
Abstract
Beryllium-doped boron clusters display essential similarities to borophene (boron sheet) with a molecular structure characterized by remarkable properties, such as anisotropy, metallicity and high conductivity. Here we have determined low-energy structures of BeBn0/− (n = 10–20) clusters by utilizing CALYPSO searching program and DFT optimization. The results indicated that most ground states of clusters prefer plane or quasi-plane structures by doped Be atom. A novel unexpected fascinating planar BeB16− cluster with C2v symmetry is uncovered which possesses robust relative stability. Furthermore, planar BeB16− offers a possibility to construct metallo-borophene nano-materials. Molecular orbital and chemical bonding analysis reveal the peculiarities of BeB16− cluster brings forth the aromaticity and the strong interaction of B-B σ-bonds in boron network.
Similar content being viewed by others
Introduction
Molecular geometric configuration and attributes of pure1,2,3 and doped boron clusters4,5,6,7,8,9 have drawn much attention in recent years. The use of boron clusters as subunits in novel bioactive architectures with potential use as drugs is of primary importance10. From a Materials Science perspective the emergence of graphene11 and synthetic two-dimensional structures as silicene12,13, germanene14, stanene15, antimonene16, bismuthene17,18 and tellurene19 have opened new pathways for modern research20,21,22. Relying on experimental and theoretical work, Hersam’s group23 have confirmed and established the synthesis of 2D boron polymorphs (borophene) characterized by anisotropy and metallicity, and paved the way to investigations leading to the discovery of novel materials. Recently, it was reported that magnesium diboride (MgB2), which consists of graphene-like honeycomb networks of sandwiched boron, shows superconductivity24. It should be noted that beryllium has the same valence electrons number with magnesium. Be-doped boron clusters appear to have significant potential candidate as layered 2D materials25,26,27,28. This certainly gives reason for more systematic investigations.
Boron is the lightest metalloid chemical element, the lowest-Z element23 with a trivalent outer shell29,30. Consequently, boron does not form closed-shell electronic structures via conventional covalent bonds31,32,33, but favors delocalized chemical bonds with electron pairs shared among three (or more) atoms instead. Recently, systematic investigations of pure boron clusters in term of the anisotropy and polymorphism have brought forth new significant findings leading to the design of new borides. A selection of characteristic architectures of pure boron clusters includes: tank tread34, wankel motor35,36,37,38,39, wheel-like40, boron nanotubes41, B12 icosahedra42, buckyballs43, fullerene44, B36 with hexagonal holes (HHs)45, naphthalene46, borospherene47 and more. Co and Rh doped B12− clusters featured half-sandwich structure has been reported by Wang and co-workers48. There followed the Co-centered boron molecular drums structure for the CoB16− cluster49. Additional work by the same group includes the Mn-centered tubular boron cluster for MnB16−, a drum and quasi-planar structure for RhB18− and the planar CoB18−50,51,52. Very recently, Cui and co-workers reported tubular structures for LiB20 and LiB20−53. These impressive findings reveal that single metal atom doping leads to new opportunities for the use of boron clusters as geometrical ligands.
Several theoretical investigations of boron clusters with doping transition-element serve as the object of discovering new materials recently54,55. The alkaline-earth metal-doped boron clusters and Be-doped ones in particular have been systematically studied56,57,58. Nevertheless, more systematic work is needed to systematize and deepen our understanding of Be-doped boron clusters. To fill the existing lacunae and bring forth new insights on medium-sized Be-doped boron clusters, we have thoroughly investigated BeBn0/− clusters.
Results and Discussion
Geometric configurations and photoelectron spectra
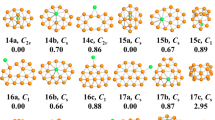
The determined low-energy BeBn0/− (n = 10–20) are showed in Figs 1 and 2. We labeled each isomer using nt/t− (t = a, b, c), therein nt stands for the neutral clusters and nt− stands for the anionic clusters. The lowest-energy structures BeBn0/− (n = 10, 12, 13, 14, 15, 16) and BeB11− are quasi-planar structures. The lowest-energy structure BeB11 shows a half-sandwich structure consisting of one half-sandwich structure composed by eleven boron atoms and one Be atom in the center. The lowest-energy structures BeB170/− like a trapezoid and its center portion appear on the convex. The lowest-energy structure of BeB18 and BeB20− are 3D cage-like structure. The lowest-energy structure BeB18− with a parallelogram located in the center displays a planar structure. The ground-state structures BeB190/− and BeB20 can be viewed as plate-like structures (in Figs S4 and S5 of Supplementary Information). The lowest-energy structures of BeBn0/− clusters are generally evolutional from the quasi-planar to 3D cage-like or plate-like structures. For plane and quasi-planar structures, the coordinate number of Be atom is interesting. The BeBn0/− (n = 10, 12, 14, 16) and BeB18− feature heptacoordinate and the BeB11− and BeBn0/− (n = 13, 15) possess octacoordinate, while the BeB170/− are quasi-planar hexacoordinate structures due to the attribute of Be atom59,60. This evident structures evolution pattern contributes to form plane clusters of BeBn0/−, which are potential two-dimensional material. The metastable of nb/b− (n = 10–13) clusters display half-sandwich architectural feature, while when the cluster sizes increase n ≥ 14, the clusters are varies cage-like, quasi-planar and plate-like structures. The nc/c− (n = 10–18) clusters display half-sandwich, plane, cage-like structures, different from the larger size isomers (n ≥ 19) are double-ring and plate-like structures.

Low-lying geometrical structures of BeBn (n = 10–20) clusters. “a” stands for the lowest-energy structures. “b” and “c” stand for the metastable state structures.

Low- lying geometrical structures of BeBn− (n = 10–20) clusters. “a” stands for the lowest-energy structures. “b” and “c” stand for the metastable state structures.
To get a deep understand to differences between different metal-doped clusters, we provide a comparison for doped boron clusters. The transition-metal doped boron clusters, NbB10− and TaB10−, are wheels structures with high coordination number4, while BeB100/− clusters are quasi-planar structures with one B-Be unit inside. For doped B12 clusters, the prior works report that half-sandwich structures VB10−, CoB12− and RhB12− clusters4,48 are different with BeB120/− clusters, which are standard quasi-planar structures featuring a triangle in the center. Compare with drum-like CoB16− cluster49 and tubular-like MnB16− cluster50, the ground state BeB160/− display quasi-plane structures. It is worth noting that adjacent alkali element Lithium doped into B20 display highly symmetrical tubular LiB200/− clusters53. We report BeB20 and BeB20− are plate-like and 3D cage-like structures, respectively. The reason for the structural differences of same-sized clusters may be doped-metals have different valence electron and atomic radius61.
Photoelectron spectra (PES) analysis, obtained via a TD–DFT approach, is of absolute importance for the assessment of the nature of the determined lowest-energy structures. We simulated the PES of BeBn− clusters and the results are displayed in Fig. 3. Our group also simulated the PES of some other cluster system using the method62,63. The PES pattern of the BeB10− possesses five peaks located at 3.26, 3.75, 4.18, 4.75 and 5.77 eV. The PES of BeB11− possesses four clear peaks at 3.45, 4.21, 4.59, and 5.01 eV, with B and C peaks forming a broad bond. For BeB12−, we observe three major peaks at 2.90, 4.21 and 4.50 eV, wherein the double-peak feature (A and B) is prominent and broad. The BeB13− PES contains five major peaks at 3.16, 3.49, 4.32, 4.75 and 5.22 eV. The relevant broad bond is found at triple-peak feature consisted of peaks B, C and D. Five peaks are observed for BeB14− at 3.33, 3.86, 4.16, 4.63 and 5.45 eV. The peaks A, B and C constitute a relatively wide bond. For BeB15− there are five major peaks at 3.46, 4.28, 4.64, 5.06 and 5.82 eV, whereas the BeB16− spectrum has only two sparse peaks at 4.08 and 5.25 eV. The well-structure spectrum of BeB17− shows five peaks at 3.90, 4.32, 4.79, 5.13 and 5.49 eV, suggesting a greater span triple-peak feature (B, C and D). A crowded spectrum pattern BeB18− has five peaks observed at 3.59, 3.98, 4.21, 5.13 and 5.57 eV, with two broad bonds. There are five peaks in the spectrum of BeB19− at 3.63, 4.73, 5.13, 5.51 and 5.82 eV, therein an unfitted bond is located at the range between 4.5 to 6.0 eV. The spectrum of BeB20− possesses five peaks at 2.59, 3.36, 4.43, 4.85 and 5.79 eV.

The simulated PES of BeBn− (n = 10–20) clusters.
Relative stabilities
We characterize the inherent stability of the BeBn0/− (n = 10–20) clusters by computing the Eb (eV), according to the following formula:
The average binding energy (Eb) of a cluster is clearly a measure of its thermodynamic stability. An increase in Eb means a higher stability. The value of neutral BeBn clusters less than the value of their anionic counterparts in Fig. S1(a), indicating that the anionic clusters feature higher thermodynamically. The trend of the curves for both neutral and anionic are gradually upward indicated that the high thermodynamic stability with the cluster size increases. The second vital physical quantity we take into account here is the Δ2E. The relevant formulae are
As inferred from Fig. S1(b), both of the neutral and anionic curves show odd-even alteration. The evident peak values generated at n = even number, suggest that clusters with the even boron atoms feature higher stability than which with odd boron atoms. Finally, we discuss the HOMO-LUMO energy gap (Egap) which provides a valuable index of the stability of clusters. Large values indicate strong chemical stability. We summarize the Egap values of the lowest-energy BeBn0/− clusters in Table 1, and the line chart is displayed in Fig. S1(c). From the latter we can clearly see some apparent local maxima: BeB11 and BeB16−, which means that they feature higher stability than the others. Consequently, based on the above analyses, we can reach a definitive conclusion that the BeB16− can seen as a “magic” cluster.
Chemical banding
To deeply perceive the bonding nature of BeB16− (C2v symmetry), we display eleven MO figures for BeB16−, including one LUMO, one HOMO and nine HOMO-n (n = 1–9) in Fig. 4 by analyzing the chemical bonding. The LUMO, HOMO, HOMO-2, HOMO-5 and HOMO-9 dominated primarily by πp and πp∗ orbitals are a direct interaction 2p orbitals of B atoms. The HOMO-n (n = 1, 4, 8) feature σp and σp∗ orbitals. The HOMO-n (n = 3, 6, 7) features σp, σp∗, σsp and σsp∗ orbitals. AdNDP analysis distributes 51 valence electrons into different regions as reflected by the occupation numbers (ONs) in Fig. 5. We divide it into three sets. The first set consists of twelve 2c-2e (1.79–1.93 |e|) localized σ-bonds. The second set consists of nine delocalized σ-bonds, which are five 3c-2e (1.79–1.86 |e|), two 4c-2e (1.72 |e|), and two 4c-2e (1.79 |e|). The five delocalized π-bonds in last set involving two 4c-2e (1.81 |e|), two 4c-2e (1.83 |e|) and one 17c-2e (2.00 |e|). It is worth nothing that the ON of the 17c-2e π-bonds maintain ON of 2.00 |e|. All values of the ONs listed above ranging from 1.72–2.00 |e| are approaching the ideal value 2.00 |e|, which means that the results we calculate is fairly credible. Furthermore, the ten π electrons conform to the 4n + 2 rule (n = 2), indicating the BeB16− cluster possesses π-aromaticity, which result to the robust relative stability for BeB16− cluster.

Molecular orbitals for BeB16− cluster corresponding to different energy level.

AdNDP analysis of BeB16− cluster.
The Wiberg bond index of BeB16−, showed in Fig. S2(a), indicate that the bond orders values of B-B (0.13–0.35) greater than the Be-B (0.06–0.11). For Fig. S2(b), the B-B bond lengths (1.54–1.80 Å) are shorter than Be-B bond lengths (1.85–2.03 Å). The results of bond orders and bond lengths show that the peripheral B-B bonds are stronger than the inner Be-B bonds. We have also performed the NPA (natural charge of atom) calculations of BeBn0/− in Fig. S3 indicate that electron transfer from Be atom to boron fragment. The NPA data of BeBn0/− (n = 10–20) clusters are summarized in Table S1. From what has been discussed, we come to the conclusion that the B-B σ-bonds and the aromaticity decide the high stability of BeB16− cluster. It is worth noting that due to planar structure and chemical bonding characteristics of BeB16− cluster, also inspired by fascinating prospect of two-dimensional monolayer metallo-borophene4, we successfully build a schematic of possibility of metallo-borophene (not optimized) based on BeB16− unit cluster presented in Fig. S6 of Supplementary Information, which indicated the BeB16− cluster is a potential motif for metallo-borophene.
Conclusions
In summary, the ground-state BeBn0/− (n = 10–20) structure obey the evolution rule: quasi-planar to 3D cage-like or plate-like structures, which the doped Be atom contributed to the plane or quasi-plane structures. We hope that the simulated PES can provide valuable guidance for future research on BeBn clusters and borophene. Based on the relative stability analysis, the BeB16− cluster characterized by enhanced stability is clearly a “magic” cluster. Chemical bonding analysis indicated that BeB16− cluster adapt π-aromaticity and the strong interaction of B-B σ-bonds which is deemed as the dominant reasons for the inherent stability of BeB16− cluster. The planar BeB16− cluster may serve as a motif for the design of a new boron-based functional material to complement the metallo-borophene effort for synthetic 2D materials development. Our present findings on Be-doped boron clusters should provide valuable information for further explorations of novel cluster architectures.
Computational Methods
We used the CALYPSO code to search the BeBn0/− (n = 10–20) clusters. The global explorations of Be-doped boron cluster system was implemented by utilizing particle swarm optimization (PSO) algorithm64,65,66. The effectiveness of this structural prediction method, has been successfully tested on the identification of ground-state structures of various systems67,68,69. To ensure high efficiency in structure predicting, we proceeded to 50 generations for each size, where each generation contains 30 structures. PSO algorithm produces sixty percent of the structures and the rest is generated randomly. The top fifty low-lying isomers were reoptimized with PBE070 functional and 6–311 + G(d)71, as performed via Gaussian 09 package72. The PES of Be-doped boron clusters was simulated utilizing TD-DFT method73. We then analyzed chemical bonding of BeB16− cluster relying on the NBO and AdNDP methods74 at the PBE0/6-311 + G(d) level to display valuable insights into the nature of the bonding by using Multiwfn75. The bond orders, bond lengths and NPA are also computed by using the same basis set and method.
References
Pham, H. T., Muya, J. T., Buendia, F., Ceulemans, A. & Nguyen, M. T. Formation of the quasi-planar B50 boron cluster: topological path from B10 and disk aromaticity. Phys. Chem. Chem. Phys. 21, 7039 (2019).
Chen, Q. et al. Planar B38 − and B37 − clusters with a double-hexagonal vacancy: molecular motifs for borophenes. Nanoscale. 9, 4550–4557 (2017).
Arasaki, Y. & Takatsuka, K. Chemical bonding and nonadiabatic electron wavepacket dynamics in densely quasi-degenerate excited electronic state manifold of boron clusters. J. Chem. Phys. 150, 114101 (2019).
Li, W. L. et al. planar boron clusters to borophenes and metalloborophenes. Nat. Rev. Chem. 1, 0071 (2017).
Li, P. F., Du, X. D., Wang, J. J., Lu, C. & Chen, H. H. Probing the Structural Evolution and Stabilities of Medium-Sized MoBn 0/− Clusters. J. Phys. Chem. C 122, 20000–20005 (2018).
Li, W. L. et al. Recent Progress on the investigations of boron clusters and boron-based materials (I): borophene. Sci. Sin. Chim. 48, 98–107 (2018).
Jian, T. et al. Probing the structures and bonding of size-selected boron and doped-boron clusters. Chem. Soc. Rev. 48, 3550 (2019).
Li, W. L. et al. Observation of highly stable and symmetric lanthanide octa-boron inverse sandwich complexes. Proc. Natl. Acad. Sci. USA 115, 30 (2018).
Chen, T. T., Li, W. L., Chen, W. J., Li, J. & Wang, L. S. La3B14 −: an inverse triple-decker lanthanide boron cluster. Chem. Commun. 55, 7864 (2019).
Hawthorne, M. F. & Maderna, A. Applications of Radiolabeled Boron Clusters to the Diagnosis and Treatment of Cancer. Chem. Rev. 99, 3421–3434 (1999).
Geim, A. K. & Novoselov, K. S. Photoelectron Spectroscopy and Ab Initio Study of B3 − and B4 − Anions and Their Neutrals. Nat. Mater. 6, 183–191 (2007).
Vogt, P. et al. Silicene: Compelling Experimental Evidence for Graphenelike Two-Dimensional Silicon. Phys. Rev. Lett. 108, 155501 (2012).
Cahangirov, S. et al. Electronic Structure of Silicene on Ag(111): Strong Hybridization Effects. Phys. Rev. B 88, 035432 (2013).
Davila, M. E., Xian, L., Cahangirov, S., Rubio, A. & Lay, G. L. Germanene: A Novel Two-Dimensional Germanium Allotrope Akin to Graphene and Silicene. New J. Phys. 16, 095002 (2014).
Zhu, F. F. et al. Epitaxial Growth of Two-Dimensional Stanene. Nat. Mater. 14, 1020–1025 (2015).
Ji, J. P. et al. Two-Dimensional Antimonene Single Crystals Grown by Van Der Waals Epitaxy. Nat. Commun. 7, 13352 (2016).
Nagao, T. et al. Nanofilm Allotrope and Phase Transformation of Ultrathin Bi Film on Si(111)–7 × 7. Phys. Rev. Lett. 93, 105501 (2004).
Reis, F. et al. Bismuthene on a SiC Substrate: A Candidate for a High-Temperature Quantum Spin Hall Material. Science 357, 287–290 (2017).
Zhu, Z. L. et al. Multivalency-Driven Formation of Te-Based Monolayer Materials: A Combined First Principles and Experimental Study. Phys. Rev. Lett. 119, 106101 (2017).
Yu, X. H., Zhang, X. M. & Yan, X. W. Stability of the Fe12O12 cluster. Nano. Res. 11, 3574–3581 (2008).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nat. Nanotechnol. 7, 699–712 (2012).
Ling, X., Wang, H., Huang, S. X., Xia, F. N. & Dresselhaus, M. S. The Renaissance of Black Phosphorus. Proc. Natl. Acad. Sci. USA 112, 4523–4530 (2015).
Mannix, A. J., Zhang, Z. H., Guisinger, N. P., Yakobson, B. I. & Hersam, M. C. Borophene as a Prototype for Synthetic 2D Materials Development. Nat. Nanotechnol. 13, 444–450 (2018).
Nagamatsu, J., Nakagawa, N., Muranaka, T., Zenitani, Y. & Akimitsu, J. Superconductivity at 39 K in Magnesium Diboride. Nature 410, 63–64 (2001).
Li, Q. S. & Jin, Q. Theoretical Study on the Aromaticity of the Pyramidal MB6 (M = Be, Mg, Ca, and Sr) Clusters. J. Phys. Chem. A 107, 7869–7873 (2013).
Adamska, L., Sadasivam, S., Foley, J. J., Darancet, P. & Sharifzadeh, S. First-Principles Investigation of Borophene as a Monolayer Transparent Conductor. J. Phys. Chem. C 122, 4037–4045 (2018).
Er, S., Wijs, G. A. & Brocks, G. DFT Study of Planar Boron Sheets: A New Template for Hydrogen Storage. J. Phys. Chem. C 113, 18962–18967 (2009).
Jiang, H. R., Lu, Z. H., Wu, M. C., Ciucci, F. & Zhao, T. S. Borophene: A Promising Anode Material Offering High Specific Capacity and High Rate Capability for Lithium-Ion Batteries. Nano Energy 23, 97–104 (2016).
Albert, B. & Hillebrecht, H. Boron: Elementary Challenge for Experimenters and Theoreticians. Angew. Chem. Int. Ed. 48, 8640–8668 (2009).
Pelaz, L. et al. B Diffusion and Clustering in Ion Implanted Si: The Role of B Cluster Precursors. Appl. Phys. Lett. 70, 2285–2287 (1997).
Qgitsu, T., Schwegler, E. & Galli, G. β-Rhombohedral Boron: At the Crossroads of the Chemistry of Boron and the Physics of Frustration. Chem. Rev. 113, 3425–3449 (2013).
Sergeeva, A. P. et al. Understanding Boron through Size-Selected Clusters: Structure, Chemical Bonding, and Fluxionality. Acc. Chem. Res. 47, 1349–1358 (2014).
Oganov, A. R. et al. Ionic High Pressure form of Elemental Boron. Phys. Rev. Lett. 457, 863–867 (2009).
Wang, Y. J. et al. Chemical Bonding and Dynamic Fluxionality of a B15 + Cluster: a Nanoscale Double-Axle Tank Tread. Phys. Chem. Chem. Phys. 18, 15774–15782 (2016).
Guajardo, G. M. et al. Unravelling Phenomenon of Internal Rotation in B13 + through Chemical Bonding Analysis. Chem. Commun. 47, 6242–6244 (2011).
Jimѐnez-Halla, J. O. C., Islas, R., Heine, T. & Merino, G. B19 −: An Aromatic Wankel Motor. Angew. Chem. Int. Ed. 49, 5668–5671 (2010).
Morene, D. et al. B18 2−: A Quasi-Planar Bowl Member of the Wankel Motor Family. Chem. Commun. 50, 8140–8143 (2014).
Cervantes-Navarro, F. et al. Stop Rotating! One Substitution Halts the B19 − Motor. Chem. Commun. 50, 10680–10682 (2014).
Merino, G. & Heine, T. And Yet It Rotates: The Starter for a Molecular Wankel Motor. Angew. Chem. Int. Ed. 51, 10226–10227 (2012).
Heina, T. & Merino, G. What Is the Maximum Coordination Number in a Planar Structure? Angew. Chem. Int. Ed. 51, 4275–4276 (2012).
Dong, X. et al. Li2B12 and Li3B12: Pediction of the Smallest Tubular and Cage-like Boron Structures. Angew. Chem. Int. Ed. 57, 4627–4631 (2018).
Lau, K. C. & Pandey, R. Highly Conductive Boron Nanotubes: Transport Properties, Work Functions, and Structural Stabilities. J. Phys. Chem. C 111, 2906–2912 (2007).
Muya, J. T. et al. Jahn-Teller Instability in Cationic Boron and Carbon Buckyballs B80 + and C60 +: A Comparative Study. Phys. Chem. Chem. Phys. 15, 2892–2835 (2013).
Muya, J. T., Gopakumar, G., Nguyen, M. T. & Ceulemans, A. The Leapfrog Principle for Boron Fullerenes: A Theoretical Study of Structure and Stability of B112. Phys. Chem. Chem. Phys. 13, 7524–7533 (2011).
Piazzi, Z. A. et al. Planar Hexagonal B36 as a Potential Basis for Extended Single-Atom Layer Boron Sheets. Nat. Commun. 5, 3113 (2014).
Sergeeva, A. P., Zubarev, D. Y., Zhai, H. J., Boldyrev, A. I. & Wang, L. S. A Photoelectron Spectroscopic and Theoretical Study of B16 − and B16 2−: An All-Boron Naphthalene. J. Am. Chem. Soc. 130, 7244–7246 (2008).
Gerardo, M. G. et al. Dynamical Behavior of Borospherene: A Nanobubble. Sci. Rep. 5, 11287 (2015).
Popov, I. A., Li, W. L., Piazza, Z. A., Boldyrev, A. I. & Wang, L. S. Complexes between Planar Boron Clusters and Transition Metals: A Photoelectron Spectroscopy and Ab Initio Study of CoB12 − and RhB12 −. J. Phys. Chem. A 118, 8098–8105 (2014).
Popov, I. A., Jian, T., Lopez, G. V., Boldyrev, A. I. & Wang, L. S. Cobalt-Centred Boron Molecular Drums with the Highest Coordination Number in the CoB16 − Cluster. Nat. Commun. 6, 8654 (2015).
Jian, T. et al. Manganese-Centered Tubular Boron Cluster-MnB16 −: A New Class of Transition-Metal Molecules. J. Chem. Phys. 114, 154310 (2016).
Jian, T. et al. Competition between Drum and Quasi-Planar Structures in RhB18 −: Motifs for Metallo-Boronanotubes and Metallo-Borophenes. Chem. Sci. 7, 7020–7027 (2016).
Li, W. L. et al. The Planar CoB18 − Cluster as a Motif for Metallo-Borophenes. Angew. Chem. Int. Ed. 55, 7358–7363 (2016).
Liang, W. Y., Das, A., Dong, X. & Cui, Z. H. Lithium Doped Tubular Structure in LiB20 and LiB20 −: A Viable Global Minimum. Phys. Chem. Chem. Phys. 20, 16202–16208 (2018).
Li, W. L. et al. Observation of a Metal-Centered B2-Ta@B18 − Tubular Molecular Rotor and a Perfect Ta@B20 − Boron Drum with the Record Coordination Number of Twenty. Chem. Commun. 53, 1587–1590 (2017).
Li, P. F. et al. A Detailed Investigation into the Geometric and Electronic Structures of CoBQ n (n = 2−10, Q = 0, −1) Clusters. New J. Chem. 41, 11208–11214 (2017).
Bai, H., Chen, Q., Zhai, H. J. & Li, S. D. Endohedral and Exohedral Metalloborospherenes: M@B40 (M = Ca, Sr) and M&B40 (M = Be, Mg). Angew. Chem. Int. Ed. 54, 941–945 (2015).
Liu, C., Si, H., Han, P. & Tang, M. S. Density Functional Theory Study on Structure and Stability of BeBn + Clusters. Rapid Commun. Mass Spectrom. 31, 1437–1444 (2017).
Guo, J. C. et al. Coaxial Triple-Layered versus Helical Be6B11 − Clusters: Dual Structural Fluxionality and Multifold Aromaticity. Angew. Chem. Int. Ed. 56, 10174–10177 (2017).
Pu, Z. F., Ge, M. F. & Li, Q. S. MB2− (M = Be, Mg, Ca, Sr, and Ba): Planar Octacoordinate Alkaline Earth Metal Atoms Enclosed by Boron Rings. Sci. China Chem. 53, 1737–1745 (2010).
Li, S. D., Miao. C. Q., Guo, J. C. & Ren, G. M. Planar Tetra-, Penta-, Hexa-, Hepta-, and Octacoordinate Silicoons: A Universal Structural Pattern. J. Am. Chem. Soc, 126, 16227–16231 (2004).
Sun, W. G., Xia, X. X., Lu, C., Kuang, X. Y. & Hermann, A. Probing the structural and electronic properties of zirconium doped boron clusters: Zr distorted B12 ligand framework. Phys. Chem. Chem. Phys. 20, 23740 (2018).
Sun, W. G. et al. Evolution of the Structural and Electronic Properties of Medium-Sized Sodium Clusters: A Honeycomb-like Na20 Cluster. Inorg. Chem. 56, 1241–1248 (2017).
Chen, B. L. et al. Insights into the effects produced by doping of medium-sized boron clusters with ruthenium. Phys. Chem. Chem. Phys. 20, 30376–30383 (2018).
Wang, H. et al. CALYPSO Structure Prediction Method and Its Wide Application. Comput. Mater. Sci. 112, 406−415 (2016).
Lv, J., Wang, Y. C., Zhu, L. & Ma, Y. M. Particle-Swarm Structure Prediction on Clusters. J. Chem. Phys. 137, 084104 (2012).
Wang, Y. C., Lv, J., Zhu, L. & Ma, Y. M. Crystal Structure Prediction via Particle-Swarm Optimization. Phys. Rev. B 82, 094116 (2010).
Lu, S. H., Wang, Y. C., Liu, H. Y., Miao, M. S. & Ma, Y. M. Self-Assembled Ultrathin Nanotubes on Diamond (100) Surface. Nat. Commun. 5, 3666 (2014).
Wang, H., Tse, J. S., Tanaka, K., Litaka, T. & Ma, Y. M. Superconductive Sodalite-like Clathrate Calcium Hydride at High Pressures. Proc. Natl. Acad. Sci. USA 24, 6463−6466 (2012).
Li, Y. W., Hao, J., Liu, H. Y., Li, Y. L. & Ma, Y. M. The Metallization and Superconductivity of Dense Hydrogen Sulfide. J. Chem. Phys. 140, 174712 (2014).
Adamo, C. & Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 110, 6158−6170 (1999).
Krishnan, R., Binkley, J. S., Seeger, R. & Pople, J. A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 72, 650−654 (1980).
Frisch, M. J. et al. Gaussian 09 (Revision C.0), Gaussian, Inc., Wallingford, CT, (2009).
Casida, M. E., Jamorski, C., Casida, K. C. & Salahub, D. R. Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. J. Chem. Phys. 108, 4439–4449 (1998).
Zubarev, D. Y. & Boldyrev, A. I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 10, 5207–5217 (2008).
Lu, T. & Chen, F. W. Multiwfn: A multifunctional wavefunction analyzer. Comput. Phys. Commun. 33, 580−592 (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 11574220 and 11874043) and the Program for Science & Technology Innovation Talents in Universities of Henan Province (No. 15HASTIT020).
Author information
Authors and Affiliations
Contributions
X.Y.K. and C.L. conceived the idea. D.L.K., W.G.S. and C.L. performed the calculations. D.L.K., W.G.S., H.X.S., B.L.C., X.X.X. and G.M. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, D., Sun, W., Shi, H. et al. Probing the structure and electronic properties of beryllium doped boron clusters: A planar BeB16− cluster motif for metallo-borophene. Sci Rep 9, 14367 (2019). https://doi.org/10.1038/s41598-019-50905-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50905-7
This article is cited by
-
Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host–guest binding: I. Standard procedure
Journal of Computer-Aided Molecular Design (2022)
-
Evaluation of restricted probabilistic cellular automata on the exploration of the potential energy surface of Be6B11−
Theoretical Chemistry Accounts (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
