
Abstract
Fermented foods play a major role in the diet of people in Africa, where a wide variety of raw materials are fermented. Understanding the microbial populations of these products would help in the design of specific starter cultures to produce standardized and safer foods. In this study, the bacterial diversity of African fermented foods produced from several raw materials (cereals, milk, cassava, honey, palm sap, and locust beans) under different conditions (household, small commercial producers or laboratory) in 8 African countries was analysed by 16S rRNA gene amplicon sequencing during the Workshop “Analysis of the Microbiomes of Naturally Fermented Foods Training Course”. Results show that lactobacilli were less abundant in fermentations performed under laboratory conditions compared to artisanal or commercial fermentations. Excluding the samples produced under laboratory conditions, lactobacilli is one of the dominant groups in all the remaining samples. Genera within the order Lactobacillales dominated dairy, cereal and cassava fermentations. Genera within the order Lactobacillales, and genera Zymomonas and Bacillus were predominant in alcoholic beverages, whereas Bacillus and Lactobacillus were the dominant genera in the locust bean sample. The genus Zymomonas was reported for the first time in dairy, cereal, cassava and locust bean fermentations.
Similar content being viewed by others

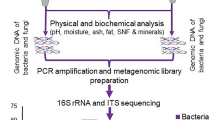
Introduction
Traditional fermented foods play a major role in the diet of numerous communities worldwide. Africa is perhaps the continent with the richest variety of fermented foods, where fermentation still plays a major role in combating food spoilage, foodborne diseases and represents a significant postharvest value addition. In fact, fermentation is still a largely home-based process used throughout the continent1. A wide variety of raw materials are traditionally fermented in different regions of Africa. As a result, fermented foods with different characteristics are produced and they have been classified in groups such as fermented non-alcoholic cereals (mainly produced from sorghum, millet and maize), starchy root crops (mainly produced from cassava), animal proteins (mainly dairy products), vegetable proteins (produced from legumes and oilseeds) and alcoholic beverages (produced from cereals, sap, honey or fruits, among other materials)1. Fermented products have been described to provide health benefits, such as protection against gastrointestinal disorders, prevention of hypertension and heart disease or protection from diabetes and osteoporosis. In addition, traditional African fermented foods contain live microorganisms that can produce health-promoting compounds, such as antimicrobials, essential nutrients or molecules with antioxidant activity, and can act as probiotic strains2,3.
Knowledge about the microbial ecology of natural food fermentations can be used to identify biomarkers to assess the quality of fermented foods and would help in the design of optimum starter cultures4. Predominant bacterial groups present in African fermented foods have been widely analysed using culture-dependant methods5, but these methods present several limitations, such as not being able to detect non-culturable populations or being unable to detect microorganisms in low numbers in complex ecosystems with dominant populations6. As an alternative, culture-independent methods, particularly amplicon sequencing, are increasingly being used to study the bacterial populations of fermented foods, although to date, few studies have focused on African foods7,8,9.
Culture-independent methods present several biases, some of which are associated with the DNA extraction procedures used. The extraction of DNA from foods can be challenging due to the structure and chemical composition of the matrices. Therefore, due to the vast variety of raw materials that are fermented, diverse DNA extraction procedures can be found in the literature10. In fact, some food matrices require pre-processing steps before DNA extraction11,12,13. In order to analyse the microbial population of different fermented samples, the application of a standardized method would be beneficial. Commercial extraction kits partially solve the bias problems, but usually they have been tested for common food matrices and cannot be applied to complex foods10. In addition, commercial kits use small amounts of sample. This can be an advantage as less material is required, but for some types of food it may not yield enough microbial DNA, as we have experienced in previous experiments in our laboratory. Here, the use of a single method for the analysis of a wide range of African fermented products was evaluated for its performance across diverse food matrices.
Despite the important information that culture-independent methods contribute to the understanding of the complete microbiome associated with foods, few African labs are currently equipped to run these types of analyses. To address this need, a workshop was recently organized in Accra, Ghana to train participants in wet and dry lab approaches to food microbiome analysis. During the workshop Analysis of the Microbiomes of Naturally Fermented Foods Training Course, a modified DNA extraction method was applied to a wide range of foods and evaluated using amplicon sequence data. Bacterial populations of the samples were analysed by amplification of the V4 regions of the 16S rRNA gene and Illumina high-throughput sequencing. This study demonstrates that the modified method is suitable to analyse the bacterial populations of fermented foods produced from different raw materials by amplicon sequencing. The bacterial composition of different types of fermented foods produced throughout Africa was described and the structure of their bacterial populations was compared based on the raw material used and the conditions in which the fermentations were performed.
Results and Discussion
DNA extraction yield from fermented foods prepared from different raw materials
To test the performance of the DNA extraction method for analysis of the microbial diversity of different fermented foods, 40 spontaneously fermented African food samples with different characteristics (different raw material, production under different conditions or country of origin) were used (Table 1). The samples analysed in this study were produced in different regions of Africa, although they originate mainly from West African countries. Samples were grouped based on the raw material used as follows: cereal, cassava, dairy, locust beans and alcoholic beverages. DNA from samples S1 to S40 was extracted by the participants during the workshop, with 37 out of 40 samples yielding enough DNA to be analysed by amplicon-based metagenomics. Only three samples, one from the cassava group (S26), one from the alcoholic beverages group (S39) and one from the cereal group (S40) did not yield a measurable quantity of DNA (Table 1).
The mean and standard deviation of the DNA concentration from all the samples was 243.72 ± 300.93 ng/µl. Higher total DNA concentrations were obtained from cereal-based samples (289.23 ± 342.07 ng/µl) and alcoholic beverages (354.63 ± 275.7 ng/µl) compared to cassava (65.33 ± 68.04 ng/µl), dairy (77.49 ± 56.09 ng/µl) and locust bean (18.72 ng/µl) fermented samples.
Bacterial diversity
Bacterial diversities of the 37 samples that yielded the required amount of DNA were analysed by 16S rRNA gene amplicon sequencing. DNA sequencing of the V4 amplicons by the Illumina MiSeq platform resulted in 3,321,898 paired-end sequence reads with an average of 89,781.03 ± 6,189.48 sequences per sample. Of these, 16.69% were discarded due to poor quality, reads not merging, or after being identified as chimeras; as a result of these steps, 2,767,337 high quality sequences were retained and analysed, with an average of 74,792.89 ± 5,069.18 sequences per sample. Distribution of reads per sample can be found in Supplementary Table S1.
Background DNA was removed by filtering out sequences assigned to chloroplast and mitochondrial taxonomic groups. Data were rarefied to 46,726 sequences per sample to avoid bias. As shown in Fig. 1(a,b), the rarefaction curves for the operational taxonomic units (OTUs) and Shannon index reach a plateau showing that the coverage was sufficient to capture the majority of the microbial diversity. Alpha diversity indexes (observed OTUs, Shannon, Faith’s phylogenetic diversity and Pielou’s evenness) were compared based on types of raw materials (cereal, cassava, dairy, locust beans and alcoholic beverages) and on the type of production process (artisanal, commercial or laboratory). Although alcoholic samples and the locust bean sample seemed to have a higher number of observed OTUs than cassava, cereal and dairy samples (Fig. 1c), no significant differences were found in any of the alpha diversity indexes compared.

Rarefaction plots of (a) Observed OTUs and (b) Shannon index for each sample; (c) Boxplots of the observed OTUs per fermented sample for each fermentation group. Spots represent outliers.
Principal coordinates analysis (PCoA) was performed to determine the effect of the raw material used and the production conditions on the microbial communities (Fig. 2). The differences based on the presence and absence of taxonomic groups were analysed using Jaccard (Fig. 2a) and unweighted UniFrac (Fig. 2b) distances. The fermentation samples grouped by the raw material correlated with the differences in the microbial populations observed when using the Jaccard (p-value 0.005) and unweighted UniFrac (p-value 0.005) distances. Most of the cereal and dairy samples clustered to one end of principal component 1 in the Jaccard (Fig. 2a) and unweighted UniFrac distances plots (Fig. 2b) (40.06% and 43.91% of variation explained, respectively), while most of the alcoholic samples cluster to the opposite end together with the locust bean sample. One of the cassava samples was grouped with the majority of the cereal and dairy samples, whilst the other was grouped with a large proportion of the alcoholic and locust bean samples. The differences between samples are mostly due to OTUs assigned to the Lactobacillus, Weissella, Acetobacter and Enterococcus genera and Enterobacteriaceae family for the Jaccard distance (vectors in Fig. 2a), and also for the unweighted UniFrac distance, but with the inclusion of the Streptococcus genera rather than the Enterobacteriaceae family (vectors in Fig. 2b). Cereal and dairy fermentations are both lactic acid fermentations, which likely explains why these samples share more OTUs than was observed with the alcoholic and locust bean fermentation samples.

Principal coordinate analysis (PCoA) biplots showing (a) Jaccard, (b) unweighted UniFrac, (c) weighted UniFrac distances with samples coloured by raw material (a,b) or production conditions (c). The percentage of variation explained by the plotted principal coordinates is indicated in the axes. The arrows indicate the 5 taxonomic groups (at genera level) that contribute most to the indexes.
In general, the most abundant bacteria in cereal and dairy fermentations were genera within the order Lactobacillales (phylum Firmicutes) such as Lactobacillus, Weissella and Streptococcus, except for samples produced under laboratory conditions (Fig. 3). Most of the OTUs within the Lactobacillus genus could not be classified at species level due to the short length of the 16S rRNA sequences. In general, most of the lactobacilli that could be classified at species level were identified as Lactobacillus fermentum. The relative abundance of the Lactobacillus species per sample can be found as Supplementary Fig. S2.

Relative abundance of the bacterial communities at the genus level. Taxonomic groups with abundance <1% were included in the group “others”. Where genera (g) could not be determined, family (f) level identification is shown. Arrows indicate samples produced under laboratory conditions.
Other genera, such as Acetobacter, were found at relatively high abundances in some of the dairy and cereal fermented samples. These observations agree with most of the previous studies that describe the microbial populations of dairy6 and cereal8,14,15 foods produced in Africa. Bacterial genera that include potential pathogenic species such as Escherichia or Clostridium (cluster I and XI) were found in several cereal and dairy samples. It should be noted that the presence of the genus Zymomonas had not previously been described in fermented dairy or cereal samples. To our knowledge, this is the first time that this genus is reported in these types of food. In the sample of fermented cassava that showed a similar diversity to the cereal and dairy samples, Lactobacillus and Weissella were the most abundant genera, whereas the other sample showed high levels of bacteria from Lactobacillus, Leuconostoc, Weissella, Bacillus and Enterobacteriaceae (Fig. 3). Previous studies indicate that lactic acid bacteria from the Lactobacillus, Weissella and Leuconostoc genera16,17 together with the genus Bacillus5,18 were highly abundant in fermented cassava. However, the high abundance of Zymomonas observed in the latter sample was not reported in other studies. Zymomonas movilis has been previously used to produce ethanol from cassava waste19 but, to our knowledge, this is the first time that this genus is reported in non-alcoholic fermented cassava. In all the alcoholic samples, Lactobacillus and Streptococcus are among the most abundant genera. The genera Zymomonas, Leuconostoc and Bacillus are highly abundant in all the alcoholic samples except one, which clustered together with the lactic acid fermented samples. Previous studies described Lactobacillus and Leuconostoc as dominant groups in palm wine20,21. Although there is scarce information regarding the abundance of Zymomonas in these types of beverages, this genus has been typically isolated from alcoholic drinks22, such as palm wine and ripening honey23. In contrast to the previous samples, the locust bean undergoes an alkaline fermentation. Our results show that Bacillus, Lactobacillus and other genera within the Lactobacillales order are the predominant genera in this sample. A high abundance of Bacillus was expected in this sample, as this genus is responsible for the fermentation of the beans24. Although high levels of lactobacilli were not expected, they have previously been isolated from this type of product25. In this sample, several genera that have not been reported before in legume fermentations, such as Zymomonas, Gemella, Brevibacillus, Jeotgalicoccus, Atopostipes and members of the family Carnobacteriaceae were found. To compare the populations between the samples whilst considering the relative abundance of the taxa, weighted UniFrac distance was used (Fig. 2c). The production conditions explained the differences between the samples (p-value 0.002). Commercial and artisanal samples did not show significant differences between them (p-value 0.274), whilst samples produced in the laboratory were different from both artisanal (p-value 0.003) and commercial (p-value 0.002) samples. The differences between samples are largely due to OTUs within Lactobacillus, Paenibacillus, Weissella, Enterococcus and Leuconostoc genera (vectors in Fig. 2c). Analysis of Composition of Microbiomes (ANCOM) was used to predict significant differences in the relative abundance at the genera level. Based on the type of raw material used, significant differences (W = 281) were found in the Lachnospiraceae UCG-006 group. Alcoholic and locust bean samples showed a higher number of sequences (median = 52 and 61, respectively) than cereal, dairy and cassava samples (median = 1, 1 and 8, respectively). Nevertheless, this taxonomic group had very low abundance (less than 0.1%) in the samples in which it was present. Based on the production conditions, statistical differences (W = 113) were observed in the genus Lactobacillus. Artisanal and commercial samples showed a higher number of sequences (median of 33753 and 45292, respectively) assigned to Lactobacillus than those samples produced under laboratory conditions (median of 8429). This can be observed per individual sample in the relative abundance graph (Fig. 3), which also shows that most of the samples produced under laboratory conditions (red arrows, Fig. 3) have higher levels of bacteria from the Enterobacteriaceae family. In a recent study, higher levels of Enterobacteriaceae and Pseudomonas were observed in fermentations performed in the laboratory compared to household fermentations8. The lower relative abundance of lactobacilli in the fermented samples produced under laboratory conditions could be explained by the use of a sterile environment and sterilized tools. The environmental microbiota has been described as playing an important role in the fermentation dynamics26, and tools used during some fermentations have been pointed to as a source of lactobacilli7,27. This should be considered when performing fermentation experiments under laboratory conditions, as spontaneous fermentations may not develop equally in the laboratory and in-field production.
Conclusion
Most of the previous reports on the bacterial diversity of African fermented foods were based on culture-dependent analysis. Nevertheless, these methods present several limitations, which are resolved by culture-independent methods. In this study, a modified method to extract DNA from different food matrices is proposed. The resultant DNA was used to analyse the bacterial diversity of traditional African fermented products by 16S rRNA gene amplicon sequencing. In this study, the genus Zymomonas has been reported for the first time in dairy, cereal, cassava and locust bean fermentations. This shows the importance of using culture-independent methods to study the bacterial communities of African fermented food. Results show that the genus Lactobacillus is less abundant in fermentations performed in laboratory conditions compared to artisanal or commercial fermentations. This is an important finding as this could affect the interpretation of results observed in model fermentations.
Materials and Methods
Sample collection and storage
In this study, 40 food samples were collected and used by the participants of the Workshop “Analysis of the Microbiomes of Naturally Fermented Foods”, held from the 5th to the 9th of February 2018 in Accra (Ghana). The fermented samples, produced from 10 different raw materials (sorghum, maize, millet, banana, cassava, locust beans, palm tree sap, teff, honey and milk), were collected from 8 different African countries (Ghana, Nigeria, Benin, Burkina Faso, Uganda, Kenya, Ethiopia and South Africa). They were obtained from rural areas (artisanal), small-scale producers (commercial) or laboratory spontaneous fermentations (laboratory) (Table 1) and kept at -20 °C until the DNA was extracted. Samples were grouped according to the type of raw material: cereals, cassava, dairy, locust beans and alcoholic beverages (Table 1).
Pre-processing, DNA extraction and DNA quantification
To extract DNA from fermented samples, the FastDNA Spin Kit for Soil (MP Biomedicals, UK) was used in conjunction with a pre-processing step which separates the microbial cells from large solid particles present in the sample. See Supplementary methods for a description of the optimization of the method. First, 20 g of the sample were mixed with 10 ml of cold ultrapure H2O by vigorous vortexing. The solid particles were removed by centrifugation at 800 × g for 1 min at 4 °C and the supernatant was retained. A further 10 ml H2O was added and the process was repeated three times in total; a final volume of approximately 30 ml supernatant was obtained. Cells were harvested from the particle-free supernatants by centrifugation at 3000 × g for 20 min at 4 °C. The supernatant was discarded and the pellet was washed three times using 1 ml PBS buffer. After centrifugation at 14,000 × g for 2 min, the pellet was resuspended in 978 µl sodium phosphate buffer and 122 µl MT buffer and incubated for 1 h at 4 °C and homogenized for 60 s at a speed setting of 6.5 m/s, using a FastPrep-24 instrument (MP Biomedicals, UK). This process was repeated three times and samples were kept on ice for 5 min between each homogenization step. Otherwise, DNA extraction was performed according to the manufacturer’s instructions. The extracted DNA was resuspended in 50 μl elution buffer.
Total DNA extracted from the fermented samples was quantified fluorometrically by a Qubit 3.0 fluorometer (Invitrogen, Carlsbad, CA) using the Qubit dsDNA BR Assay Kit (Invitrogen), or the Qubit dsDNA HS Assay Kit (Invitrogen) when the concentration of DNA was <10 ng/µl.
Illumina high-throughput sequencing
16S rRNA gene PCR amplification and sequencing was performed by Novogene (HK) Company Limited (Hong Kong). The V4 hypervariable region of the 16S rRNA gene was amplified using specific primers 515 F and 806R28. All PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs, USA). The libraries generated with NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, England) were sequenced using paired-end Illumina sequencing (2 × 250 bp) on the HiSeq. 2500 platform (Illumina, USA).
Sequence analysis
Sequencing data, for the 37 samples that yielded a perceptible concentration of DNA (>0.2 ng/μl), were analysed using the Quantitative Insights Into Microbial Ecology 2 (QIIME2) 2018.8 software29. The demultiplexed paired-end reads were filtered of substitution and chimera errors and merged using DADA230. Bacterial taxonomic assignment was performed at 97% similarity using a Naive Bayes classifier trained on the Silva version 132 99% OTU database31, where the sequences have been trimmed to only include 250 bases from the V4 region bound by the 515 F/806 R primer pair. Alpha diversity was analysed using observed OTUs, Shannon, Pielou’s evenness, and Faith’s phylogenetic diversity indexes. Rarefaction curves were computed using OTUs and Shannon index. Jaccard, unweighted and weighted UniFrac distances were used to generate beta diversity PCoA biplots, which were visualised using the Emperor tool32.
Statistical analysis
Wilcoxon sign test (2-tailed) was used to compare the DNA yielded by the two extraction methods tested using IBM SPSS Statistics Version 22.0. software (IBM Corp., USA). Significant differences in alpha diversity between groups were calculated using the alpha-group-significance script in QIIME2, which performs the Kruskal-Wallis test. Differences in beta diversity between groups were analysed using PERMANOVA including pairwise test (Anderson, 2001). Significant differences in the bacterial community structure amongst the groups were evaluated by Analysis of Composition of Microbiomes (ANCOM)33. A p-value ≤ 0.05 was considered statistically significant.
Data Availability
The datasets generated and analysed during the current study are available in the SRA database under the accession number PRJNA532858.
References
Olasupo, N., Odunfa, S. & Obayori, O. Ethnic African Fermented Foods in Fermented foods and beverages of the world 335–364 (CRC press, 2010).
Franz, C. M. et al. African fermented foods and probiotics. International Journal of Food Microbiology 190, 84–96, https://doi.org/10.1016/j.ijfoodmicro.2014.08.033 (2014).
Tamang, J. P., Shin, D.-H., Jung, S.-J. & Chae, S.-W. Functional properties of microorganisms in fermented foods. Frontiers in Microbiology 7, https://doi.org/10.3389/fmicb.2016.00578 (2016).
Walsh, A. M., Crispie, F., Claesson, M. J. & Cotter, P. D. Translating Omics to Food Microbiology. Annual Review of Food Science and Technology 8, 113–134, https://doi.org/10.1146/annurev-food-030216-025729 (2017).
Tamang, J. P. Diversity of Fermented Foods in Fermented foods and beverages of the world 41–84 (CRC press, 2010).
Cocolin, L., Alessandria, V., Dolci, P., Gorra, R. & Rantsiou, K. Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. International Journal of Food Microbiology 167, 29–43, https://doi.org/10.1016/j.ijfoodmicro.2013.05.008 (2013).
Parker, M. et al. Naturally fermented milk from northern Senegal: Bacterial community composition and probiotic enrichment with Lactobacillus rhamnosus. Frontiers in Microbiology 9, 2218, https://doi.org/10.3389/fmicb.2018.02218 (2018).
Ezekiel, C. N. et al. High-throughput sequence analyses of bacterial communities and multi-mycotoxin profiling during processing of different formulations of Kunu, a traditional fermented beverage. Frontiers in Microbiology 9, 3282, https://doi.org/10.3389/fmicb.2018.03282 (2018).
Gabaza, M. et al. Lactococci dominate the bacterial communities of fermented maize, sorghum and millet slurries in Zimbabwe. International Journal of Food Microbiology 289, 77–87, https://doi.org/10.1016/j.ijfoodmicro.2018.09.001 (2019).
Keisam, S., Romi, W., Ahmed, G. & Jeyaram, K. Quantifying the biases in metagenome mining for realistic assessment of microbial ecology of naturally fermented foods. Scientific Reports 6, 34155, https://doi.org/10.1038/srep34155 (2016).
Escobar-Zepeda, A., Sanchez-Flores, A. & Quirasco Baruch, M. Metagenomic analysis of a Mexican ripened cheese reveals a unique complex microbiota. Food Microbiology 57, 116–127, https://doi.org/10.1016/j.fm.2016.02.004 (2016).
Turpin, W., Humblot, C. & Guyot, J.-P. Genetic screening of functional properties of lactic acid bacteria in a fermented pearl millet slurry and in the metagenome of fermented starchy foods. Applied and Environmental Microbiology 77, 8722–8734, https://doi.org/10.1128/AEM.05988-11 (2011).
Wei, Y.-j et al. High-throughput sequencing of microbial community diversity in soil, grapes, leaves, grape juice and wine of grapevine from China. PloS one 13, e0193097, https://doi.org/10.1371/journal.pone.0193097 (2018).
Houngbédji, M. et al. Occurrence of lactic acid bacteria and yeasts at species and strain level during spontaneous fermentation of mawè, a cereal dough produced in West Africa. Food Microbiology 76, 267–278, https://doi.org/10.1016/j.fm.2018.06.005 (2018).
Mukisa, I. M. et al. The dominant microbial community associated with fermentation of Obushera (sorghum and millet beverages) determined by culture-dependent and culture-independent methods. International Journal of Food Microbiology 160, 1–10, https://doi.org/10.1016/j.ijfoodmicro.2012.09.023 (2012).
Adesulu-Dahunsi, A., Sanni, A. & Jeyaram, K. Rapid differentiation among Lactobacillus, Pediococcus and Weissella species from some Nigerian indigenous fermented foods. LWT - Food Science and Technology 77, 39–44, https://doi.org/10.1016/j.lwt.2016.11.007 (2017).
Kostinek, M. et al. Characterisation and biochemical properties of predominant lactic acid bacteria from fermenting cassava for selection as starter cultures. International Journal of Food Microbiology 114, 342–351, https://doi.org/10.1016/j.ijfoodmicro.2006.09.029 (2007).
Ahaotu, N. N. et al. Influence of soy fortification on microbial diversity during cassava fermentation and subsequent physicochemical characteristics of garri. Food Microbiology 66, 165–172, https://doi.org/10.1016/j.fm.2017.04.019 (2017).
Amutha, R. & Gunasekaran, P. Production of ethanol from liquefied cassava starch using co-immobilized cells of Zymomonas mobilis and Saccharomyces diastaticus. Journal of Bioscience and Bioengineering 92, 560–564, https://doi.org/10.1016/S1389-1723(01)80316-9 (2001).
Amoa-Awua, W. K., Sampson, E. & Tano-Debrah, K. Growth of yeasts, lactic and acetic acid bacteria in palm wine during tapping and fermentation from felled oil palm (Elaeis guineensis) in Ghana. Journal of Applied Microbiology 102, 599–606, https://doi.org/10.1111/j.1365-2672.2006.03074.x (2007).
Ouoba, L. I. et al. The microbiology of Bandji, palm wine of Borassus akeassii from Burkina Faso: identification and genotypic diversity of yeasts, lactic acid and acetic acid bacteria. Journal of Applied Microbiology 113, 1428–1441, https://doi.org/10.1111/jam.12014 (2012).
Sahm, H., Bringer-Meyer, S. & Sprenger, G. A. The genus Zymomonas. The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses, 201–221 (2006).
Swings, J. & De Ley, J. The biology of Zymomonas. Bacteriological Reviews 41, 1 (1977).
Adedeji, B. S. et al. Bacterial species and mycotoxin contamination associated with locust bean, melon and their fermented products in south-western Nigeria. International Journal of Food Microbiology 258, 73–80, https://doi.org/10.1016/j.ijfoodmicro.2017.07.014 (2017).
Parkouda, C. et al. The microbiology of alkaline-fermentation of indigenous seeds used as food condiments in Africa and Asia. Critical Reviews in Microbiology 35, 139–156, https://doi.org/10.1080/10408410902793056 (2009).
Wang, X., Du, H., Zhang, Y. & Xu, Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Applied and Environmental Microbiology 84, e02369–17, https://doi.org/10.1128/aem.02369-17 (2017).
Grounta, A., Doulgeraki, A. I. & Panagou, E. Z. Quantification and characterization of microbial biofilm community attached on the surface of fermentation vessels used in green table olive processing. International Journal of Food Microbiology 203, 41–48, https://doi.org/10.1016/j.ijfoodmicro.2015.03.001 (2015).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America 108, 4516–4522, https://doi.org/10.1073/pnas.1000080107 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335, https://doi.org/10.1038/nmeth.f.303 (2010).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods 13, 581, https://doi.org/10.1038/nmeth.3869 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41, D590–D596, https://doi.org/10.1093/nar/gks1219 (2013).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience 2, 16, https://doi.org/10.1186/2047-217X-2-16 (2013).
Mandal, S. et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecology in Health and Disease 26, https://doi.org/10.3402/mehd.v26.27663 (2015).
Acknowledgements
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC) via a Global Challenge Research Fund Data and Resources award and Institute Strategic Programmes for Food Innovation and Health (BB/R012512/1) and its constituent project BBS/E/F/000PR10343 and Gut Microbes and Health (BB/R012490/1). M. D. was the beneficiary of a Clarin COFUND outgoing grants (ACA17–16) co-funded by the 7th Work Package of the European Union, Marie Curie Actions and the FICyT Foundation.
Author information
Authors and Affiliations
Contributions
M.D. performed the bioinformatic and statistical analysis and drafted the manuscript. M.D. and L.K. organized and supervised the experiments. N.A. developed the modified method. A.A., E.A., T.A., N.B.A., O.A., A.B.O., G.R.B., J.I., A.P., M.H., D.K., I.M.M. and G.M. provided samples and performed the DNA extractions. A.P., S.M.J. and W.O.C. performed laboratory fermentations. L.K., N.A., O.A., A.B.O., G.R.B., J.I., A.P., M.H., D.K., I.M.M., G.M., A.A., E.A., T.A., N.B.A, S.M.J., W.O.C., E.B., D.J.H., C.M., J.N., D.A.O., W.A.A., M.O., M.J.M., F.A.O. and A.N. critically revised, read and approved the final manuscript with final check by M.D.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diaz, M., Kellingray, L., Akinyemi, N. et al. Comparison of the microbial composition of African fermented foods using amplicon sequencing. Sci Rep 9, 13863 (2019). https://doi.org/10.1038/s41598-019-50190-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50190-4
This article is cited by
-
Microbial Diversity in African Foods and Beverages: A Systematic Assessment
Current Microbiology (2024)
-
Traditional Fermentation Affects the Nutraceutical Properties of Parkia biglobosa Seeds
Plant Foods for Human Nutrition (2023)
-
Development of mixed starter culture for the fermentation of Ethiopian honey wine, Tej
Scientific Reports (2022)
-
African fermented foods: overview, emerging benefits, and novel approaches to microbiome profiling
npj Science of Food (2022)
-
Pyrosequencing scrutiny of bacterial and fungal communities in two Sudanese sorghum-based fermented foods
Annals of Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
