
Abstract
We studied the effects of intestinal inflammation on pentylenetetrazole (PTZ)-induced seizures in mice and the effects thereon of some antiepileptic and anti-inflammatory treatments to establish if a link may exist. The agents tested were: alpha-lactoalbumin (ALAC), a whey protein rich in tryptophan, effective in some animal models of epilepsy and on colon/intestine inflammation, valproic acid (VPA), an effective antiepileptic drug in this seizure model, mesalazine (MSZ) an effective aminosalicylate anti-inflammatory treatment against ulcerative colitis and sodium butyrate (NaB), a short chain fatty acid (SCFA) normally produced in the intestine by gut microbiota, important in maintaining gut health and reducing gut inflammation and oxidative stress. Intestinal inflammation was induced by dextran sulfate sodium (DSS) administration for 6 days. Drug treatment was started on day 3 and lasted 11 days, when seizure susceptibility to PTZ was measured along with intestinal inflammatory markers (i.e. NF-κB, Iκ-Bα, COX-2, iNOS), histological damage, disease activity index (DAI) and SCFA concentration in stools. DSS-induced colitis increased seizure susceptibility and while all treatments were able to reduce intestinal inflammation, only ALAC and NaB exhibited significant antiepileptic properties in mice with induced colitis, while they were ineffective as antiepileptics at the same doses in control mice without colitis. Interestingly, in DSS-treated mice, VPA lost part of its antiepileptic efficacy in comparison to preventing seizures in non-DSS-treated mice while MSZ remained ineffective in both groups. Our study demonstrates that reducing intestinal inflammation through ALAC or NaB administration has specific anticonvulsant effects in PTZ-treated mice. Furthermore, it appears that intestinal inflammation may reduce the antiepileptic effects of VPA, although we confirm that it decreases seizure threshold in this group. Therefore, we suggest that intestinal inflammation may represent a valid antiepileptic target which should also be considered as a participating factor to seizure incidence in susceptible patients and also could be relevant in reducing standard antiepileptic drug efficacy.
Similar content being viewed by others


Introduction
Neuroinflammation is clearly linked to seizure and epilepsy pathophysiology with a bidirectional interaction, since increased brain inflammation has been associated with the occurrence of seizures and epileptogenesis and, on the other hand, seizures themselves can induce inflammatory responses in the brain1,2,3. Many studies have indicated that inflammatory mediators and pathways can be considered a suitable target for the development of novel drugs effective in the treatment of epilepsy; furthermore, mounting evidence suggests that these mediators can also be considered reliable biomarkers4,5. Finally, a role for neuroinflammation has also been demonstrated for epilepsy comorbidities6.
Less is known about the role of peripheral inflammation and whether it has a role in the pathophysiology of epilepsy7,8. As an example, it is known that administration of the pro-inflammatory bacterial endotoxin lipopolysaccharide (LPS) can lower seizure threshold or increase seizures in some animal epilepsy models9,10,11. However, it is known that seizures are highly associated with some systemic diseases characterized by peripheral inflammation such as systemic lupus erythematosus, Behcet’s disease (blood vessel inflammation) and Chron’s disease12. This latter, along with other inflammatory bowel diseases, have been suggested to be an increased risk factor for neurological complications including seizures and epilepsy13,14. Accordingly, it was previously reported that intestinal inflammation in animal epilepsy models lowers seizure threshold, most likely through increased circulating levels of some cytokines or other inflammatory mediators7,15.
Alpha-lactoalbumin (ALAC) is a whey protein rich in tryptophan; it is the major protein in human colostrum (20–25% of total protein vs 4% of total protein in milk) and has been described to have several physiological functions in the neonatal period16. ALAC has been demonstrated to be effective in some animal models of epilepsy and epileptogenesis17,18 and patients with epilepsy19,20. Furthermore, it is also known to inhibit colon/intestine inflammation and more generally to contribute to the development and maintenance of gastrointestinal physiological functions16,21.
We herein describe the effects of ALAC on intestinal inflammation induced by dextran sulfate sodium (DSS) administration22 and examine its anticonvulsant effects in the pentylenetetrazol (PTZ) mouse model18. Moreover, we compared the effects of ALAC with those of a valproate (VPA), an established anti-seizure drug in this animal model23 and with those of an effective anti-inflammatory treatment against DSS-induced colitis (mesalazine; MSZ)24 as well as with sodium butyrate (NaB), a short chain fatty acid (SCFA) with beneficial effects on the intestinal and other tissues, including the brain25,26,27,28.
Material and Methods
Animals
Experiments were performed in 10 week-old BALB/c AnNHsd male mice (25 ± 2 g), which were purchased from Charles River (Italy). They were housed in cages (at the most, 5 mice per cage) under stable conditions of humidity (60 ± 5%) and temperature (21 ± 2 °C), kept under a reversed light/dark (12/12 h) cycle (light on at 19:00 h). All animals had free access to water and standard chow diet VRF1 (purchased from Special Diet Service-SDS). Procedures involving animals and their care were conducted in conformity with international and national law and policies (EU Directive 2010/63/EU). The procedures reported here were approved by the Institutional Committee on the Ethics of Animal Experiments (CSV) and by the Ministero della Salute. As indicated by the animal welfare protocol, all efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data.
Induction of colitis and treatments
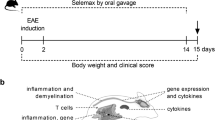
Experimental colitis was induced in mice by administration of 4% wt:vol DSS (36–50 kDa, MP Biomedical, Italy) in drinking water ad libitum from day 1 until day 6 followed by DSS-free water from day 7 until day 13 (end of experimental protocol; Fig. 1). Mice were randomly divided into the ten following groups (n = 40 per group) in which, when drug treatment (in drinking water18) was required, the drug was always given from day 3 until day 13: (1) control mice (CTR group) receiving tap water for all the experimental time; (2) mice receiving DSS from day 1 until day 6 (DSS group); (3) DSS-fed mice receiving ALAC (Kolfarma srl, Italy) at a dose of 375 mg/kg/os (DSS-ALAC); (4) DSS-fed mice receiving valproic acid (Sigma-Aldrich, Italy) at a dose of 600 mg/kg/os (DSS-VPA) (5) DSS-fed mice receiving sodium butyrate (Sigma-Aldrich, Italy) at a dose of 100 mg/kg/os (DSS-NaB) and (6) DSS-fed mice receiving mesalazine (MSZ; Sigma-Aldrich, Italy) at a dose of 15 mg/kg/os (DSS-MSZ group). The last four groups (7) ALAC (8) VPA (9) NaB and (10) MSZ received from day 3 until day 13 their respective treatments as in the DSS-fed groups. At day 13, animals were sacrificed by an i.p. injection of a mixture of ketamine/xylazine followed by cervical dislocation. For histological and western blotting analysis, only mice (at least 5 mice in each group) not receiving PTZ were used.

Schematic representation of experimental protocol. Drugs were given in both control (CTRL) and dextran sulfate sodium (DSS)-treated groups starting on day 3 for 10 consecutive days in the drinking water.
Evaluation of pentylentetrazol-induced seizure susceptibility
All animals were treated with various doses of pentylentetrazol (PTZ; 30–100 mg/kg i.p.) at 15 mg intervals with at least 4 doses in each group (n = 7) to calculate the CD50 (the dose calculated inducing convulsions in 50% of animals)29. The doses of these drugs were injected in a volume of 0.1 ml/10 g of body weight of the mouse. The animals were then placed in isolated cages and observed 30 min after the administration of all drugs. A threshold convulsion was considered as an episode of clonic spasms lasting for at least 5 s. The absence of this threshold convulsion over 30 min indicated that the animal was protected from the convulsant-induced seizures30.
Evaluation of experimental colitis and gut permeability
In all mice, their weight, presence of blood and gross stool consistency were determined daily, as previously described22. Each score was determined as follows:1) change in weight (0: weight loss <1% compared with the starting weight; 1: weight loss between 1 and 5%; 2: weight loss between 6 and 15%; 4: weight loss >15%); (2) stool blood (0: negative; 2: positive; 4: gross bleeding); and (3) stool consistency (0: normal; 2: loose stools; 4: diarrhea). Briefly, the disease activity index (DAI) was determined by combining the scores from these three categories and dividing that number by 322.
An in vivo intestinal permeability assay was performed on mice using the fluorescein isothiocyanate-labeled dextran (FITC-dextran) method, as previously described31. Briefly, food and water were withdrawn for 6 h and mice (n = 5, each group) were administered by gavage with FITC-labeled dextran 4000 (60 mg/100 g body weight; Sigma-Aldrich, Milan, Italy), as a permeability tracer. After 24 hours, blood of all animals was collected by intracardiac puncture and centrifuged (3000 rpm for 15 min at RT). Then, plasma FITC-dextran concentration was determined (excitation, 485 nm; emission, 535 nm; HTS-7000 Plus-plate-reader; Perkin Elmer, Wellesley, MA, USA), using a standard curve.
Histological analysis and scoring of colon sections
At day 13, mice were sacrificed, tissues were collected, and colon length was measured. Distal sections were stored in 10% formalin for histological analysis. Following hematoxylin and eosin (H&E) staining, colon sections were analyzed in a blinded manner by two independent observers for the evaluation of the histopathological score. The samples were given a score of 0–4 (where 0 = none; 1 = mild; 2 = moderate; 3 = severe; 4 = very severe) for epithelial injury, mononuclear infiltrate, and polymorphonuclear infiltrate22.
Western blot analysis
Colons were homogenized on ice-cold lysis buffer (20 mM Tris–HCl (pH 7.5), 10 mM NaF, 150 mM NaCl, 1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, leupeptin and trypsin inhibitor 10 g/mL; 0.25/50 mg tissue). After 1 h, tissue lysates were obtained by centrifugation at 20,000 g for 15 min at 4 °C. Protein concentrations were estimated by the Bio-Rad protein assay using bovine serum albumin as standard.
Colon lysate proteins (70 g) were dissolved in Laemmli sample buffer, boiled for 5 min and separated on SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (240 mA for 40 min at room temperature). The filter was then blocked with 1 × phosphate buffer saline (PBS) and 3% non-fat dried milk for 40 min at room temperature and probed with anti-inducible nitric oxide synthase (iNOS) antibody (dilution 1:1000; cat. no. 610204, BD Bioscience), or anti-cyclooxygenase (COX)-2 (dilution 1:1000; cat. no. 610431, BD Bioscience, from Becton Dickinson, Buccinasco, Italy), or anti-NF-κB p65 (dilution 1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, United States), or anti-Iκ-Bα (dilution 1:500; Santa Cruz Biotechnology, Inc.) antibody for cytosolic and nuclear lysates, or anti-Occludin (dilution 1:500, cat. no. sc-492, Santa Cruz Biotechnology, Santa Cruz, CA, USA), in 1 × PBS, 3% non-fat dried milk, and 0.1% Tween 20 at 4 °C overnight. The secondary antibody was incubated for 1 h at room temperature. Subsequently, the blot was extensively washed with PBS, developed using enhanced chemiluminescence detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to the manufacturer’s instructions, and the immune complex visualized by Image Quant (GE Healtcare, Milan, Italy). Nitrocellulose filter gels were repeatedly stripped for different antibodies. The protein bands were scanned and densitometrically analyzed with a model GS-700 imaging densitometer (Bio-Rad Laboratories, Milan, Italy). To ascertain that blots were loaded with equal amounts of protein lysates, they were also incubated in the presence of the antibody against the β-actin protein (dilution 1:15000, Sigma–Aldrich, Milan, Italy).
GC-MS propionate and butyrate quantification
Fecal samples (n = 6 for each group) were obtained at experimental day 7 (the day after the last DSS administration) and 13 (end of all treatments), weighted (1 g) and suspended in 1 mL of saline and vortexed. After centrifugation at 4 °C, 14000 rpm for 15 min, 500 μL of supernatant was combined with 20 μL phosphoric acid (85%). Then 500 μL of ethyl acetate was added and samples were centrifugated again at 4 °C 14000 rpm for 30 min. This step was repeated three times. A quantity of the pooled extract containing the acidified propionate and butyrate were transferred into a 2-mL glass vial and loaded onto an Agilent Technologies (Santa Clara, CA, USA) 7890 gas chromatograph (GC) system with automatic loader/injector. The GC column was an Agilent DB-WAX Ultra Inert with the length 30 m, internal diameter 0.25 mm and film thickness 0.25 μm. The GC was programmed to achieve the following run parameters: the temperature of the inlet was 250 °C and the volume of injection was 1 μL. The temperature program was as follows: the initial oven temperature at 50 °C held for 1 min, ramped to 250 °C at 8 °C/min. The carrier gas was helium with a constant linear velocity of 1.5 mL/min. Pure fatty acid standards (Sigma-Aldrich) were also prepared to produce a calibration curve. Each sample was analyzed two times on the same day.
Statistical analysis
Statistical comparison between groups of mice was made using Fisher’s Exact Probability Test (incidence of the clonic seizure phase). The percentages of animals exhibiting clonic seizures were plotted against the corresponding doses by a computer construction of the dose- effect curves for calculation of CD50 (±95% confidence limits). The CD50 values for each compound were calculated using a computer program of the method of Litchfield and Wilcoxon, as previously described30. At least 32 animals were used to calculate each CD50 value. All other data are presented as means ± SEM. The statistical analyses were performed with the use of Graph‐Pad Prism (Graph‐Pad Software). For all the experimental data, we evaluated group differences with two‐way ANOVA followed by Tukey’s multiple comparison test. Statistical significance was set at p < 0.05.
Results
Evaluation of pentylentetrazol-induced seizure susceptibility
Mice in all groups were treated with various doses of PTZ and CD50 values were calculated for each group and reported in Fig. 2A. In the control mice group, the calculated PTZ CD50 was 61.96 mg/kg and in the mice without DSS-induced colitis, only VPA (600 mg/kg) was able to significantly increase the CD50 (89.94 mg/kg) whereas the other treatments (ALAC, NaB and MSZ) were not effective. On the other hand, DSS-induced colitis significantly decreased the PTZ CD50 (43.68 mg/kg) relative to control non-DSS treated mice, indicating a decreased seizure threshold. Mesalazine treatment in these DSS-treated mice did not modify susceptibility to PTZ-induced seizures while, notably, the previously ineffective doses of ALAC (375 mg/kg) and NaB (100 mg/kg) were significantly effective in increasing the CD50 in comparison to the DSS-control group with ALAC, maintaining the CD50 to about the same value as that obtained in CTRL mice (not DSS treated) and with NaB only slightly lower. Finally, VPA was still effective in increasing PTZ CD50 in DSS-treated mice, however its effects were much lower in comparison to that obtained in non DSS-treated mice. The CD50 in this latter group was 89.94 mg/kg with an increase in comparison to control of 27.98 mg/kg and in the DSS-treated mice group this increase was of 24.29 mg/kg and therefore similar.

Treatment effects on. (A) PTZ-induced seizures measured by determination of CD50 (±95% confidence interval) in control and DSS (intestinal inflammation) mice groups (n = 28 for each group), VPA was the only effective drug in control mice while after induction of colitis, ALAC and NaB become effective; MSZ was not effective and VPA appeared less effective than in control mice; (B) colon length (n = 10 for each group) was shortened by DSS and recovered by all treatments with the exception of VPA. (C) Disease Activity Index; (n = 30 for each group) and gut permeability. (D) FITC Dextran; n = 5 for each group) were normalized by all treatments. (E) Occludin expression (n = 6 for each group), was normalized by ALAC and NaB treatments, significantly increased by MSZ but not modified by VPA. Data are means ± SEM and * indicates p < 0.05. ALAC = alpha-lactoalbumin; NaB = sodium butyrate; CTRL = control; DSS = dextran sulfate sodium; MSZ = mesalazine; PTZ = pentylenetetrazol; VPA = valproic acid.
Evaluation of experimental colitis and gut permeability
Drug treatments in all control groups did not affect the DAI (data not shown). However, all treatments in a similar fashion, reduced DAI showing significant protective effects against DSS-induced colitis (Fig. 2B). Similarly, DSS-dependent tissue shortening (Fig. 2C) was recovered by all treatments with the exception of VPA, and gut permeability as measured by FITC dextran was increased by DSS and recovered by all treatments (Fig. 2D). No differences were observed between drug-treated groups vs CTRL group.
Finally, occludin expression was significantly (p < 0.005; F(1,50) = 8.30) reduced by DSS treatment and surprisingly also by MSZ treatment (p < 0.01; F(4,50) = 3.78) in control animals. While in DSS-treated groups, ALAC and NaB treatments recovered occludin expression to control levels, MSZ significantly (p < 0.001; F(4,50) = 7.79) increased it to about 75% of control and VPA, instead, had no effect (Fig. 2E).
Histological analysis and scoring of colon sections
Control colon sections showed an intact epithelium, well‐defined crypt length, no edema in the mucosa and submucosa, and no ulcers or erosions (Fig. 3B). In contrast, colon tissue from the DSS-treated group showed severe inflammatory lesions throughout the mucosa and loss of crypt architecture. All treatments were able to protect colonic mucosa structure, ameliorating the mucosal integrity and crypt structure and improving the epithelial surface compared with the DSS group (Fig. 3B). The beneficial effects mediated by treatments were also confirmed by evaluation of the histopathological score of distal colon sections stained with H&E (Fig. 3A). On a scale, ALAC and NaB were the treatments with the highest protecting score (both about 0.8), followed by VPA and MSZ (both about 1.2).

Drug effects on colon histology (n = 6 for each group). (A) All treatments were able to protect colonic mucosa structure, ameliorating the mucosal integrity and crypt structure and improving the epithelial surface compared with the DSS-treated group. (B) Control colon sections showed an intact epithelium, well‐defined crypt length, no edema in the mucosa and submucosa, and no ulcers or erosions. In contrast, colon tissue from the DSS group showed severe inflammatory lesions throughout the mucosa and loss of crypt architecture. Data are means ± SEM and * indicates p < 0.05. ALAC = alpha-lactoalbumin; Na Bu = sodium butyrate; CTRL = control; DSS = dextran sulfate sodium; MSZ = mesalazine; PTZ = pentylenetetrazol; VPA = valproic acid.
Western blot analysis for inflammatory parameters
Protein concentrations of colon lysate were evaluated with specific antibodies in order to study the effects of DSS and drug treatments on inflammatory proteins. NF-κB expression was increased 4-fold by DSS-treatment (p < 0.001; F(1,50) = 11.57) and accordingly Iκ-Bα was decreased 2-fold (P < 0.003; F(1,50) = 9.83) while drug treatments in non-DSS-treated animals had no effects on NF-κB (Fig. 4A). At odds with this, Iκ-Bα expression was significantly (p < 0.004 F(4,50) = 4.26) reduced by ALAC and NaB treatments in CTRL animals while VPA and MSZ treatment had no effects in this experimental group. On the other hand, in DSS-treated mice, MSZ was not effective while all other treatments were able to counteract the induced increase of NF-κB. In this experimental group, Iκ-Bα expression was normalized by all treatments and MSZ, in this case, was the most effective on this parameter, being significantly different in comparison to all other DSS-treated groups and even significantly higher than CTRL group (Fig. 4B).

Western blot analysis of inflammatory parameters (n = 6 for each group). (A) NF-κB; (B) Iκ-Bα; (C) COX-2 and (D) iNOS. All drugs are able to rescue DSS inflammatory effects. Data are means ± SEM and *indicates p < 0.05. ALAC = alpha-lactoalbumin; NaB = sodium butyrate; COX-2 = cyclooxygenase type 2; CTRL = control; DSS = dextran sulfate sodium; iNOS = inducible nitric oxide synthase; MSZ = mesalazine; PTZ = pentylenetetrazol; VPA = valproic acid.
COX-2 expression was ~2-fold increased by DSS treatment (p < 0.002; F(1,50) = 9.87), and notably significantly reduced by VPA and MSZ treatment in non-DSS-treated mice. However, ALAC, NaB and VPA were able to significantly normalize DSS effects (p < 0.007; F(4,50) = 4.05) while, surprisingly, MSZ was not effective in this case (Fig. 4C).
iNOS concentration was ~2-fold increased by DSS-treatment (p < 0.006; F(1,50) = 7.98) while drug treatments did not alter its expression in control animals while completely preventing its increase in DSS-treated groups with no clear differences between drugs (Fig. 4D).
GC-MS propionate and butyrate quantification
Propionate and butyrate concentrations in feces were measured both at experimental day 7 (1 day after the end of DSS administration) and on experimental day 13 (before mice sacrifice and sampling, see Fig. 5). DSS treatment, in both cases, virtually abolished (p < 0.008; F(1,50) = 56) the production of these SCFAs in the intestine which were nearly undetectable in stool samples. None of the treatments modified propionate and butyrate concentrations in control animals at day 7 (after 4 days) while at day 13 (at 10 days of treatment) butyric acid concentration was significantly increased by all treatments with the exception of MSZ (Fig. 5). In DSS-treated experimental groups, all drugs were effective at rescuing SCFA production, this effect was already significant at experimental day 7 (with VPA exception) and on experimental day 14, while propionate was still about half the amount of the control group for all drugs, and butyrate levels were not significantly different from control. Significant effects were observed between drug groups; it can be noted that NaB treatment was significantly (p < 0.001; F(4,50) = 5.17) more effective than VPA and MSZ (Fig. 5B).

Drug effects (n = 6 for each group) on propionic and butyric acids levels in stools both at day 7 and 14 after initiation of DSS treatment which, per se, nearly completely abolished production of both acids by the gut-microbiota (dysbiosis) and treatments differently affected acid levels, generally increasing their amounts. Data are means ± SEM and *indicates p < 0.05. ALAC = alpha-lactoalbumin; NaB = sodium butyrate; CTRL = control; DSS = dextran sulfate sodium; MSZ = mesalazine; PTZ = pentylenetetrazol; VPA = valproic acid.
Discussion
The role of intestinal inflammation in reducing seizure threshold or aggravating seizures has been previously reported7,15. In this paper, we confirmed the link between gut inflammation and epilepsy in a different colitis animal model (i.e. DSS model). In addition, we demonstrated that drugs able to recover intestinal inflammation indeed had an effect on seizure threshold, whereas intestinal inflammation may reduce the efficacy of previously effective drugs. In particular, we showed that the reduction of intestinal inflammation through ALAC treatment as well as Na butyrate treatment resulted in a strong antiseizure effect. Furthermore, our data support the view that intestinal inflammation may reduce the antiepileptic effects of VPA and confirmed that it lowers seizure threshold.
The mechanisms by which intestinal inflammation can lead to a higher seizure susceptibility has not yet been completely clarified. It has been previously demonstrated that induction of colitis by 2,4,6-trinitrobenzene sulfonic acid (TNBS) increases cytokine levels (IL-1β and TNF-α) in the hippocampus and that TNF-α is the most important in mediating microglia activation and this was linked to a higher seizure susceptibility15. In our study, all treatments were in fact able to reduce intestinal damage and inflammation and an unclear proportionality can be defined within this set of experiments; indeed, a dose-response analysis and an in-depth evaluation of several parameters could try to define the exact mechanisms involved. However, these results can be interpreted with a specific action of ALAC and NaB in counteracting the proconvulsant role of intestinal inflammation which could be mediated by several agents spanning from the protective role to the involvement of other factors. For example, ALAC may exert its effect by an increase in tryptophan levels and its metabolites such as kynurenic acid which may penetrate the brain and act on N-methyl-D-aspartate (NMDA)-type glutamate receptors, as previously suggested17,18. Similarly, NaB may have also acted directly on the brain, considering its ability to modulate several processes28. In contrast, MSZ was not effective on seizure threshold while it was able to protect the intestine, similarly to ALAC and NaB, indicating that removing inflammation per se might not necessarily be enough to obtain an effect on the CNS. On the other hand, VPA effects were notably dampened by colitis even if the drug was also able to reduce intestinal inflammation; this latter anti-inflammatory effect by VPA was already reported both on the nervous system32,33 and peripheral organs34. The apparently reduced VPA efficacy may have several explanations; for example, VPA absorption may have been reduced by intestinal inflammation and therefore its final brain concentration may have been lower. Nevertheless, it is particularly relevant that in the case of an intestinal inflammation, as could happen in clinical practice, the antiepileptic effect of VPA could be reduced and this may lead to the appearance or aggravation of uncontrolled seizures in patients with epilepsy.
Recently, the gut-brain axis and the role of microbiota in brain diseases including epilepsy has gathered attention with mounting evidence of a strict connection; for example, it is known that chronic restraint stress or social defeat stress are able to trigger dysbiosis along with a possible involvement of the gut-microbiota-brain axis35,36,37 and microbiota alteration due to stress was involved in the facilitation of kindling in rats38. It was then suggested that dysbiosis and/or microbiome reshaping may be relevant in several areas of epilepsy research including traumatic brain injury or biomarkers. Furthermore, the well-recognized antiepileptic ketogenic diet was convincingly demonstrated to have a modulating effect on gut microbiota with this latter aspect being necessary for its action39,40. DSS-induced colitis has as a consequence, an alteration of gut microbiota (i.e. dysbiosis) which is also confirmed in our experiments indirectly by the strong reduction in the amount of propionic and butyric acids in the stools41.
In our study, ALAC and NaB better restored SCFA levels and were the most effective drugs on seizure threshold in the PTZ mouse model although it remains to be determined what the contribution of microbiota were to increased seizure susceptibility. In fact, ALAC and NaB were both more effective compared with VPA and MSZ on a number of parameters in reducing intestinal inflammation, probably with an impact on microbiota and on circulating inflammatory mediators linked to peripheral inflammation. Therefore, the CNS effects observed may be linked to more than one single parameter. It is intriguing, however, to believe that seizures may arise from an inflamed intestine and that drugs acting on this inflammation may prove to be effective antiepileptics, even if the lack of MSZ effects suggest that a general intestinal anti-inflammatory action is not sufficient, but that a more specific intervention leading to probable restoration of a healthy intestine is more effective.
In conclusion, our data suggest that in patients with epilepsy, the intestine and its physiopathological state may also contribute to the disease and in some cases an appropriate drug intervention aimed at restoring a “normal” gut function may have beneficial anticonvulsant effects. In addition, it has to be underlined that an alteration of intestinal function can reduce drug effects and that a specific clinical evaluation should be always performed. Finally, it will be crucial to further understand whether other mediators than known inflammatory factors are involved in the link between inflammation and epilepsy and studying the role of gut microbiota is currently one of the most promising research areas confirming its relevance as a potential therapeutic target for this condition.
Data Availability
Data may be available upon request.
References
Vezzani, A., Aronica, E., Mazarati, A. & Pittman, Q. J. Epilepsy and brain inflammation. Exp. Neurol. 244, 11–21 (2013).
Librizzi, L., Noè, F., Vezzani, A., De Curtis, M. & Ravizza, T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann. Neurol. 72, 82–90 (2012).
Vezzani, A. et al. Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234 (2016).
van Vliet, E. A., Aronica, E., Vezzani, A. & Ravizza, T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 44, 91–111 (2018).
Aronica, E. et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 58, 27–38 (2017).
Mazarati, A. M., Lewis, M. L. & Pittman, Q. J. Neurobehavioral comorbidities of epilepsy: Role of inflammation. Epilepsia 58, 48–56 (2017).
Riazi, K., Galic, M. A. & Pittman, Q. J. Contributions of peripheral inflammation to seizure susceptibility: Cytokines and brain excitability. Epilepsy Res. 89, 34–42 (2010).
Yuen, A. W. C., Keezer, M. R. & Sander, J. W. Epilepsy is a neurological and a systemic disorder. Epilepsy Behav. 78, 57–61 (2018).
Russo, E. et al. Early molecular and behavioral response to lipopolysaccharide in the WAG/Rij rat model of absence epilepsy and depressive-like behavior, involves interplay between AMPK, AKT/mTOR pathways and neuroinflammatory cytokine release. Brain. Behav. Immun. 42, 157–168 (2014).
Sayyah, M., Javad-Pour, M. & Ghazi-Khansari, M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: Involvement of proinflammatory factors: Nitric oxide and prostaglandins. Neuroscience 122, 1073–1080 (2003).
Auvin, S. et al. Inflammation in rat pups subjected to short hyperthermic seizures enhances brain long-term excitability. Epilepsy Res. 86, 124–130 (2009).
Devinsky, O., Schein, A. & Najjar, S. Epilepsy associated with systemic autoimmune disorders. Epilepsy Curr. 13, 62–68 (2013).
Morís, G. Inflammatory bowel disease: An increased risk factor for neurologic complications. World J. Gastroenterol. 20, 1228–1237 (2014).
Casella, G. et al. Neurological disorders and inflammatory bowel diseases. World J. Gastroenterol. 20, 8764–8782 (2014).
Riazi, K. et al. Microglial activation and TNF production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. 105, 17151–17156 (2008).
Lonnerdal, B. Biochemistry and physiological function of human milk proteins. Am. J. Clin. Nutr. 42, 1299–1317 (1985).
Citraro, R. et al. Preclinical activity profile of α-lactoalbumin, a whey protein rich in tryptophan, in rodent models of seizures and epilepsy. Epilepsy Res. 95, 60–69 (2011).
Russo, E. et al. Protective activity of α-lactoalbumin (ALAC), a whey protein rich in tryptophan, in rodent models of epileptogenesis. Neuroscience 226, 282–288 (2012).
Mainardi, P., Leonardi, A. & Albano, C. Potentiation of brain serotonin activity may inhibit seizures, especially in drug-resistant epilepsy. Med. Hypotheses 70, 876–879 (2008).
Errichiello, L. et al. A proof-of-concept trial of the whey protein alfa-lactalbumin in chronic cortical myoclonus. Mov. Disord. 26, 2573–2575 (2011).
Yamaguchi, M., Takai, S., Hosono, A. & Seki, T. Bovine milk-derived α-lactalbumin inhibits colon inflammation and carcinogenesis in azoxymethane and dextran sodium sulfate-treated mice. Biosci. Biotechnol. Biochem. 78, 672–679 (2014).
Simeoli, R. et al. An orally administered butyrate-releasing derivative reduces neutrophil recruitment and inflammation in dextran sulphate sodium-induced murine colitis. Br. J. Pharmacol. 174, 1484–1496 (2017).
Töllner, K., Twele, F. & Löscher, W. Evaluation of the pentylenetetrazole seizure threshold test in epileptic mice as surrogate model for drug testing against pharmacoresistant seizures. Epilepsy Behav. 57, 95–104 (2016).
Zhang, H. & Chen, W. Interleukin 6 inhibition by triptolide prevents inflammation in a mouse model of ulcerative colitis. Exp. Ther. Med. 14, 2271–2276 (2017).
Citraro, R. et al. Role of Histone Deacetylases (HDACs) in Epilepsy and Epileptogenesis. Curr. Pharm. Des. 23, 5546–5562 (2018).
Reddy, S. D., Clossen, B. C. & Reddy, D. S. Epigenetic Histone Deacetylation Inhibition Prevents the Development and Persistence of Temporal Lobe Epilepsy. J. Pharmacol. Exp. Ther. 364, jpet.117.244939 (2017).
Lee, C. et al. Sodium butyrate inhibits the NF-kappa B signaling pathway and histone deacetylation, and attenuates experimental colitis in an IL-10 independent manner. Int. Immunopharmacol. 51, 47–56 (2017).
Stilling, R. M. et al. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 99, 110–132 (2016).
De Luca, G. et al. Amino acid levels in some brain areas of inducible nitric oxide synthase knock out mouse (iNOS−/−) before and after pentylenetetrazole kindling. Pharmacol. Biochem. Behav. 85, 804–812 (2006).
De Sarro, G. et al. Seizure susceptibility to various convulsant stimuli of knockout interleukin-6 mice. Pharmacol. Biochem. Behav. 77, 761–766 (2004).
Cani, P. D. et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103 (2009).
Chen, S. et al. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-ΚB pathway dependent of HDAC3. J. Neuroinflammation 15, 150 (2018).
Dambach, H. et al. Glia and epilepsy: Experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia 55, 184–192 (2014).
Amirzargar, M. A. et al. Anti-inflammatory Effects of Valproic Acid in a Rat Model of Renal Ischemia/Reperfusion Injury: Alteration in Cytokine Profile. Inflammation 40, 1310–1318 (2017).
Zhang, J. C. et al. Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: a possible role of gut-microbiota-brain axis. Transl. Psychiatry 7, e1138 (2017).
Wong, M. L. et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 21, 797–805 (2016).
Pujia, A. M. et al. The probiotics in dentistry: a narrative review. Eur. Rev. Med. Pharmacol. Sci (2017).
Medel-Matus, J.-S., Shin, D., Dorfman, E., Sankar, R. & Mazarati, A. Facilitation of kindling epileptogenesis by chronic stress may be mediated by intestinal microbiome. Epilepsia Open 3, 290–294 (2018).
Olson, C. A. et al. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 173, 1728–1741.e13 (2018).
Zhang, Y. et al. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 145, 163–168 (2018).
Munyaka, P. M., Rabbi, M. F., Khafipour, E. & Ghia, J. E. Acute dextran sulfate sodium (DSS)-induced colitis promotes gut microbial dysbiosis in mice. J. Basic Microbiol. 56, 986–998 (2016).
Acknowledgements
The Italian Ministry of Health is kindly acknowledged for economical support by the Bando Ricerca Finalizzata 2013. Dr. Giovanni Politi is kindly acknowledged for his technical support.
Author information
Authors and Affiliations
Contributions
E.R., G.D.S., P.S. and P.M. participated in the conception and design of the study; E.R., C.D.C., A.L., V.N., C.G., L.d.C.M., C.A. and R.C. participated at the acquisition and analysis of data; E.R., A.C., C.D.C., P.S., A.Con. and G.D.S. contributed to drafting of a significant portion of the manuscript and/or figures.
Corresponding author
Ethics declarations
Competing Interests
Rita Citraro has received research funding from Kolfarma Srl. Paolo Mainardi is an employee of Kolfarma Srl. Pasquale Striano has received research grants or compensation from GW pharma (UK) and Kolfarma, Eisai pharma. Emilio Russo has received research grants or compensation from Eisai pharma. All other authors have nothing to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Caro, C., Leo, A., Nesci, V. et al. Intestinal inflammation increases convulsant activity and reduces antiepileptic drug efficacy in a mouse model of epilepsy. Sci Rep 9, 13983 (2019). https://doi.org/10.1038/s41598-019-50542-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50542-0
This article is cited by
-
Gut microbiota profile in CDKL5 deficiency disorder patients
Scientific Reports (2024)
-
Microglia in Microbiota-Gut-Brain Axis: A Hub in Epilepsy
Molecular Neurobiology (2024)
-
Effect of dietary supplementation with Lactobacillus helveticus R0052 on seizure thresholds and antiseizure potency of sodium valproate in mice
Psychopharmacology (2024)
-
Drug-Resistant Epilepsy and Gut-Brain Axis: an Overview of a New Strategy for Treatment
Molecular Neurobiology (2023)
-
The Microbiota–Gut–Brain Axis and Epilepsy
Cellular and Molecular Neurobiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
