
Abstract
This study investigated the potential of single nucleotide polymorphisms as predictors of survival in two cohorts comprising 417 metastatic colorectal cancer (mCRC) patients treated with the FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) regimen. The rs4806668G > T of the ribosomal protein gene RPL28 was associated with shorter progression-free survival and overall survival by 5 and 9 months (P = 0.002), with hazard ratios of 3.36 (P < 0.001) and 3.07 (P = 0.002), respectively. The rs4806668T allele was associated with an increased RPL28 expression in transverse normal colon tissues (n = 246, P = 0.007). RPL28 expression was higher in colorectal tumors compared to paired normal tissues by up to 124% (P < 0.001) in three independent datasets. Metastatic cases with highest RPL28 tumor expression had a reduced survival in two datasets (n = 88, P = 0.009 and n = 56, P = 0.009). High RPL28 was further associated with changes in immunoglobulin and extracellular matrix pathways. Repression of RPL28 reduced proliferation by 1.4-fold to 5.6-fold (P < 0.05) in colon cancer HCT116 and HT-29 cells. Our findings suggest that the ribosomal RPL28 protein may influence mCRC outcome.
Similar content being viewed by others


Introduction
Metastatic colorectal cancer (mCRC) presents a 5-year relative survival just above 10%1. There are many treatment options for these patients including irinotecan-based chemotherapy. Particularly, the FOLFIRI regimen is composed of irinotecan (CPT-11) used in combination with 5-fluorouracil (5-FU) and folinic acid alone or with targeted therapies2. Both irinotecan and 5-FU are potent on actively replicating cancer cells. SN-38, the active metabolite of irinotecan, is an inhibitor of topoisomerase I (TOP1). It prevents DNA ligation by directly binding the TOP1-DNA complex, leading to replication arrest, double-strand breaks and cell death3. 5-FU is a pyrimidine analog that exerts its effect by inhibiting the thymidylate synthetase and DNA synthesis. 5-FU can also be incorporated into RNA during synthesis and interferes with protein synthesis4. The clinical response to FOLFIRI-based regimens is variable with dose-limiting toxicities occurring in a significant proportion of patients5,6. Several markers in pharmacokinetic pathways have been linked to severe toxicities. For instance, the UGT1A1*28 polymorphism was established as a predictive marker of severe neutropenia, explained by a decreased UGT1A1 expression. Because this gene encodes the main hepatic enzyme responsible for SN-38 inactivation and elimination, reduced expression leads to greater exposure to SN-38 and an increased risk of severe neutropenia7,8. By contrast, clinical genetic-based evidence to predict therapeutic response of mCRC patients is limited9. Several genes were found to be associated with response to irinotecan and 5-FU in vitro, but their potential value as markers of treatment response has not been addressed in patients. These genes are related to several pathways such as detoxification of reactive oxygen species, cellular responses to stress, DNA damage recognition or DNA repair. In this study, we tested the hypothesis that genetic variability in these genes may be associated with survival of mCRC patients treated with FOLFIRI-based regimen.
Results
The RPL28 rs4806668G > T variant is associated with survival of mCRC patients
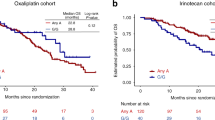
A screening of a total of 105 haplotype-tagging single nucleotide polymorphisms (htSNPs) covering the genetic variability of 17 candidate genes, which were previously associated with drug response in vitro or in patients (Additional file 1: Supplementary Tables 1 and 2) was performed in the discovery cohort comprising 167 mCRC Canadian patients. We identified 21 and 14 htSNPs associated (P < 0.1) with PFS and OS, respectively (Additional file 1: Supplementary Tables 3 and 4). In the replication cohort of 250 mCRC Italian patients, one of these htSNPs remained significantly associated (P < 0.05) with survival outcomes. The RPL28 rs4806668G > T variant was associated with a reduced PFS in the Canadian cohort (hazard ratio (HR) = 3.23, P = 0.013), the Italian cohort (HR = 3.28, P = 0.021) and in combined cohorts (HR = 3.36, P < 0.001) using a recessive model (Fig. 1a). This marker was also linked to shorter OS with HR of 3.09 (P = 0.032), 3.05 (P = 0.030) and 3.07 (P = 0.002) in the Canadian, Italian and combined cohorts, respectively, using a recessive model (Fig. 1b). Overall, there were 9 carriers of rs4806668TT genotype. Kaplan-Meier analysis revealed that median PFS was reduced by 5 months for homozygous TT carriers compared to heterozygotes or non-carriers (9 months versus 4 months, P = 0.002, Fig. 1c), and OS reduced by 9 months (23 months versus 14 months, P = 0.002, Fig. 1d).

RPL28 rs4806668G > T polymorphism is associated with survival of mCRC patients treated with FOLFIRI. (a,b) Cox proportional hazards models adjusted for age and co-treatment (Canadian cohort, n = 167) and for age (Italian cohort, n = 250) showed association of rs4806668G > T with progression-free survival (PFS) and overall survival (OS) using a recessive genetic model. Tumor site did not have a statistically significant association with mCRC outcome. (c,d) Univariate Kaplan-Meier survival curves for PFS and OS according to rs4806668G > T genotype in combined cohorts. The percentage survival according to genotypes is shown under the graphs. HRadj, adjusted hazard ratio; htSNPs, haplotype-tagging single nucleotide polymorphisms; CI, confidence interval.
The RPL28 rs4806668G > T variant affects RPL28 gene expression that is increased in tumor tissues
The RPL28 rs4806668G > T is located in the 5′-untranslated (5′-UTR) region of the RPL28 locus and is in strong linkage disequilibrium in the CEU population (European population, LD with r² > 0.80) with six other SNPs, located upstream of the 5′-UTR (Fig. 2a,b). Four of these SNPs are predicted to affect transcription factor binding (score 2a or 2b) according to RegulomeDB (Fig. 2b). Genotypic frequencies of rs4806668G > T was found to be highly variable among ethnic groups with 1–3% of homozygous TT in populations of Asian and European origins and reaching 58% in the African population (Fig. 2c). To explore the potential impact of rs4806668G > T and its linked SNPs on RPL28 gene expression, data from healthy donors of the GTEx project were used. An increased RPL28 expression was observed for carriers of the variant allele rs4806668T (P = 0.007) in the transverse colon, as well as for variant alleles of SNPs in linkage disequilibrium (LD) with the rs4806668G > T (Fig. 3a). By contrast, expression of the nearby gene TMEM238 was not affected. Data are summarized in Supplementary Table 5 (Additional file 1). In the TCGA cohort, RPL28 expression was significantly higher by 124% (P < 0.001) in colon tumors compared to paired normal tissues (Fig. 3b). In mCRC cases from dataset GSE49355, RPL28 expression was increased by 35% (P < 0.01) in primary tumors and by 34% (P < 0.01) in hepatic metastases compared to paired normal colon tissues (Fig. 3c). Similarly, in the GSE50760 dataset, RPL28 expression was higher by 27% (P < 0.05) in primary tumors and by 30% (P < 0.05) in liver metastatic tissues, compared to paired normal tissues (Fig. 3d).

Rs4806668 is located in the promoter region of the RPL28 locus and is linked to several other polymorphisms (SNPs). (a) Localization of the RPL28 rs4806668G > T variant and its associated SNPs in strong linkage disequilibrium (r² > 0.80 in the European population). (b) Position of the rs4806668G > T marker and its linked SNPs relative to the translation start site of RPL28. Scores are from RegulomeDB and represent the probability for a SNP to be functional. N/A, not available. (c) Frequencies of RPL28 rs4806668G > T among different ethnic groups (Ensembl GRCh38 release 91).

Relationship between RPL28 variants, tissue type and gene expression. (a) RPL28 rs4806668G > T and linked SNPs are associated with an increased gene expression in transverse colon tissues of healthy individuals from the GTEx cohort. (b) RPL28 expression is higher in primary tumor relative to paired normal colorectal tissues (n = 50 pairs) from the TCGA cohort. (c) RPL28 expression is higher in primary colorectal tumors and liver metastases relative to paired normal colorectal tissues (n = 10 pairs) from the GSE49355 dataset. (d) RPL28 expression is higher in primary colorectal tumors and liver metastases relative to paired normal colorectal tissues (n = 18 pairs) from the GSE50760 dataset. A.U., arbitrary units; FPKM, fragments per kilobase million; N, normal tissue; PT, primary tumor tissue; M, liver metastases. *P < 0.05; **P < 0.01; ***P < 0.001.
High RPL28 expression is associated with reduced survival of mCRC patients, altered gene expression and affects colon cancer cells growth and survival in vitro
Based on the dichotomization at the median expression levels, mCRC patients from the TCGA cohort with high tumor expression of RPL28 had a reduced survival compared to those with low levels (n = 88, P = 0.009; Fig. 4a). This observation was validated in mCRC cases from the dataset GSE17538 (n = 56, P = 0.009; Fig. 4b). Dichotomization at the optimal expression level cut-off also lead to significantly reduced survival for high RPL28 expression groups, in both TCGA (P = 0.004) and GSE17538 (P < 0.001) datasets (Additional file 1: Supplementary Fig. 1). A differential gene expression and pathway enrichment analysis further indicated that over 804 genes were significantly altered (FDR < 0.05) in association with RPL28 expression (high versus low, median separation) in cases of the TCGA cohort, including 285 down-regulated and 519 up-regulated genes (Additional file 2: Supplementary Table 6). From up-regulated genes, ten pathways related to immunoglobulins were enriched (adjusted P-value < 0.05) in cases with high RPL28, whereas five pathways related to extracellular matrix (ECM) or collagen were enriched from down-regulated genes (adjusted P-value < 0.05) (Fig. 4c). In these pathways, 17 genes comprised of mostly immunoglobulin genes were significantly upregulated (FDR < 0.05) in high RPL28 cases, and a total of 20 genes, mainly collagen genes, were significantly down regulated (FDR < 0.05) (Fig. 4c). In vitro investigation further indicated significantly reduced proliferation by 1.4-fold to 5.6-fold (P < 0.05) and reduced viable cell ratios by 15 to 75% (P < 0.05) associated with knockdown of RPL28 in HCT116 and HT-29 colon cell lines (Fig. 5).

RPL28 expression level in colorectal tumor tissues is associated with survival and changes in tumor transcriptome. (a) Kaplan-Meier curves for high and low RPL28 expression groups (median separation) of stage IV mCRC individuals from the TCGA cohort (n = 88). The percentage survival according to RPL28 expression group is shown under the graph. (b) Kaplan-Meier curves for high and low RPL28 expression groups (median separation) of stage IV mCRC individuals from the GSE17538 dataset (n = 56). The percentage survival according to RPL28 expression group is shown under the graph. The median and the optimal cut-off values of gene expression were highly similar (Additional file 1: Supplementary Fig. 1). (c) Significantly enriched pathways from differential gene expression in advanced stage IV mCRC cases with high RPL28 expression compared to those with low RPL28 expression from the TCGA cohort. Pathways in blue are enriched with down-regulated genes whereas those in red are enriched with up-regulated genes. Genes belonging to enriched pathways are shown on the right.

RPL28 expression levels influence proliferation of colorectal cancer cells. (a,b) Efficient knock-down of RPL28 protein expression in HCT116 and HT-29 exposed to specific siRNAs (siRPL28) relative to cells transfected with scrambled non-targeting (NT) negative control (siCTR). A representative cropped Western blot is shown for each cell line and a quantification of expression levels based on densitometry of blots from three independent experiments are shown. Full-length blots are shown in Additional file 1: Supplementary Fig. 2. (c–f) The knock-down of RPL28 impairs cell proliferation. Viable cells were monitored up to 96 hours after siRNA transfection in (c) HCT116 and (e) HT-29 (n = 3 in duplicate). Cell proliferation was measured by MTS assays 96 hours post-transfection in (d) HCT116 and (f) HT-29 (n ≥ 3 in triplicate). Data are presented as mean ± S.E.M. *P < 0.05; ***P < 0.001.
Discussion
We report that the germline variant rs4806668G > T in RPL28 and tumor expression of this gene are associated with survival of mCRC patients. The rs4806668TT genotype was associated with reduced PFS and OS in two independent cohorts of mCRC. This polymorphism was also associated with an increased expression of RPL28 in colon tissue of healthy donors, and its expression was further increased in tumor tissues compared to paired normal tissues. In two other independent cohorts of mCRC, higher RPL28 tumor expression level was associated with poor survival.
Our findings reveal that the RPL28 rs4806668G > T is associated with PFS and OS in mCRC cases treated with first-line FOLFIRI regimen. FOLFIRI is one of the most common chemotherapy backbones for mCRC first-line treatment, however; only few studies investigated the potential of germline polymorphisms to predict its efficacy in several cohorts. Xu et al. identified the combination of UGT1A1*28 and UGT1A1*6 as predictive of reduced overall survival in two cohorts of Asian mCRC cases receiving irinotecan-based regimen in second- and third-line treatment10. A meta-analysis in Caucasians concluded that UGT1A1*28 cannot be considered as a reliable predictor of survival in CRC patients treated with irinotecan-based regimen11. Most studies including a discovery and a replication set focused on genes involved in targeted therapies – mainly bevacizumab – administered along with FOLFIRI, and reported markers of survival in several genes including IL6, CD39, CXCR4 and MKNK112,13,14,15,16. RPL28 is one of the 79 members of the ribosomal proteins (RPs) family. These proteins are constitutive components of the large subunit (RPL) and small subunit (RPS) of the ribosome primarily responsible for protein synthesis, which is greatly perturbed in tumor cells17. In addition to their role in ribosomal biogenesis and protein production, RPs possess ribosome-independent functions, especially in tumorigenesis, immune signaling and development18. This was notably demonstrated in breast, ovarian and pancreatic cancers as well as in osteosarcoma19,20,21,22. In CRC, a study showed that a higher level of the ribosomal RPS7 protein was associated with improved PFS and OS in 92 stage IV mCRC patients23. Another study of 200 CRC patients revealed a reduced OS associated with a higher level of RPS15A protein24. RPL28 upregulation was associated with reduced OS after azacytidine treatment in patients with myelodysplasia and related neoplasms25. These studies pointed toward potential functions of ribosome-free RPs that might engage an oncogenic role or suppressing tumor cell proliferation, depending on the RPs proteins involved18. A recent study suggested that RPL28 was part of a hub of genes correlated to microsatellite instability status in CRC, indicating a potential role for this protein in defective DNA mismatch repair system associated with CRC26.
Various RPs are overexpressed in different cancer cells and are associated with the development and progression of malignant cancers. The RPL28 htSNP rs4806668G > T and its two tightly linked SNPs (rs3810168T > C and rs3745272C > G) were associated with an increased RPL28 expression in transverse colon of healthy individuals and were predicted to be functional based on RegulomeDB data, in support of a causal link with at least one of these SNPs. In the transverse colon, RPL28 expression level appeared similar for GT and TT carriers and higher than in GG carriers of rs4806668. However, an allele dose effect was observed for the linked rs3810168T > C, rs73617860T > G and rs73617855T > A, with higher expression for minor homozygous carriers, in line with the recessive model of the association in our cohorts. An upregulation of RPL28 was further noted in tumors compared to paired normal tissues suggesting an oncogenic role of this ribosomal protein in CRC. In support of this notion, repression of RPL28 was shown to significantly impair cell proliferation of HCT116 and HT-29 colon cancer cell models, suggesting that RPL28 higher expression may be associated to a more aggressive phenotype. Knockdown of other RPs such as RPL9 and RPS24 suppressed colon carcinoma cell growth in vitro and human CRC xenografts in nude mice27,28. Similarly, overexpression of RPS7 led to reduced proliferation of a panel of 4 CRC cell lines including HCT116 and HT-29, suggested to be mediated through repression of HIF-1α and glycolysis23.
Our analysis of transcriptomic changes associated with high RPL28 tumor expression in mCRC patients suggested an upregulation of several genes of immunoglobulins, proposing the involvement of the complement system. Although the complement is recognized as an important actor of the immune system that contributes to the destruction of cancer cells, recent data have indicated a tumor-promoting role29,30. Several studies support that malignant cells efficiently activate complement and that metastatic pathways are triggered by imbalanced complement activation and inflammation31. Recently, RPL28 expression was inversely associated to viral peptide presentation by the major histocompatibility complex class I, suggesting a potential role of this ribosomal protein in the immune system processing32. Immunosuppressive properties were proposed for RPS19, also increased in colon cancer cells and released from apoptotic tumor cells33. It was further suggested that RPS19 immunosuppressive role in cancer involved its interaction with the complement receptor C5aR1 (CD88), promoting tumor growth in a transgenic model of breast cancer34. Another study reported increased cell invasiveness in response to C5a-induced secretion of metalloproteinases from C5aR1-expressing cancer cells and degradation of extracellular matrix (ECM), known to contribute to neoplastic progression35,36. Indeed, we observed a down-expression of several ECM genes in high compared to low RPL28 expressing primary tumors. A role for ECM components such as collagen, fibronectin or matrix metalloproteinases was clearly established in cancer development and metastatic spread37. A down-regulation of genes related to ECM adhesion and remodeling was also observed between hepatic CRC metastases and primary colorectal tumors38. As a lower adhesion is thought to favor metastasis and dissemination, a down-regulation of ECM related genes in the high RPL28/low survival group of patients might be the sign of a higher susceptibility for cancer cells to spread more efficiently and quickly from the primary tumor39. Overall, it may be envisioned that the changes in gene expression associated with RPL28 activate cancer-related signaling pathways and exert tumor-promoting activities in mCRC patients.
A limitation of this study was that we did not apply correction for multiple testing in the polymorphism association study due to its exploratory nature. However, we studied two independent cohorts to corroborate initial findings, limiting the possibility of false-positive associations. We also included multiple datasets, including the large GTEx and TCGA cohorts, to study associations with gene expression. We acknowledge that our study design comprising only FOLFIRI-treated patients did not allow to clarify the predictive value of the positive marker and that further investigations are needed to explain the molecular mechanisms by which RPL28 affects survival and its possible interplay with ECM and immunoglobulin pathways. Given the high frequency of the rs4806668TT genotype in Africans, it would be of interest to evaluate further the association of the rs4806668G > T with clinical outcome in this population. Finally, the lack of data regarding microsatellite instability in our datasets precluded further analysis of its influence on mCRC outcome in relation to RPL28 status. One further limitation was our inability to account for treatment status in relation to RPL28 expression in the public datasets40.
In conclusion, we reported the germline variant RPL28 rs4806668G > T, associated with increased gene expression, as a novel marker of survival of mCRC patients treated with the FOLFIRI regimen. We further established that high RPL28 tumor expression is linked to a reduced survival of mCRC patients, possibly through an enhanced tumor growth and a remodelling of the expression of ECM and immunoglobulin pathways. This study highlights a potential role of the ribosomal protein RPL28 in colon tumorigenesis and as a possible prognostic survival marker of mCRC patients.
Methods
Characteristics of cohorts
Two independent cohorts of mCRC patients were included in the study. They were previously described in details, including the study eligibility criteria and genomic DNA isolation procedure41,42. Briefly, the discovery cohort consisted of 167 patients all treated with FOLFIRI-based regimen recruited in three centers of Eastern Canada from 2003 to 2012. These patients received FOLFIRI and 75 were also co-treated with bevacizumab or an experimental drug. The replication cohort enrolled 250 mCRC patients recruited in 13 centers of Northeastern Italy between 2002 and 2005 all treated with FOLFIRI. Clinical outcomes for this study were progression-free survival (PFS, defined as the time between the beginning of FOLFIRI treatment and the first evidence of disease progression, including death, or the last follow-up) and overall survival (OS, defined as the time between the beginning of FOLFIRI treatment and death from any cause). Characteristics of both cohorts are summarized in Table 1. Ethnicity was self-reported with the vast majority identified as Caucasian or White. All patients provided written informed consent and the research protocol was approved by the local ethics committees (CHU de Québec ethics committee for the Canadian cohort and Comitato Etico Indipendente - Centro di Riferimento Oncologico di Aviano for the Italian cohort). This study was conducted in accordance with the declaration of Helsinki.
Genetic analysis
Single Nucleotide Polymorphisms (SNPs) with a minor allele frequency higher than 5% were identified from the European (CEU) population of the International HapMap project for the selected candidate genes (Additional file 1: Supplementary Table 1). Haplotype-tagging SNPs (htSNPs) in high linkage disequilibrium (LD, r² > 0.80) in the CEU population were selected with Haploview v4.2 (Broad Institute, Cambridge, MA, United States)43. A complete list of htSNPs and their associated SNPs is provided as supplementary data (Additional file 1: Supplementary Table 2). All htSNPs were genotyped using the Sequenom iPLEX matrix-assisted laser desorption/ionization time-of-flight mass spectrometry technology (Sequenom, San Diego, CA, United States). Genotypic frequencies and LD data were retrieved through Ensembl GRCh38 release 91 - December 2017 (https://www.ensembl.org/index.html) for European (CEU), Chinese (CHB), Japanese (JPT) and African (YRI) populations. RegulomeDB v1.1 was used to investigate whether polymorphisms may have a functional impact44.
Statistical analysis
Hardy-Weinberg equilibrium was verified using PLINK v1.0745 and SNPs deviating from the equilibrium (P < 0.05) were excluded from further analysis. Associations between htSNPs and clinical outcomes were tested using Cox proportional hazards model adjusted for age and co-treatment in the Canadian and combined cohorts and for age in the Italian cohort. Additive, dominant and recessive models were fitted independently for each htSNP. Analyses were carried out using SAS version 9.4 (SAS Institute Inc., Cary, NC, United States) and R v3.2.2 Survival package. Genetic variants were initially tested in the Canadian cohort and those with a P-value < 0.1 were genotyped in the Italian cohort. A marker was considered validated when associated to survival using the same genetic model in both cohorts (P < 0.05). Kaplan-Meier curves and log-rank test P-values were estimated with GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, United States). Groups based on gene expression for Kaplan-Meier analysis were separated at the median or at the optimal cut-off values of gene expression, defined as the point with the most significant log-rank test split determined by Cutoff Finder version 2.146.
Expression data
Normalized gene expression and P-value data from the Genotype-Tissue Expression (GTEx) project (Analysis Release V7) were obtained through the GTEx portal (https://www.gtexportal.org/home/) on May 29, 2019. The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) was used to obtain data of Colon Adenocarcinoma (TCGA-COAD) and Rectum Adenocarcinoma (TCGA-READ) projects, accessed through the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/) on October 2, 2018. HTseq counts (raw counts) for stage IV TCGA-COAD and TCGA-READ individuals were retrieved with TCGAbiolinks package (version 2.9.5) in R and processed for TMM normalization with edgeR package (version 3.22.3). Differential gene expression between individuals with high and low RPL28 expression levels based on the median separation was then performed using edgeR package and exact test method. Ensembl gene ID were converted into gene symbols with g:Profiler47. Pathway enrichment analysis with significantly differentially expressed genes (FDR < 0.05) and log2(FC) lower than −1.2 or higher than 1.2 was performed using Reactome 2016 library in Enrichr48,49. Expression and clinical data publicly available for dataset GSE49355, GSE50760 and GSE17538 were obtained through GEO database with GEOquery package (version 2.48.0)38,50,51. Groups were compared using a two-tailed paired t-test or a one-way ANOVA followed by a post-hoc Dunnett’s test in GraphPad Prism 5 software.
In vitro experiments
HCT116 and HT-29 cell lines were obtained from the American Type Culture Collection (Rockville, MD, United States) and grown as recommended. Cells were passaged less than 15 times between thawing and completion of experiments. Cells were reverse-transfected in 96-well or 24-well plates with a final concentration of 20 nM siRNA (Dharmacon, Chicago, IL, USA) using DharmaFECT2 Transfection Reagent as per the manufacturer’s recommendations. Two siRNA targeting RPL28 (D-011145-02 or siRPL28-A and D-011145-03 or siRPL28-B) and a scrambled non-targeting negative control (D-001206-14 further referred as siCTR) were tested. Viable cells were counted at 24 h, 48 h, 72 h and 96 h post-transfection using trypan blue and an automated cell counter (Bio-Rad, Mississauga, ON, Canada). For each time point, fold over control was calculated by dividing the viable cell count by the viable cell count of siCTR at 24 h. Experiments were carried out three independent times in duplicate. Viable cell proliferation at 96 h was assessed by an MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI, United States) followed by measurement of absorbance at 490 nm. Viable cell proliferation was expressed as the ratio of absorbance for each condition over absorbance for siCTR. At least three independent experiments were performed in triplicate. At 96 h, conditions were compared using a one-way ANOVA followed by a post-hoc Dunnett’s test in GraphPad Prism 5 software. For protein expression, cells were washed with PBS 72 hours post-transfection and harvested in lysis buffer [0.05 M Tris-HCl pH 7.4, 0.15 M NaCl, 1% (w/v) Igepal CA-630 (Sigma-Aldrich), 1 mM dithiothreitol, and Complete protease inhibitor cocktail (Roche, Laval, QC, Canada)], lysed on a rotation unit for 30 min and cleared by centrifugation for 15 min at 13000 g. Cell lysates (5 μg) were resolved on a 12% polyacrylamide gel and transferred to a nitrocellulose membrane. RPL28 protein was detected using a rabbit polyclonal anti-RPL28 antibody (Proteintech, Group, 16649-1-AP, Rosemont, IL, United States) and GAPDH, used as a loading control, with a mouse monoclonal anti-GAPDH antibody (G8795, Sigma-Aldrich). RPL28 protein levels were determined by densitometry scanning of band intensity and normalized by the corresponding GAPDH band intensity on immunoblots using ImageJ (https://imagej.nih.gov/ij/). Experiments were carried out three independent times.
Data Availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
Brenner, H. & Chen, C. The colorectal cancer epidemic: challenges and opportunities for primary, secondary and tertiary prevention. Br J Cancer 119, 785–792 (2018).
Fujita, K., Kubota, Y., Ishida, H. & Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J Gastroenterol 21, 12234–12248 (2015).
Smith, N. F., Figg, W. D. & Sparreboom, A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro 20, 163–175 (2006).
Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3, 330–338 (2003).
Gupta, E. et al. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res 54, 3723–3725 (1994).
Hu, Z. Y., Yu, Q., Pei, Q. & Guo, C. Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res 16, 3832–3842 (2010).
Barbarino, J. M., Haidar, C. E., Klein, T. E. & Altman, R. B. PharmGKB summary: very important pharmacogene information for UGT1A1. Pharmacogenet Genomics 24, 177–183 (2014).
Beutler, E., Gelbart, T. & Demina, A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 95, 8170–8174 (1998).
Rodrigues, D., Longatto-Filho, A. & Martins, S. F. Predictive Biomarkers in Colorectal Cancer: From the Single Therapeutic Target to a Plethora of Options. Biomed Res Int 2016, 6896024 (2016).
Xu, C. et al. UGT1A1 gene polymorphism is associated with toxicity and clinical efficacy of irinotecan-based chemotherapy in patients with advanced colorectal cancer. Cancer Chemother Pharmacol 78, 119–130 (2016).
Liu, X., Cheng, D., Kuang, Q., Liu, G. & Xu, W. Association between UGT1A1*28 polymorphisms and clinical outcomes of irinotecan-based chemotherapies in colorectal cancer: a meta-analysis in Caucasians. PLoS One 8, e58489 (2013).
Matsusaka, S. et al. Prognostic Impact of IL6 Genetic Variants in Patients with Metastatic Colorectal Cancer Treated with Bevacizumab-Based Chemotherapy. Clin Cancer Res 22, 3218–3226 (2016).
Berger, M. D. et al. A Polymorphism within the Vitamin D Transporter Gene Predicts Outcome in Metastatic Colorectal Cancer Patients Treated with FOLFIRI/Bevacizumab or FOLFIRI/Cetuximab. Clin Cancer Res 24, 784–793 (2018).
Tokunaga, R. et al. Prognostic Effect of Adenosine-related Genetic Variants in Metastatic Colorectal Cancer Treated With Bevacizumab-based Chemotherapy. Clin Colorectal Cancer 18, e8–e19 (2019).
Berger, M. D. et al. Impact of genetic variations in the MAPK signaling pathway on outcome in metastatic colorectal cancer patients treated with first-line FOLFIRI and bevacizumab: data from FIRE-3 and TRIBE trials. Ann Oncol 28, 2780–2785 (2017).
Matsusaka, S. et al. CXCR4 polymorphism predicts progression-free survival in metastatic colorectal cancer patients treated with first-line bevacizumab-based chemotherapy. Pharmacogenomics J 17, 543–550 (2017).
Loreni, F., Mancino, M. & Biffo, S. Translation factors and ribosomal proteins control tumor onset and progression: how? Oncogene 33, 2145–2156 (2014).
Zhou, X., Liao, W. J., Liao, J. M., Liao, P. & Lu, H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol 7, 92–104 (2015).
Dave, B. et al. Role of RPL39 in Metaplastic Breast Cancer. J Natl Cancer Inst 109 (2017).
Wei, F. et al. Ribosomal protein L34 promotes the proliferation, invasion and metastasis of pancreatic cancer cells. Oncotarget 7, 85259–85272 (2016).
Luo, S. et al. Highly expressed ribosomal protein L34 indicates poor prognosis in osteosarcoma and its knockdown suppresses osteosarcoma proliferation probably through translational control. Sci Rep 6, 37690 (2016).
Shi, J. et al. Biological Function of Ribosomal Protein L10 on Cell Behavior in Human Epithelial Ovarian Cancer. J Cancer 9, 745–756 (2018).
Zhang, W. et al. RPS7 inhibits colorectal cancer growth via decreasing HIF-1alpha-mediated glycolysis. Oncotarget 7, 5800–5814 (2016).
Chen, J. et al. Ribosomal protein S15A promotes malignant transformation and predicts poor outcome in colorectal cancer through misregulation of p53 signaling pathway. Int J Oncol 48, 1628–1638 (2016).
Monika Belickova, M. et al. Up-regulation of ribosomal genes is associated with a poor response to azacitidine in myelodysplasia and related neoplasms. Int J Hematol 104, 566–573 (2016).
Yu, C. et al. Identification of key genes and pathways involved in microsatellite instability in colorectal cancer. Mol Med Rep 19, 2065–2076 (2019).
Baik, I. H. et al. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-kappaB signaling axis. Int J Oncol 49, 1953–1962 (2016).
Wang, Y. et al. RPS24 knockdown inhibits colorectal cancer cell migration and proliferation in vitro. Gene 571, 286–291 (2015).
Macor, P., Capolla, S. & Tedesco, F. Complement as a Biological Tool to Control Tumor Growth. Front Immunol 9, 2203 (2018).
Pio, R., Ajona, D. & Lambris, J. D. Complement inhibition in cancer therapy. Semin Immunol 25, 54–64 (2013).
Reis, E. S., Mastellos, D. C., Ricklin, D., Mantovani, A. & Lambris, J. D. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol 18, 5–18 (2018).
Wei, J. et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol Cell 73, 1162–1173 e1165 (2019).
Kondoh, N., Schweinfest, C. W., Henderson, K. W. & Papas, T. S. Differential expression of S19 ribosomal protein, laminin-binding protein, and human lymphocyte antigen class I messenger RNAs associated with colon carcinoma progression and differentiation. Cancer Res 52, 791–796 (1992).
Markiewski, M. M. et al. The Ribosomal Protein S19 Suppresses Antitumor Immune Responses via the Complement C5a Receptor 1. J Immunol 198, 2989–2999 (2017).
Pickup, M. W., Mouw, J. K. & Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15, 1243–1253 (2014).
Nitta, H. et al. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep 32, 1715–1719 (2014).
Crotti, S. et al. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J Cell Physiol 232, 967–975 (2017).
Del Rio, M. et al. Specific extracellular matrix remodeling signature of colon hepatic metastases. PLoS One 8, e74599 (2013).
Miyaki, M. et al. Increased cell-substratum adhesion, and decreased gelatinase secretion and cell growth, induced by E-cadherin transfection of human colon carcinoma cells. Oncogene 11, 2547–2552 (1995).
Kamal, Y., Schmit, S. L., Hoehn, H. J., Amos, C. I. & Frost, H. R. Transcriptomic differences between primary colorectal adenocarcinomas and distant metastases reveal metastatic colorectal cancer subtypes. Cancer Res (2019).
Levesque, E. et al. Refining the UGT1A haplotype associated with irinotecan-induced hematological toxicity in metastatic colorectal cancer patients treated with 5-fluorouracil/irinotecan-based regimens. J Pharmacol Exp Ther 345, 95–101 (2013).
Toffoli, G. et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol 24, 3061–3068 (2006).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007).
Budczies, J. et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 7, e51862 (2012).
Reimand, J. et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44, W83–89 (2016).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97 (2016).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Kim, S. K. et al. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol Oncol 8, 1653–1666 (2014).
Smith, J. J. et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 138, 958–968 (2010).
Acknowledgements
We would like to thank all study participants and clinical staff involved in the study. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 05/29/19. The results shown here are in part based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/) and we thank Julien Prunier for his help with the retrieving of these data. This work was supported by the Canadian Institutes of Health Research (CIHR, FRN-42392) and the Canada Research Chair in Pharmacogenomics (Tier I) to C.G. A.L. was supported by graduate scholarships from the “Fonds d’enseignement et de recherche” of the Faculty of pharmacy and “Fonds de Recherche du Québec - Santé”. E.L. received a CIHR clinical-scientist phase II award and Prostate Cancer Canada Rising Star Award (RS2013-55).
Author information
Authors and Affiliations
Contributions
A.L. performed the experiments; A.L., L.V. and D.S. performed statistical analysis; E.L., E.C., E.D.M., L.V., D.J., F.C., A.B., M.D.A., G.T. and C.G. participated in the recruitment of patients and obtaining DNA; A.L. and E.P.A. performed bioinformatics analysis; A.L., M.R. and C.G. wrote the manuscript; G.T. and C.G. designed the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Labriet, A., Lévesque, É., Cecchin, E. et al. Germline variability and tumor expression level of ribosomal protein gene RPL28 are associated with survival of metastatic colorectal cancer patients. Sci Rep 9, 13008 (2019). https://doi.org/10.1038/s41598-019-49477-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49477-3
This article is cited by
-
Using iTRAQ-Based Quantitative Proteomics Analysis to Identify Differentially Expressed Proteins Related to Larval Development of Portunus trituberculatus
Journal of Ocean University of China (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
