-rotation of the LSD around a horizontal axis traversing F100 and P176, which causes a maximal displacement of ~3 Å (at S123Cα). Owing to the slight differences between hFB moieties B and D (see previous section), the A·B and C·D complexes also vary. Superposition of the respective MP moieties reveals that while the chock elements of the hFBs coincide, the cleft-distal parts of the inhibitors are rotated relative to each other by ~12° about a horizontal axis traversing T150 and V201, which causes a displacement of ~11 Å maximally (at P126; see Fig. 3C). This is reminiscent of the complex between the otherwise unrelated MP thermolysin from Bacillus thermoproteolyticus and its specific inhibitor IMPI from the greater wax moth Galleria mellonella45. The latter likewise follows a wedge-like inhibition mechanism involving a few structural elements on the inhibitor surface. In the two complexes in the asymmetric unit, the inhibitor moieties are rotated by ~5° relative to each other by visual inspection.

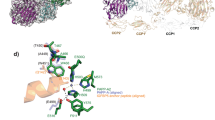
Structure of the astacin complex with cleaved human fetuin-B. (A) Ribbon-type plot in cross-eye stereo of the crayfish astacin·hFB complex A·B. The inhibitor is colored as in Fig. 2A, astacin is in salmon, and the catalytic zinc ion is shown as a magenta sphere for reference. The orientation results from that of Fig. 2A after a horizontal downward rotation of ~40°. This corresponds to a ~90°-rotation downward with respect to the standard orientation of astacin (see49). (B) Close-up view of (A) depicting the principal residues engaged in complex formation of astacin (pink ribbon and carbons; residue numbers in dark red) and hFB (tip of CY1 LBL as an orange ribbon, LNK as a red ribbon, CY2 hairpin I as a turquoise ribbon, and CY2 hairpin II as a green ribbon) with carbons in pale blue (residue numbers in blue). (C) Superposition of the enzyme moieties of the two astacin·hFB complexes A·B (astacin in sky blue, CY1 in yellow, LNK in red, CY2 in white, and CTR in pink) and C·D (astacin in blue, CY1 in orange, LNK in green, CY2 in gray, and CTR in purple) reveals flexibility of domain CY1.
In the hFB·astacin complex A·B (Fig. 3A,B), the interface spans 1,245 Å2, with a theoretical solvation energy gain on complex formation (ΔiG according to46) of −20.1 kcal/mol and a significant complexation significance score (CSS according to46) of 0.50. The interface involves 35 residues of hFB and 41 of astacin, which make 14 hydrogen bonds, two ionic interactions, and hydrophobic interactions between 27 pairs of residues (see Table 2). Astacin segments involved in the complex are S62-V68, Q80-H102, S126-Y133, and Y149-W158. Participating hFB regions include I108-E111 from the LBL of CY1 and Y148-P155 from helix α2 plus the downstream CPDCP-trunk, which from M149 to D153 runs along the non-primed side of the cleft in the direction and orientation of a substrate (Fig. 3B). In particular, D153 binds the catalytic zinc of astacin according to an aspartate-switch mechanism, as well as the general base/acid E93 and Y149. After D153, the polypeptide performs a ~90°-kink outward and leaves the cleft, which explains why fetuin-B is not cleaved at bond D153-C154. This contrasts with standard-mechanism inhibitors47 of MPs, such as the aforementioned IMPI from which a reactive-site loop is inserted like a substrate across the entire cleft of thermolysin. Thus, the reactive-site bond that links residues in sub-site positions S1 and S1’ (for peptidase active-site cleft sub-site nomenclature, see48,49) is cleaved45.
Assisting the CY1 LBL and the CPDCP-trunk, the tip of hairpin I of CY2 spanning Q198-F206 blocks sub-sites on the primed side of the cleft (Fig. 3B). This hairpin contains segment Q198-WVVG-P203, which conforms to the hallmark sequence motif of inhibitory fetuins (QWVXGP36), and V200 and V201 nestle into sub-sites S2’ and S3’, respectively. Finally, the tip of CY2 hairpin II (S240-F249) further contributes to the complex by blocking outmost cleft sub-sites and attaching to the right surface of the enzyme (Fig. 3B). Overall, these interactions conform to the raised-elephant-trunk mechanism of astacin inhibition previously described in detail for mFB36. Thus, the present structure suggests that the excised CTR is not required for inhibition.
As expected from a comparison of the isolated inhibitor moieties (see previous section), superposition of the peptidases of the two complexes in the asymmetric unit reveals that the segments engaged in inhibition (tip of CY1 LBL, LNK with CPDCP-trunk and hairpins I and II of CY2), as well as the rest of CY2 (Fig. 3C), appear well-aligned. Hence, the aforementioned interactions at the A·B enzyme·inhibitor interface are also found in the C·D complex with just a couple of exceptions (Table 2). In contrast, the rest of CY1 with the attached CTR fragment significantly deviates in both complexes owing to a ~12°-rotation about K144, which causes a displacement of ~11 Å maximally, at the cleft-distal edge around G77Cα and S127Cα (Fig. 3C). In line with these differences, in complex C·D the interface spans 1,421 Å2, with a ΔiG of −20.4 kcal/mol and a CSS of 0.43. A total of 40 residues of hFB and 43 of astacin participate in the interface through 13 hydrogen bonds, two ionic interactions and hydrophobic interactions between 26 pairs of residues (see Table 2). Taken together, these findings support an ancillary role for CY1 in inhibition and, possibly, a tethering role for the missing CTR to fix the relative orientation of CY1 and CY2, although crystal packing may also play a role.
Inhibitory studies in vitro
To assess the role of the CTR and the arrangement between CY1 and CY2 in peptidase inhibition, we first determined the apparent inhibition constant Ki of intact hFB in front of crayfish astacin and human meprin α and β as a control of the generally valid inhibition of astacins by hFB, and found values in the subnanomolar range (Fig. 1D). These values were indicative of very potent inhibition and compared well with those of mFB (see Fig. 3 in36). Next, we isolated astacin-cleaved hFB (see above), which entailed treatment with urea to dissociate the inhibitor from the peptidase, and determined its Ki values in front of the aforementioned enzymes. As a control, we also determined these parameters for intact hFB subjected to the same chaotropic treatment (Fig. 1F). We found that urea slightly decreased the inhibitory power of intact hFB.However, the cleaved inhibitor still evinced potent inhibition with Ki values in the nanomolar range (Fig. 1D,F). We conclude that the CTR and the relative orientation of CY1 and CY2 play a negligible role in the inhibition of astacin MPs.
Comparison with the mouse fetuin-B·crayfish astacin complex
Despite the lack of almost the entire CTR in the human inhibitor, but consistently with a sequence identity of 67% based on the CY1-LNK-CY2 domains involved in enzyme·inhibitor contact (Fig. 4A), the general inhibition modes of CTR-nicked mFB and CTR-depleted hFB are very similar and generally encompass the same segments, which have been outlined above and extensively described in36. However, significant differences can be found on detailed comparison.

Comparison of human and mouse fetuin-B. (A) Structure-assisted sequence alignment of the human and mouse orthologs, identical residues (62%) are in magenta. Residues of CY1, LNK, CY2, CTR are over orange, salmon, turquoise and purple backgrounds, respectively. Predicted signal peptides are over a gray background. The lines above and below the alignment indicate missing residues in the astacin-complex structures (dashed lines), as well as β-strands (arrows labelled β1-β10) and α-helices (zigzags labelled α1-α3) of human and mouse fetuin-B, respectively. Residues connected by disulfide bonds are denoted by encircled numbers (①–⑥), C216-C224 (hFB) and C217-C224 (mFB) are tentatively assigned as the respective LBL segments are disordered. (B) Superposition of the two structures of hFB (Cα-traces of protomers B and D in turquoise and yellow, respectively), unbound mFB (Cα-trace in red), and astacin-bound mFB (Cα-trace in plum). The orientation corresponds to that of Fig. 2B. (C) Close-up view of (B) at the respective interfaces of hFB (ribbons in turquoise and carbons in light blue; residue labels in dark blue) and mFB (ribbons and carbons in plum; residue labels in magenta) with astacin (ribbons and carbons in brown and yellow, respectively; brown labels). The orientation corresponds to that of Fig. 3B.
First, in contrast with hFB, the two astacin·mFB complexes within the asymmetric unit evince only minor variations, mainly in the CTRs, as reflected by an rmsd of 0.85 Å. In the mouse complex, the complex interface spans 1,059 Å2 and gives a ΔiG of −10.8 kcal/mol and a CSS of 0.33. The interface involves 33 residues of fetuin-B and 38 of astacin, which perform ten hydrogen bonds, two ionic interactions and 20 hydrophobic interactions36. All these values are significantly lower than those for the two astacin·hFB interfaces (see above and Table 2), thus pointing to looser interaction in the mouse complex. This difference in inhibitory action is apparently reflected by the disparate serum concentrations of mFB (156 ± 3 µg/ml) and hFB (5 ± 1 µg/ml)50.
Second, superposition of the entire complexes reveals rmsd values of 1.9 Å and 2.2 Å for mFB compared with hFB complexes A·B and C·D, respectively. If the respective peptidase moieties are superposed, the leftmost part of the chock comprising LNK and the LBL tip of CY1 (I108-F110 in hFB and M111-Y113 in mFB) neatly coincide for their main chains and most interactions are equivalent, including those of the CPDCP-trunk aspartate (D153 in hFB and D156 in mFB) (Fig. 4B). However, the rest of the fetuin-B moieties diverge substantially. Within the coil connecting α3 and β7 of CY2, deviations of ~10 Å and ~8 Å are detected between mFB K186Cα and topologically equivalent K185Cα from hFB molecules B and D, respectively. Similarly, mFB R130Cα and equivalent S127Cα at the tip of CY1 hairpin II are ~7 Å apart in both hFB molecules B and D. Even within the better fitting parts of the complexes, the first two residues of the second turn of LNK helix α2 of hFB (Y148-M149) penetrate the non-primed side of the cleft of astacin more deeply than the equivalent pair of mFB, H151-T152 (Fig. 4B).
Third, both hairpins I and II of CY2 on the primed side of the cleft significantly diverge. While the former has the same length in both fetuins and likewise comprises the hallmark motif QWVXGP (Q199-WVSG-P204 in mFB), in hFB it intrudes the astacin cleft more deeply owing to a relative ~90°-rotation of the Cα-C bond of the tryptophan. This causes the side chain of the downstream valine to enter the S1’ sub-site while in mFB it merely performs a hydrophobic interaction with disulfide C64-C84 on the top of the active-site cleft (Fig. 4B). In addition, hairpin II is eight residues shorter in hFB and, while slightly flexible at its tip (H245-R246), it is clearly resolved in the final Fourier map. In contrast, it is disordered and untraceable for four and six residues in the two mFB complexes. Thus, interactions of hFB with Y177-D178 and W158 of astacin that could not be detected in the mouse complex were defined here (Fig. 4B and Table 2). Interestingly, mFB stands alone with its long CY2 hairpin II, which uniquely contains seven-to-nine residues more than fetuin-Bs from other animals, including the evolutionarily closely related naked mole rat (see Suppl. Fig. 3 in36). This suggests that the other orthologs will probably have a structured CY2 hairpin II and bind astacins more similarly to hFB than to mFB.
Conclusions
We crystallized the complex between hFB and crayfish astacin in the presence of an excess of the MP, which proteolytically removed most of the CTR but retained the complex-formation and inhibitory capacities of the fetuin. As found for the mouse ortholog, hFB blocked astacin following a raised-elephant-trunk mechanism by inserting like a chock into the active-site cleft but sparing sub-sites S1 and S1’. This explains why hFB was not cleaved at the inhibitory loops in contrast with standard-mechanism inhibitors.
The two astacin·hFB complexes in the asymmetric unit evinced significant differences in the relative arrangement of CY1 and CY2. This differs from the mouse complex, which was only nicked at the CTR upon incubation with an equivalent excess of astacin and for which the two complexes in the asymmetric unit were similar. This indicates that the proteolytic susceptibility of the human and mouse fetuin-B orthologs vary despite close relatedness based on high sequence identity. In the mouse complex, the interaction surface and the theoretical solvation energy gain on complex formation were significantly smaller than in the human CTR-depleted complex, which points to a looser enzyme·inhibitor interaction of the intact mouse inhibitor.
Biochemical assays in vitro revealed that intact hFB and mFB inhibited crayfish astacin and human meprin α and β with comparable potency, with Ki values in the subnanomolar range. This suggests similar inhibition mechanisms for both full-length orthologs and indicates that the unstructured CTR has a function in structural maintenance of a particular relative disposition between CY1 and CY2 in fetuins. This fixed arrangement probably limits the adaptive capacity of the intact inhibitor to produce an optimal interaction surface with the target MP as large as that of the cleaved form. Notably, the Ki values of cleaved hFB in front of crayfish astacin and meprins α and β were still in the nanomolar range.
Materials and Methods
Protein production and purification
Protein hFB with a C-terminal hexahistidine-tag, thus spanning residues C16-P382-IEGRHHHHHH, was expressed and secreted to the extracellular medium by mammalian ExpiCHO-S cells according to the manufacturer’s instructions (ThermoFisher Scientific) as previously reported35. Human meprin β was obtained as a zymogen as described51 and activated with trypsin52. Protein mFB with a C-terminal hexahistidine-tag was cloned in vector pFASTBac1 and expressed in baculovirus-transduced High Five cells as described for meprin35,51,53. Human meprin α was produced as described53. All proteins used for crystallization were purified by nickel-nitrilotriacetic-affinity and size-exclusion chromatography steps as published33,35. Astacin was purified from the digestive fluid of the European freshwater crayfish Astacus astacus as reported10.
Crystallization and diffraction data collection
Crystallization assays were set up following the sitting-drop vapor diffusion method at the joint IBMB/IRB Automated Crystallography Platform of Barcelona Science Park. A Tecan robot (Tecan Trading) was used to prepare reservoir solutions, and a Cartesian Microsys 4000 XL robot (Genomic Solutions) or a Phoenix nanodrop robot (Art Robbins Instruments) dispensed nanocrystallization drops on 96 × 2-well Swissci Polystyrene MRC Crystallization Plates (Molecular Dimensions). Plates were stored at 4 °C or 20 °C in thermostatic crystal farms (Bruker AXS). The astacin·hFB complex only crystallized after incubating the inhibitor (at 7.5 mg/mL) with six-fold molar excess of the peptidase in 10 mM Tris·HCl, 140 mM sodium chloride, pH 6.8. Crystals were obtained at 20 °C in 200 nL:100 nL drops with protein complex solution and 20% [w/v] polyethylene glycol 3,350, 0.2 M sodium tartrate dibasic as reservoir solution. Crystals were cryo-protected through quick immersion in drops containing reservoir solution implemented with 20% [v/v] glycerol and flash vitrified in liquid nitrogen. They were mounted on a MD2M diffractometer (Maatel) by a CATS Automatic Sample Changer (Irelec) at beam line XALOC54 of the ALBA synchrotron in Cerdanyola (Catalonia, Spain) and kept at 100 K in the cryostream of an Oxford Cryosystems 700 series. Diffraction data was collected on a Pilatus 6 M pixel detector (Dectris) and processed with programs XDS55 and XSCALE56, and converted to the MTZ-format used by the CCP4 suite of programs57 with XDSCONV. Crystals belonged to space group P61, contained two peptidase·inhibitor complexes per asymmetric unit (molecules A·B and C·D, respectively), and diffracted to 3.0 Å resolution (see Table 1 for data processing statistics).
Structure solution and refinement
The structure of the astacin·hFB complex was solved by maximum-likelihood-scored molecular replacement with the PHASER program58. Initial searches with the entire astacin·mFB complex (PDB 6HT936) were unsuccessful. Subsequent searches with the separate astacin and mFB moieties yielded chemically reasonable solutions upon visual inspection with a log-likelihood gain of 2,714 at final Eulerian angles and fractional cell coordinates (α, β, γ, x, y, z) 8.4, 120.3, 180.2, −0.274, 0.249, −0.003 and 178.5, 121.1, 159.3, 0.172, −0.239, −0.026 for astacin; and 359.7, 108.4, 176.7, −0.248, 0.331, −0.045 and 234.8, 111.1, 165.6, 0.453, 0.149, 0.109 for fetuin-B. However, this solution showed severe clashes between CY1 domains. Thus, a final search was performed with astacin plus the CY2 domain and LNK of mFB (S146-E267) of the latter solution and the mFB CY1 domain (R29-V145) separately. These calculations gave two solutions at 5.0, 0.3, 355.0, 0.000, 0.000, 0.001 and 270.1, 16.4, 270.5, 0.883, 0.985, −0.002 for the first searching model and two more for the second searching model at 291.6, 1.0, 128.0, −0.001, −0.008, 0.166 and 270.5, 7.3, 328.3, 0.837, 0.893, 0.162. The crystal packing was acceptable, and the log-likelihood gain was 2,508. Thereafter, automated model building was used to input the sequence of hFB and density modification was carried out with twofold averaging with the AUTOBUILD routine of program suite PHENIX59, which resulted in a Fourier map that was used for manual model building with the COOT program60. The latter alternated with crystallographic refinement with PHENIX61 and BUSTER/TNT62 under inclusion of non-crystallographic symmetry restraints and translation/libration/screw-rotation refinement until completion of the model. The final refined model comprised residues A1-S199 and A1-L200 of astacin molecules A and C, respectively, plus a zinc cation each; segments A25-P215 + S229-E258 + R364-S376 and N27-P215 + S229-S259 + T365-A375 of hFB molecules B and D, respectively; and 31 solvent molecules (see Table 1 for the final model statistics). Residues N37 of molecules B and D had the glycan structure NAG(FUC)-NAG-BMA attached (for sugar moiety nomenclature, see the legend of Table 1). Residues N136 of molecules B and D showed only two and one NAG moieties attached, respectively. The quality of the final model was assessed with the wwPDB X-ray Structure Validation Service (https://validate.wwpdb.org 63).
Proteolytic susceptibility of human and mouse fetuin-B
Both inhibitors were incubated with astacin in 150 mM sodium chloride, 50 mM Tris·HCl, pH 7.4. N-terminal sequencing by Edman degradation was performed by Proteome Factory AG (Berlin, Germany). Immunoblot analysis was performed as reported35 using a polyclonal hFB antibody as reported50 and a penta-His-antibody (Qiagen, Hilden, Germany). Cleaved hFB was isolated from the complex by astacin affinity chromatography. To this aim, astacin was covalently coupled to a HiTrap NHS-Activated HP affinity column (GE Life Sciences, Freiburg, Germany) according to the manufacturer’s specifications. Protein hFB was applied to the column and cleaved by addition of further astacin. The cleaved hFB was eluted by addition of 6 M urea, desalted and concentrated. The non-cleaved hFB control was treated similarly except for cleavage by astacin. The residual inhibitory capacities of both samples against astacin and human meprins α and β were determined.
Inhibition assays in vitro
Inhibition of meprin α, meprin β and astacin by intact and astacin-cleaved hFB was determined in vitro in a Varioskan Flash 3001 spectral plate reader with SKANIT 2.4.3.RE software (ThermoFisher Scientific, Dreieich, Germany) with a fluorescence-based activity assay as previously described for mFB36. Enzyme concentrations were determined by absorbance at 280 nm (εastacin = 42,800 M−1 cm−1; εmeprin α = 106,520 M−1 cm−1; εmeprin β = 113,385 M−1 cm−1) and assays were performed in triplicate at 37 °C in 100 µl final volume, with 150 mM sodium chloride, 50 mM Tris·HCl, pH 7.4 and 0.01% Brij-35 as buffer. Cleavage reactions were initiated by the addition of 20–30 µM Ac-R-E(Edans)-D-R-Nle-V-G-D-D-P-Y-K(Dabcyl)-NH2 (Biosyntan, Berlin, Germany) for meprins α and β, and 170–180 µM Dansyl-P-K-R-P-W-V-OH (PANATecs, Heilbronn, Germany) for astacin, both predissolved in dimethyl sulfoxide (final concentration 0.4%). Initial velocities were recorded for at least 600 s (100 times for 100 ms at intervals of 15 s). Thereafter, 1.5 µl of proteinase K (Sigma-Aldrich) at 20 mg/ml or 1 µl astacin at 100 µM were added for complete substrate turnover. The latter was monitored and subsequently calculated using the formula v = [S] × m/ΔF, where [S] is the substrate concentration, m the [F/t] slope of initial linear substrate turnover, and ∆F the maximal fluorescence intensity corresponding to complete turnover. Kinetic parameters of inhibition (Ki) were determined using Morrison’s equation64.
Bioinformatics
Structure figures were prepared with the CHIMERA program65. Structure superimpositions were performed with SSM66 within COOT. Protein interfaces were analyzed with PISA46 (www.ebi.ac.uk/pdbe/pisa). The area of the interface of a complex was taken as half the sum of the buried surface areas of either molecule. Sequence identities were calculated with SIM with default parameters (https://web.expasy.org/cgi-bin/sim/sim.pl?prot). Signal peptides were predicted with SIGNALP v. 5.0 at http://www.cbs.dtu.dk/services/SignalP-5.067. The final coordinates of the crayfish astacin·human fetuin-B complex are available from the PDB at www.rcsb.org (code 6SAZ).
References
Blobel, C. P. Remarkable roles of proteolysis on and beyond the cell surface. Curr. Opin. Cell Biol. 12, 606–612, https://doi.org/10.1016/S0955-0674(00)00139-3 (2000).
Klein, T., Eckhard, U., Dufour, A., Solis, N. & Overall, C. M. Proteolytic cleavage-mechanisms, function, and “omic” approaches for a near-ubiquitous posttranslational modification. Chem. Rev. 118, 1137–1168, https://doi.org/10.1021/acs.chemrev.7b00120 (2018).
Arolas, J. L., Goulas, T., Cuppari, A. & Gomis-Rüth, F. X. Multiple architectures and mechanisms of latency in metallopeptidase zymogens. Chem. Rev. 118, 5581–5597, https://doi.org/10.1021/acs.chemrev.8b00030 (2018).
Yan, C. & Boyd, D. D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 211, 19–26, https://doi.org/10.1002/jcp.20948 (2007).
Hadler-Olsen, E., Fadnes, B., Sylte, I., Uhlin-Hansen, L. & Winberg, J.-O. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 278, 28–45, https://doi.org/10.1111/j.1742-4658.2010.07920.x (2011).
Khan, A. R. & James, M. N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Prot. Sci. 7, 815–836, https://doi.org/10.1002/pro.5560070401 (1998).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632, https://doi.org/10.1093/nar/gkx1134 (2018).
Cerdà-Costa, N. & Gomis-Rüth, F. X. Architecture and function of metallopeptidase catalytic domains. Prot. Sci. 23, 123–144, https://doi.org/10.1002/pro.2400 (2014).
Bode, W., Gomis-Rüth, F. X., Huber, R., Zwilling, R. & Stöcker, W. Structure of astacin and implications for activation of astacins and zinc-ligation of collagenases. Nature 358, 164–167, https://doi.org/10.1038/358164a0 (1992).
Gomis-Rüth, F. X., Stöcker, W., Huber, R., Zwilling, R. & Bode, W. Refined 1.8 Å X-ray crystal structure of astacin, a zinc-endopeptidase from the crayfish Astacus astacus L. Structure determination, refinement, molecular structure and comparison with thermolysin. J. Mol. Biol. 229, 945–968, https://doi.org/10.1006/jmbi.1993.1098 (1993).
Gomis-Rüth, F. X., Trillo-Muyo, S. & Stöcker, W. Functional and structural insights into astacin metallopeptidases. Biol. Chem. 393, 1027–1041, https://doi.org/10.1515/hsz-2012-0149 (2012).
Stöcker, W. & Gomis-Rüth, F. X. In Proteases: structure and function. (eds Brix, K. & Stöcker, W.) 235–263 (Springer Verlag, 2013), https://doi.org/10.1007/978-3-7091-0885-7.
Bond, J. S. Proteases: History, discovery, and roles in health and disease. J. Biol. Chem. 294, 1643–1651, https://doi.org/10.1074/jbc.TM118.004156 (2019).
Bode, W., Gomis-Rüth, F. X. & Stöcker, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ´metzincins´. FEBS Lett. 331, 134–140, https://doi.org/10.1016/0014-5793(93)80312-i (1993).
Stöcker, W. et al. The metzincins - Topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Prot. Sci. 4, 823–840, https://doi.org/10.1002/pro.5560040502 (1995).
Gomis-Rüth, F. X. Structural aspects of the metzincin clan of metalloendopeptidases. Mol. Biotech. 24, 157–202, https://doi.org/10.1385/MB:24:2:157 (2003).
Gomis-Rüth, F. X. Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem. 284, 15353–15357, https://doi.org/10.1074/jbc.R800069200 (2009).
Pérez-Silva, J. G., Español, Y., Velasco, G. & Quesada, V. The Degradome database: expanding roles of mammalian proteases in life and disease. Nucleic Acids Res. 44, D351–D355, https://doi.org/10.1093/nar/gkv1201 (2016).
Kessler, E., Takahara, K., Biniaminov, L., Brusel, M. & Greenspan, D. S. Bone morphogenetic protein-1: the type I procollagen C-proteinase. Science 271, 360–362, https://doi.org/10.1126/science.271.5247.360 (1996).
Shimell, M. J., Ferguson, E. L., Childs, S. R. & O’Connor, M. B. The Drosophila dorsal-ventral patterning gene tolloid is related to human bone morphogenetic protein 1. Cell 67, 469–481, https://doi.org/10.1016/0092-8674(91)90522-Z (1991).
Sterchi, E. E., Stöcker, W. & Bond, J. S. Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Aspects Med. 29, 309–328, https://doi.org/10.1016/j.mam.2008.08.002 (2008).
Becker-Pauly, C. et al. Proteomic analyses reveal an acidic prime side specificity for the astacin metalloprotease family reflected by physiological substrates. Mol. Cell. Proteomics 10, M111.009233, https://doi.org/10.1074/mcp.M111.009233 (2011).
Ambort, D. et al. Specific processing of tenascin-C by the metalloprotease meprin b neutralizes its inhibition of cell spreading. Matrix Biol. 29, 31–42, https://doi.org/10.1016/j.matbio.2009.08.007 (2010).
Vazeille, E. et al. Role of meprins to protect ileal mucosa of Crohn’s disease patients from colonization by adherent-invasive E. coli. PloS one 6, e21199, https://doi.org/10.1371/journal.pone.0021199 (2011).
Burkart, A. D., Xiong, B., Baibakov, B., Jiménez-Movilla, M. & Dean, J. Ovastacin, a cortical granule protease, cleaves ZP2 in the zona pellucida to prevent polyspermy. J. Cell Biol. 197, 37–44, https://doi.org/10.1083/jcb.201112094 (2012).
Körschgen, H. et al. Intracellular activation of ovastacin mediates pre-fertilization hardening of the zona pellucida. Mol. Hum. Reprod. 23, 607–616, https://doi.org/10.1093/molehr/gax040 (2017).
Floehr, J., Dietzel, E., Schmitz, C., Chappell, A. & Jahnen-Dechent, W. Down-regulation of the liver-derived plasma protein fetuin-B mediates reversible female infertility. Mol. Hum. Reprod. 23, 34–44, https://doi.org/10.1093/molehr/gaw068 (2017).
Stöcker, W., Breit, S., Sottrup-Jensen, L. & Zwilling, R. α2-Macroglobulin from hemolymph of the freshwater crayfish Astacus astacus. Comp. Biochem. Physiol. B 98, 501–509, https://doi.org/10.1016/0305-0491(91)90244-8 (1991).
Kruse, M. N. et al. Human meprin α and β homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem. J. 378, 383–389, https://doi.org/10.1042/BJ20031163 (2004).
Zhang, Y., Ge, G. & Greenspan, D. S. Inhibition of bone morphogenetic protein 1 by native and altered forms of α2-macroglobulin. J. Biol. Chem. 281, 39096–39104, https://doi.org/10.1074/jbc.M601362200 (2006).
Lee, H. X., Ambrosio, A. L., Reversade, B. & de Robertis, E. M. Embryonic dorsal-ventral signaling: secreted frizzled-related proteins as inhibitors of tolloid proteinases. Cell 124, 147–159, https://doi.org/10.1016/j.cell.2005.12.018 (2006).
Bijakowski, C. et al. Sizzled is unique among secreted frizzled-related proteins for its ability to specifically inhibit bone morphogenetic protein-1 (BMP-1)/tolloid-like proteinases. J. Biol. Chem. 287, 33581–33593, https://doi.org/10.1074/jbc.M112.380816 (2012).
Dietzel, E. et al. Fetuin-B, a liver-derived plasma protein is essential for fertilization. Dev. Cell 25, 106–112, https://doi.org/10.1016/j.devcel.2013.03.001 (2013).
Stöcker, W. et al. Mammalian gamete fusion depends on the inhibition of ovastacin by fetuin-B. Biol. Chem. 395, 1195–1199, https://doi.org/10.1515/hsz-2014-0189 (2014).
Karmilin, K. et al. Mammalian plasma fetuin-B is a selective inhibitor of ovastacin and meprin metalloproteinases. Sci. Rep. 9, 546, https://doi.org/10.1038/s41598-018-37024-5 (2019).
Cuppari, A. et al. Structure of mammalian plasma fetuin-B and its mechanism of selective metallopeptidase inhibition. IUCrJ 6, 317–330, https://doi.org/10.1107/S2052252519001568 (2019).
Tsai, P. L., Chen, C. H., Huang, C. J., Chou, C. M. & Chang, G. D. Purification and cloning of an endogenous protein inhibitor of carp nephrosin, an astacin metalloproteinase. J. Biol. Chem. 279, 11146–11155, https://doi.org/10.1074/jbc.M310423200 (2004).
Bode, W. et al. The 2.0 Å X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 7, 2593–2599 (1988).
Stubbs, M. T. et al. The refined 2.4 Å X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. EMBO J. 9, 1939–1947 (1990).
Rawlings, N. D. & Barrett, A. J. Evolution of proteins of the cystatin superfamily. J. Mol. Evol. 30, 60–71 (1990).
Lee, C., Bongcam-Rudloff, E., Sollner, C., Jahnen-Dechent, W. & Claesson-Welsh, L. Type 3 cystatins; fetuins, kininogen and histidine-rich glycoprotein. Front. Biosci. 14, 2911–2922 (2009).
Floehr, J. et al. Association of high fetuin-B concentrations in serum with fertilization rate in IVF: a cross-sectional pilot study. Hum. Reprod. 31, 630–637, https://doi.org/10.1093/humrep/dev340 (2016).
Dall, E., Fegg, J. C., Briza, P. & Brandstetter, H. Structure and mechanism of an aspartimide-dependent peptide ligase in human legumain. Angew. Chem. Int. Ed. Engl. 54, 2917–2921, https://doi.org/10.1002/anie.201409135 (2015).
Guevara, T. et al. Proenzyme structure and activation of astacin metallopeptidase. J. Biol. Chem. 285, 13958–13965, https://doi.org/10.1074/jbc.M109.097436 (2010).
Arolas, J. L., Botelho, T. O., Vilcinskas, A. & Gomis-Rüth, F. X. Structural evidence for standard-mechanism inhibition in metallopeptidases from a complex poised to resynthesize a peptide bond. Angew. Chem. Int. Ed. Engl. 50, 10357–10360, https://doi.org/10.1002/anie.201103262 (2011).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797, https://doi.org/10.1016/j.jmb.2007.05.022 (2007).
Laskowski, M. Jr. & Kato, I. Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626, https://doi.org/10.1146/annurev.bi.49.070180.003113 (1980).
Schechter, I. & Berger, A. On the size of active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162, https://doi.org/10.1016/j.bbrc.2012.08.015 (1967).
Gomis-Rüth, F. X., Botelho, T. O. & Bode, W. A standard orientation for metallopeptidases. Biochim. Biophys. Acta 1824, 157–163, https://doi.org/10.1016/j.bbapap.2011.04.014 (2012).
Denecke, B. et al. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem. J. 376, 135–145, https://doi.org/10.1042/BJ20030676 (2003).
Becker, C. et al. Differences in the activation mechanism between the alpha and beta subunits of human meprin. Biol. Chem. 384, 825–831, https://doi.org/10.1515/BC.2003.092 (2003).
Fridrich, S., Karmilin, K. & Stöcker, W. Handling metalloproteinases. Curr. Protoc. Protein Sci. 83, 21.16.21–21.16.20, https://doi.org/10.1002/0471140864.ps2116s83 (2016).
Becker-Pauly, C. et al. The α and β subunits of the metalloprotease meprin are expressed in separate layers of human epidermis, revealing different functions in keratinocyte proliferation and differentiation. J. Invest. Dermatol. 127, 1115–1125, https://doi.org/10.1038/sj.jid.5700675 (2007).
Juanhuix, J. et al. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 21, 679–689, https://doi.org/10.1107/S160057751400825X (2014).
Kabsch, W. XDS. Acta Crystallogr. sect. D 66, 125–132, https://doi.org/10.1107/S0907444909047337 (2010).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. sect. D 66, 133–144, https://doi.org/10.1107/S0907444909047374 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. sect. D 67, 235–242, https://doi.org/10.1107/S0907444910045749 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674, https://doi.org/10.1107/S0021889807021206 (2007).
Terwilliger, T. C. et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. sect. D 64, 61–69, https://doi.org/10.1107/S090744490705024X (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. sect. D 66, 486–501, https://doi.org/10.1107/S0907444910007493 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. sect. D 68, 352–367, https://doi.org/10.1107/S0907444912001308 (2012).
Smart, O. S. et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. sect. D 68, 368–380, https://doi.org/10.1107/S0907444911056058 (2012).
Berman, H., Henrick, K. & Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980–980, https://doi.org/10.1038/nsb1203-980 (2003).
Morrison, J. F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286, https://doi.org/10.1016/0005-2744(69)90420-3 (1969).
Pettersen, E. F. et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612, https://doi.org/10.1002/jcc.20084 (2004).
Krissinel, E. & Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. sect. D 60, 2256–2268, https://doi.org/10.1107/S0907444904026460 (2004).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423, https://doi.org/10.1038/s41587-41019-40036-z (2019).
Acknowledgements
We are grateful to Joan Pous and Xandra Kreplin from the joint IBMB/IRB Automated Crystallography Platform and Roman Bonet from the Protein Purification service for assistance during crystallization and purification experiments, respectively. We also thank Benedikt King for additional kinetics measurements We further acknowledge the help by local contacts from the ALBA synchrotron. Funding for data collection was provided in part by ALBA. This study was supported in part by grants from German, Spanish and Catalan public agencies (DFG FA1518/1-1; DFG JA562/16 and FL1033/1; IZKF Aachen Medical Faculty Research Fund; JGU Research Fund; BFU2015-64487R; MDM-2014-0435; Fundació “La Marató de TV3” 201815 and 2017SGR3). The Structural Biology Unit of IBMB is a “María de Maeztu” Unit of Excellence of the Spanish Ministry of Science, Innovation and Universities. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
F.X.G.-R., W.S. and W.J.-D. designed and/or coordinated the research. T.G., H.K., A.C., C.S., M.K., I.Y., J.F. and F.X.G.-R. performed experiments and/or data analysis. F.X.G.-R. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guevara, T., Körschgen, H., Cuppari, A. et al. The C-terminal region of human plasma fetuin-B is dispensable for the raised-elephant-trunk mechanism of inhibition of astacin metallopeptidases. Sci Rep 9, 14683 (2019). https://doi.org/10.1038/s41598-019-51095-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51095-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
