
Abstract
Previous studies have demonstrated that patients with Crohn’s disease (CD) in remission do not exhibit an improvement in gut microbiota composition, which might trigger relapses. The present study investigated the dysbiosis and mucins production in CD patients during remission. We performed an analytical cross-sectional single center study, which recruited 18 CD patients and 18 healthy controls (CG) residing in the same home, meaning that all of the participants experienced the same environmental factors, with similar hygiene status, diet, pollution and other common lifestyle characteristics that may influence the composition of the gut microbiota. When compared to healthy controls, the CD patients exhibited lower microbial α-diversity (p = 0.047), a greater abundance of the Proteobacteria phylum (p = 0.037) and a reduction in the Deltaproteobacteria class (p = 0.0006). There was also a reduction in the Akkermansia (p = 0.002) and Oscillospira (p = 0.024) genera and in the proportion of the yeast Saccharomyces cerevisiae (p = 0.01). Additionally, CD patients in remission presented increased neutral (p = 0.001) and acid mucin (p = 0.002) concentrations. The reductions in the proportions of Oscollospira and Akkermansia genera, sulfate-reducing bacteria and Saccharomyces cerevisiae, observed in the CD group, may account for the increased mucins production observed in these patients.
Similar content being viewed by others


Introduction
Crohn’s disease (CD) is characterized by chronic inflammation of the gastrointestinal (GI) tract, and is associated with an increase in the production of inflammatory cytokines, such as interleukins (IL) and tumor necrosis factor alpha (TNF-α)1. The disease involves complex interactions among the host immune system, intestinal mucosa and gut microbiota. Interestingly, the gut microbiome has been receiving more attention, and is thought to play a more important role than previously thought2. For example, alterations in microbial composition of the intestines, or dysbiosis, and damage to the intestinal mucosal barrier can lead to frequent clinical manifestations, such as diarrhea and weight loss3,4. The severity of the intestinal inflammation has been associated with the largest number of colonization sites of gut microbiota with lipopolysaccharide (LPS) endotoxin activity3,4,5.
The colonic mucus barrier is considered the first line of defense against antigens and bacteria present in the intestinal lumen. It is composed of glycoproteins, trefoil factors and mucins6. In fact, it was previously reported that changes in the secretion patterns of mucins may be a primary event in CD or secondary to the observed inflammation7. Two types of mucins produced by the GI tract include, neutral and acid. In the upper GI tract, neutral mucins are predominantly secreted, while acid mucins prevail in the colon5. Additionally, studies have demonstrated that mucins content and expression are important for modulating short chain fatty acid (SCFA) synthesis, affecting the anti-inflammatory and immunological roles of these compounds3,4. Despite the apparent importance of mucins in the GI tract, the role of these proteins during the CD remission phase has not yet been deeply investigated.
Several studies, over the last 10 years, have shown that dysbiosis occurs in Inflammatory Bowel Diseases (IBD)8,9,10,11,12. More specifically, in IBD, the global composition of the gut microbiota contains specific pathogens that may be relevant to the etiology and pathogenesis of the disease. With regards to the gut microbiota of CD patients, it has been reported that there is an overall reduction in microbial diversity13, evidenced by alterations in the relative abundance of specific bacterial taxa6 and fungal communities, when compared to healthy individuals14.
Predominant changes described in literature regarding the gut microbiota composition of individuals with IBD and CD, include: alterations in the proportion of Bacteroides and Firmicutes, an increase in the percentage of Gammaproteobacteria15,16 and Enterobacteriales (i.e. Escherichia coli in CD with ileal disease), and a decrease in Clostridiales (i.e. Faecalibacterium prausnitzii)17. Furthermore, it has been observed that the amount of the fungi Candida albicans is increased in patients with CD18, whereas Saccharomyces cerevisiae is more abundant in non-inflamed mucosa18.
In CD, there are frequent relapses after periods of remission which are not entirely well understood. In fact, it is plausible that clinical remission is not accompanied by a reestablishment in the microbial balance of the GI tract, which might trigger future relapses. It is important to point out that there are no previous studies have evaluated of the gut microbiota and mucins production during CD remission. Therefore, the present study sought to compare and contrast the GI tract microbiomes and mucin expression patterns in CD patients during remission and healthy controls.
Results
Study population characteristics
Between June 2016 and May 2017, 36 subjects were studied, 18 patients with CD and 18 healthy controls. The subjects in the control group (CG) resided in the same house to the corresponding CD patient, sharing the same environmental status, like hygiene habitus, diet, pollution and other factors. The baseline characteristics of groups are show in detail in Table 1. According to Montreal classification, A2, L2 and B1 phenotypes were the commonest. Perianal involvement was present in 44.4%. and endoscopic remission were present in 72.3% of the CD patients. Regarding CG, one individual used probiotic for seven days, between 2–3 months prior to study.
Histological and histochemical analysis
Both mucins concentrations were increased in CD group. Mucins neutral was 39.62 CD vs 33.01 CG (p = 0.001) and mucin acid was 46.03 CD vs 39.62 CG (p = 0.002) (Fig. 1).

The histochemical image from neutral and acid mucins. Both mucins concentrations were increased in CD group (Mucins neutral CG vs CD p = 0.001; and mucin acid CG vs CD p = 0.002). (A) Neutral mucin normal from CG. (B) Neutral mucin of inactive CD. (C) Acid mucin normal from CG. (D) Acid mucin of inactive CD. (Mucins neutral CG vs CD p = 0.001; and mucin acid CG vs CD p = 0.002).
Microbial patterns in Crohn’s disease group and control group
To evaluate the gut microbiota diversity between CD and CG groups, PERMANOVA analysis was carried out. The results showed that the distribution and abundances of two groups (Supplementary Table 1S), are different (p < 0.01)
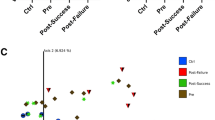
In addition, α diversity among all samples were compared, therefore CD showed more species diversity variability profile than CG, based on Shannon index (Fig. 2). In general, the individuals residing in different houses (CG) group have a range Shannon index (approximately 3.2 up to 4) while CD group showed a range Shannon index (approximately 1.8 up to 4.6) (Fig. 2). Patients with CD showed a lower microbial α diversity compared with the CG as reflected by Shannon indexes (p = 0.047). Regarding α diversity, between groups, it was observed that the control group (CG) have a very similar alpha diversity while CD group do not show this homogeneity (Fig. 2B). The analysis of the gut microbiota showed no significant change in the proportion of the two most abundant microbial phyla: Firmicutes and Bacteroidetes between CD and CG groups (Fig. 3). Contrarily, the CD group showed greater abundance of the phylum Proteobacteria (CG 3.5% ± 0.47 vs CD 7.4% ± 1.4; p = 0.037 unpaired test and p = 0.016 paired test) (Fig. 4A) and lower of the phylum Verrucomicrobia (CG 0.40% ± 0.19 vs CD 0.02% ± 0.02; p = 0.014 unpaired test and p = 0.003 paired test) (Fig. 4B).

Microbial patterns in Crohn’s disease group and control group. (A) PLS-DA scores plot for the genera shows a clear differentiation between CG and CD groups. (B) Comparison of α diversity by Shannon indexes between. Each color point corresponds α diversity of one subject. CD showed a lower microbial α diversity compared with the CG (p = 0.047).

The relative abundance of bacterial phyla. Comparison of metagenomics analysis of bacterial phyla from gut microbiota in CG (n = 18 subject) and CD (n = 18 subject). The data obtained from sequencing of the hyper-variable region (V3–V4) of the bacterial 16S rRNA gene.

Comparison (paired and unpaired) relative abundance of between the CG and CD groups. (A) Proteobaceria (CG 3.5% ± 0.47 vs CD 7.4% ± 1.4; *p = 0.037 unpaired test and p = 0.016 paired test). (B) Verrucomicrobia (CG 0.40% ± 0.19 vs CD 0.02% ± 0.02; *p = 0,014 unpaired test and p = 0.003 paired test). (C) Deltaproteobacteria (CG 1.23% ± 0.33 vs CD 0.4% ± 0.21; *p = 0.0006 unpaired test and p = 0.007 paired test). (D) Akkermansia (CG 0.68% ± 0.27 vs CD 0.04% ± 0.02; p = 0.002 unpaired test and p = 0.005 paired test). (E) Oscillospira (CG 1.20% ± 0.33 vs CD 0.55% ± 0.15; *p = 0.024 unpaired test and p = 0.025 paired test).
Deltaproteobacteria class, which contains most of the Sulfate-reducing bacteria, was reduced in the CD group. The relative abundance of Deltaproteobacteria class was high in CG group compared with CD (CG 1.23% ± 0.33 vs CD 0.4% ± 0.21; p = 0.0006 unpaired test and p = 0.007 paired test) (Fig. 4C). In addition, there was reduction of the beneficial genera Akkermansia (CG 0.68% ± 0.27 vs CD 0.04% ± 0.02; p = 0.002 unpaired test and p = 0.005 paired test) (Fig. 4D) and Oscillospira (CG 1.20% ± 0.33 vs CD 0.55% ± 0.15; p = 0.024 unpaired test and p = 0.025 paired test) (Fig. 4E) when compared to CG19. Although the analysis of the gut microbiota in this study showed a reduction in the genera Dialister and Bifidobacterium in the CD group (Supplementary Figure 1S), we did not find significant changes in the proportion of the species that have already been correlated with active CD like Bifidobacterium adolescents, Dialister invisus and Faecalibacterium prausnitzii20.
Saccharomyces cerevisiae quantification
Saccharomyces cerevisiae may exhibit a protective role in the inflammatory process. Our data showed that the proportion of Saccharomyces cerevisiae was significantly lower in CD group when compared with CG group (CG 0.02% ± 0.007 vs CD 0.005% ± 0.002 p = 0.01 unpaired test and p = 0.008 paired test) (Fig. 5) by quantification using qPCR.

Quantification of Saccharomyces cerevisiae by qPCR. Comparison of Saccharomyces cerevisiae quantification between CD group and CG group (CG 0.02% ± 0.007 vs CD 0.005% ± 0.002 *p = 0.01 unpaired test and p = 0.008 paired test).
The significant findings of this study are summarized in Fig. 6.

Graph abstract. CD patients, in remission disease, presented an increase of inflammation and intestinal permeability. In addition, CD patients also presented an increase in the amount of the neutral and acid mucins that is associated with a globally disturbed microbiota, with a reduction of the class Deltaproteobacteria, the genera Oscillospira and Akkermansia, and S. cerevisiae.
Discussion
The results of the present study demonstrated that the GI tract of CD patients in remission still display dysbiosis, that is characterized by reduced microorganism diversity. Additionally, when compared to the control group, which resided in the same home, there was an observed increase in the amount of Proteobacteria and a decrease in Verrucomicrobia. The CD group also presented increased mucins production.
According to Rothschild et al.21, individuals that experience the same environmental factors tend to have a similar gut microbiota compositions. Additionally, it is known that microbiota vary according to geography, cultural habits, age, and lifestyle factors that include diet, smoking, physical activity as well as others. Thus, in order to avoid potential biases when comparing CD patients to control group all of the study subjects must be exposed to the same environment8,22,23,24. In the present study, our control group consisted of people who were considered healthy, resided in the same home, had a similar hygiene status, consumed identical diets, and were, in general, exposed to equivalent environmental and other common lifestyle factors that could influence the composition of the gut microbiota.
The inflammation caused by CD, frequently results in damage to the intestinal wall7. The colonic mucus barrier provides a protective layer against potential antigens, pathogenic bacteria and metabolites produced by microorganisms present in the intestinal lumen. Additionally, the goblet cells produce mucins, which are connected by disulfide bridges and impede bacteria from penetrating the intestinal lumen6. It is important to mention that individuals with CD produce higher levels of mucins, that could attenuate the inflammatory response. Interestingly, the results herein showed that, even during remission CD patients also exhibit augmented mucin production, despite most of these patients not displaying any signs of inflammation, as revealed through colonoscopies.
The novelty of our study comes from integrating the gut microbiota composition and mucin production and evaluating how these changes influence the pathology of CD during remission. In this sense, it is tempting to speculate that the modulation of the gut microbiota may stimulate mucin production. In fact, we observed that the Oscillospira and Akkermansia genera were reduced in the CD group, when compared to the controls. Interestingly, the bacteria belonging to these two genera have been associated with the degradation of mucins and reductions in the proportions of these genera result in increased intestinal permeability25,26. Moreover, both neutral and acid mucin concentrations were increased in the CD group during remission.
With regards to other types of microorganisms, sulfate-reducing bacteria (SRB) obtain their energy through the reduction of sulfate to hydrogen sulfide (H2S). The SRB produced H2S can induce cytotoxity in human intestinal epithelial cells, promoting cellular damage and even cell death25,27. Most SRB are members of the Desulfovibrio genus in the Deltaproteobacteria class, and were found to be reduced in the CD group (p = 0.0166) of the present study.
The reduction in the proportion of the Oscollospira; Akkermansia genera and SRBs detected in the CD group during remission, may account for the increased mucin production, and may represent a compensatory mechanism for handling the CD-mediated inflammation. The CD group during remission presented a different gut microbiota when compared to both the healthy subjects (HS) and patients with active CD, apparently representing an intermediate microbiota composition.
In addition, a previous study reported that CD patients also have higher proportions of fungi in their intestines, with increased C. albicans and decreased S. cerevisiae content28. Herein we showed that CD patients in remission have reduced S. cerevisiae levels when compared with the control group. This result is similar to what has been observed in patients with active CD29. This is also relevant to inflammation, since a previous study with mice demonstrated that S. cerevisiae supplementation promoted a significant reduction in TNF-α expression and an increase in IL-10 production30. Through this anti-inflammatory effect S. cerevisiae can contribute to the regulation of the inflammatory process in patients with CD29, and its reduced presence may make these individuals more susceptible to relapses.
Our study has some limitations. First, it is a cross-sectional study and these results are observational. Secondly, we have not studied patients with active CD. Thirdly, the fungal analysis was limited to just one species (S. cerevisiae).
In conclusion, during remission CD patients present increased amounts of the neutral and acid mucins that are accompanied by a global dysbiosis. In particular, we identified a reduction in the Deltaproteobacteria class, Oscillospira and Akkermansia genera, and S. cerevisiae. While these results are encouraging future studies need to further investigate the relationship between the gut microbiota, and mucin production in CD. Furthermore, studies focusing on patients with active CD, CD in remission and healthy subjects need to be undertaken to further validate the results presented here.
Methods
Study design and population
We did an analytical cross-sectional single center study (IBD clinics of Campinas State University – Unicamp – Brazil), with CD patients and healthy controls. Inclusion criteria were CD patients with diagnosis confirmed by means of clinical, endoscopic and histological criteria and for control group adults residents in the same house, without previous history of chronic disease. Exclusion criteria included individuals that used antibiotics or probiotics during the previous 2 months. Disease activity in CD patients was assessed by the CDAI score31 and endoscopic findings. CDAI score under 150 were considered inactive disease (clinical remission).
Clinical data, disease classification, medications and comorbidities were collected on the same day of colonoscopy procedure. Fecal samples were collected by the subjects at home using Sarstedt tubes (Sarstedt, Nümbrecht, Germany) filled with a preservative buffer and brought to the IBD clinics within 24 hours after defecation. Stool samples were stored in frozen at -80 °C for microbiota analyses.
Metagenome profile
Total DNA of fecal samples was extracted with the Stool PSP Spin DNA kit (STRATEC Biomedical AG, Germany), an integrated system for collecting, transporting and storing feces samples and subsequent DNA purification.
To profiling microbiota composition, the hyper-variable region (V3-V4) of the bacterial 16S rRNA gene was amplified by following the Illumina 16S Metagenomic Sequencing Library Preparation guide32 which uses the following sequence: 338F - 5′-TCGTCGGCAGCGTCAG ATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG -3 and 785R - 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′ (2 × 300 bp paired‐end an insert size of ~550 bp).
Bioinformatic and statistical data analysis
The fastq sequences were analysed using DADA2 tool as describe by Callahan et al.33, that allows to recover single-nucleotide resolved Amplicon Sequence Variants (ASVs) from amplicon data. The default parameters were used to improve the overall quality of the sequences, the reads were filtered and trimmed using the “filterAndTrim” function implemented in DADA2 as described in https://benjjneb.github.io/dada2/tutorial.html. Low quality bases at the end of the reads were removed and the truncLen option was set to 280 and 220 to trim the forward and reverse fastq files respectively. Moreover, the sequences were also trimmed at the 5′ end using the trimLeft option set to 17 and 21 for the forward and reverse reads respectively. The taxonomic assignment was subsequently performed using the naïve Bayesian classifier method implemented in DADA2 using as reference the SILVA database. Finally, the final phylogenetic tree of the ASVs was obtained using the function AlignSeq implemented in DECIPHER34 R package to create the multiple sequence alignment and the Fast Tree program35.
Statistical analysis was performed on R (Version 3.4.4) using the following R packages: phyloseq (version 1.24.0) to facilitate the import, storage, analysis, and graphical display of microbiome census data36. Data were pre-processed filtering features with less than 10 read counts and present in less than 2 samples36. Vegan (version 2.4.2) for PERMANOVA analysis37. Shannon diversity indices and bar plot graphical were generated by using the R package ggplot2. The longitudinal microbiome studies was carried out from q2-longitudinal, a software plugin for the QIIME 2 microbiome analysis platform (https://qiime2.org)38.
Staining techniques for neutral and acid mucins
The biopsies of large bowel mucosa content of the neutral and acid mucins were determined individually to modify histochemical Periodic Acid Schiff (PAS)39 and Alcian Blue (AB) techniques. The slides were read under an ordinary optical microscope with a final magnification of 200×. The histological parameters were analyzed qualitatively and quantitatively by a pathologist with experience of diseases, of the digestive tract who was unaware of the origin of the material and the objectives of the study. The neutral mucins stained magenta, while the acid mucins stained blue.
Image processing, computer-assisted
The images selected were captured on a video camera that had been coupled to an optical microscope. These images were processes and analyzed using the NIS-Elements (Nikon Corporation. Instruments Company, Japan) software, installed in a computer with good image processing capacity. By means of colored histograms in RGB system (Red, Green, Blue) the software determined the color intensity in number of pixels in each field selected and transformed the final data into percentage expressions by analyzed fields. The final value in the segments with and without intestinal transit was the mean of the values found from evaluating three different fields.
Quantification of Saccharomyces cerevisiae by qPCR
For quantification of Saccharomyces cerevisiae the qPCR was performed using the primers and probe described by Mallant-Hent et al.40. Briefly, reaction was performed in 12 μl total volume containing 1x Universal Master Mix (Applied Biosystems), 150 nmol of both primers (5′-GAA ATG CCA CCG TGA ATG C and 5′-CTT TGG TGG TGA TCC TCT ATG ATT G), 100 nmol of the probe (FAM-TGG CAC CAT GAA CCC TAG CGT CGT T-TAMRA), and 120 ng of DNA extracted from stool samples. This reaction was performed on the QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems - Life Technologies Corp., USA). For the quantification, a standard curve was performed with Saccharomyces cerevisiae (strain was kindly donated by Laboratory of Enzymology and Molecular Biology of Microorganisms/State university of Campinas).
Statistical analyses
Sample size
based on pilot study data, the sample size calculation was done having as the main variable the relative contribution of Proteobacteria percentage. Considering the paired group (pilot with 6 subjects in each group, effect size = 0.90), assuming α in 5% and β in 5% (power 95%) 12 subjects were necessary in each group. The Software used for the calculation sample size was G*Power version 3.1.2. (Program written, concept and design by Franz, Universitat Kiel, Germany, freely available windows application software)41.
The inclusion of the CD patients versus control groups were expressed as medians and percentiles (interquartile range (IQR), 25–75%) for continuous variables and as frequency for categorical variables. For the qualitative variables, the Fischer exact test and the chi-square test (χ2) were select. The Mann-Whitney U-test (non-parametric distribution) was used to compare continuous variables between categories. The significance level adopted was 5% for all statistical tests (p-value < 0.05). Statistical analyses were used according to SSPS v.20.0 software (IBM Inc., Armonk, NY, USA).
Ethical statement
During routine visits, subjects who agreed in participating in the study signed up an informed consent form. All methods were performed in accordance with the relevant guidelines and regulations. The study was approved by Institutional Ethics Review Board at Unicamp, in Campinas, Brazil, under reference number 885.749/14.
References
Tsukahara, T. et al. Tumor Necrosis Factor α Decreases Glucagon-Like Peptide-2 Expression by Up-Regulating G-Protein–Coupled Receptor 120 in Crohn Disease. The American Journal of Pathology 185, 185–196, https://doi.org/10.1016/j.ajpath.2014.09.010 (2015).
Dovrolis, N. et al. Gut Microbial Signatures Underline Complicated Crohn’s Disease but Vary Between Cohorts; An In Silico Approach. Inflamm Bowel Dis 25, 217–225, https://doi.org/10.1093/ibd/izy328 (2019).
Shen, Z. et al. Update on intestinal microbiota in Crohn’s disease 2017: Mechanisms, clinical application, adverse reactions, and outlook: Intestinal microbiota in Crohn’s disease. Journal of Gastroenterology and Hepatology 32, 1804–1812, https://doi.org/10.1111/jgh.13861 (2017).
Rohr, M. et al. Inflammatory Diseases of the Gut. Journal of Medicinal Food 21, 113–126, https://doi.org/10.1089/jmf.2017.0138 (2018).
Magro, D. O. et al. Changes in serum levels of lipopolysaccharides and CD26 in patients with Crohn’s disease. Intestinal Research 15, 352–357, https://doi.org/10.5217/ir.2017.15.3.352 (2017).
Keli, E. et al. Diversion-related experimental colitis in rats. Diseases of the Colon and Rectum 40, 222–228 (1997).
Niv, Y. Mucin Genes Expression in the Intestine of Crohn’s Disease Patients: a Systematic Review and Meta-analysis. J Gastrointestin Liver Dis 25, 351–357, https://doi.org/10.15403/jgld.2014.1121.253.niv (2016).
Pascal, V. et al. A microbial signature for Crohn’s disease. Gut 66, 813–822, https://doi.org/10.1136/gutjnl-2016-313235 (2017).
Tamboli, C. P., Neut, C., Desreumaux, P. & Colombel, J. F. Dysbiosis in inflammatory bowel disease. Gut 53, 1–4 (2004).
Nagalingam, N. A. & Lynch, S. V. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 18, 968–984, https://doi.org/10.1002/ibd.21866 (2012).
Abraham, C. & Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140, 1729–1737, https://doi.org/10.1053/j.gastro.2011.02.012 (2011).
Saleh, M. & Elson, C. O. Experimental inflammatory bowel disease: insights into the host-microbiota dialog. Immunity 34, 293–302, https://doi.org/10.1016/j.immuni.2011.03.008 (2011).
Tedjo, D. I. et al. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Scientific Reports 6, https://doi.org/10.1038/srep35216 (2016).
Limon, J. J., Skalski, J. H. & Underhill, D. M. Commensal Fungi in Health and Disease. Cell Host Microbe 22, 156–165, https://doi.org/10.1016/j.chom.2017.07.002 (2017).
Kostic, A. D., Xavier, R. J. & Gevers, D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499, https://doi.org/10.1053/j.gastro.2014.02.009 (2014).
McIlroy, J., Ianiro, G., Mukhopadhya, I., Hansen, R. & Hold, G. L. Review article: the gut microbiome in inflammatory bowel disease-avenues for microbial management. Alimentary Pharmacology & Therapeutics 47, 26–42, https://doi.org/10.1111/apt.14384 (2018).
O’Brien, A. D. et al. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 226, 694–696 (1984).
Chiaro, T. R. et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Science Translational Medicine 9, eaaf9044, https://doi.org/10.1126/scitranslmed.aaf9044 (2017).
Alam, A. et al. The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat Microbiol 1, 15021, https://doi.org/10.1038/nmicrobiol.2015.21 (2016).
Alhagamhmad, M. H., Day, A. S., Lemberg, D. A. & Leach, S. T. An overview of the bacterial contribution to Crohn disease pathogenesis. J Med Microbiol 65, 1049–1059, https://doi.org/10.1099/jmm.0.000331 (2016).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215, https://doi.org/10.1038/nature25973 (2018).
Conlon, M. A. & Bird, A. R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 7, 17–44, https://doi.org/10.3390/nu7010017 (2014).
O’Toole, P. W. & Jeffery, I. B. Gut microbiota and aging. Science 350, 1214–1215, https://doi.org/10.1126/science.aac8469 (2015).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227, https://doi.org/10.1038/nature11053 (2012).
Szabo, C. et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 171, 2099–2122, https://doi.org/10.1111/bph.12369 (2014).
Konikoff, T. & Gophna, U. Oscillospira: a Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol 24, 523–524, https://doi.org/10.1016/j.tim.2016.02.015 (2016).
Coutinho, C. M. L. M. et al. Sulphate-reducing bacteria from ulcerative colitis patients induce apoptosis of gastrointestinal epithelial cells. Microb Pathog 112, 126–134, https://doi.org/10.1016/j.micpath.2017.09.054 (2017).
De Filippis, F., Vitaglione, P., Cuomo, R., Berni Canani, R. & Ercolini, D. Dietary Interventions to Modulate the Gut Microbiome-How Far Away Are We From Precision Medicine. Inflamm Bowel Dis 24, 2142–2154, https://doi.org/10.1093/ibd/izy080 (2018).
Sokol, H. et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048, https://doi.org/10.1136/gutjnl-2015-310746 (2017).
Jawhara, S. et al. Modulation of intestinal inflammation by yeasts and cell wall extracts: strain dependence and unexpected anti-inflammatory role of glucan fractions. PLoS One 7, e40648, https://doi.org/10.1371/journal.pone.0040648 (2012).
Best, W. R., Becktel, J. M., Singleton, J. W. & Kern, F. Jr. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. 70, 439–444 (1976).
Illumina 16S metagenomic sequencing library preparation (Illumina Technical Note 15044223). 2014).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583, https://doi.org/10.1038/nmeth.3869 (2016).
Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R (The R Journal, 2016).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490, https://doi.org/10.1371/journal.pone.0009490 (2010).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
vegan: Community Ecology Package. R package version 2.5-2 (https://CRAN.R-project.org/package=vegan, 2018).
Bokulich, N. A. et al. q2-longitudinal: Longitudinal and Paired-Sample Analyses of Microbiome Data. mSystems 3, https://doi.org/10.1128/mSystems.00219-18 (2018).
Gilks, C. B., Reid, P. E., Clement, P. B. & Owen, D. A. Simple procedure for assessing relative quantities of neutral and acidic sugars in mucin glycoproteins: its use in assessing cyclical changes in cervical mucins. J Clin Pathol 41, 1021–1024 (1988).
Mallant-Hent, R. C. et al. Correlation between Saccharomyces cerevisiae DNA in intestinal mucosal samples and anti-Saccharomyces cerevisiae antibodies in serum of patients with IBD. World Journal of Gastroenterology 12, 292–297 (2006).
Faul, F., Erdfelder, E., Lang, A.-G. & Buchner, A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods 39, 175–191 (2007).
Acknowledgements
We also acknowledge the financial, INCT (National Institute of Science and Technology for Diabetes and Obesity) 465693/2014-8 and CEPID/Fapesp 201307607-8.
Author information
Authors and Affiliations
Contributions
D.O.M., A.S., M.J.A.S. and C.S.R.C. designed the study; D.O.M., A.S., D.G.; L.V.P. and C.S.R.C. did data collection; A.S., D.G., F.M.G., S.H.M.S. and W.J.F.L. Microbiota profile; M.J.A.S., N.V. and S.T. critical revision of the manuscript; C.A.R.M. Mucin analysis. D.O.M., A.S. and W.J.F.L. statistical analysis; D.O.M., A.S., M.J.A.S. and C.S.R.C. drafted the article and all authors gave final revision and permission for publication.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that the research was conducted in the absence of any competing interests (financial and non-financial interests).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magro, D.O., Santos, A., Guadagnini, D. et al. Remission in Crohn’s disease is accompanied by alterations in the gut microbiota and mucins production. Sci Rep 9, 13263 (2019). https://doi.org/10.1038/s41598-019-49893-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49893-5
This article is cited by
-
The role of goblet cells in Crohn’ s disease
Cell & Bioscience (2024)
-
Epigenetics and the role of nutraceuticals in health and disease
Environmental Science and Pollution Research (2023)
-
Meta-analysis defines predominant shared microbial responses in various diseases and a specific inflammatory bowel disease signal
Genome Biology (2022)
-
Altered fecal microbiota composition in individuals who abuse methamphetamine
Scientific Reports (2021)
-
Kono-S anastomosis for Crohn’s disease: a systemic review, meta-analysis, and meta-regression
Surgery Today (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
