
Abstract
Hepatitis C virus (HCV) is characterized by high genetic variability, which is manifested both at the inter-host and intra-host levels. However, its role in the clinical course of infection is less obvious. The aim of the present study was to determine the genetic variability of HCV HVR1 (hypervariable region 1) of genotype 1b and 3 in plasma of blood donors in the early seronegative stage of infection (HCV-RNA+, anti-HCV−) and in samples from chronically infected patients using next-generation sequencing. Sequencing errors were corrected, and haplotypes inferred using the ShoRAH software. Genetic diversity parameters (intra-host number of variants, number of nucleotide substitutions and diversity per site) were assessed by DNA SP and MEGA. During the early infection, the number of variants were significantly lower in subjects infected with genotype 3 than with genotype 1b (p < 0.02). Similarly, intra-host number of variants, number of nucleotide substitutions and diversity per site were lower in genotype 3 chronic infection (p < 0.0005). In addition, early infection was characterized by significantly lower HVR1 variability values (p < 0.04) when compared to chronic infection for both genotypes. It seems that the observed differences in HVR1 variability represent an inherent property of particular viral genotypes.
Similar content being viewed by others


Introduction
Hepatitis C virus (HCV) is characterized by high genetic variability, resulting from both high error rate of viral RNA polymerase and immune pressure of the infected host. Consequently, HCV intra-host population displays high diversity reflected by the concomitant presence of multiple variants. Typically, after initial reduction upon the virus transmission to a new host, known as transmission bottleneck, HCV diversity expands as patient is progressing to chronicity1,2. Genetic variability of intra-host population, driven by immune pressure, allows the virus to escape from both humoral and cellular immune responses, gaining fitness advantage which may result in viral persistence3,4. Thus, the composition and dynamics of HCV population during the early phase of infection may determine its outcome5,6,7,8. HCV inter-patient variability is manifested mainly by the existence of different genotypes, classified from 1 to 79. Despite the fact that they represent the same species, their genome may differ by up to 31–33%10. Interestingly, the question whether HCV genotypes are characterized by different intra-host variability remains unresolved. This is partly due to technical limitations of methods utilized to analyze viral diversity, such as DNA heteroduplex gel shift method, single-strand conformation polymorphism or bulk clonal sequencing11,12,13. Their sensitivity for minor variant detection is low, with the variant frequency detection limit no better than 3–15% in most of the studies1,14. More recently, novel methods allowed for in-depth analysis of viral diversity such as single-genome or next-generation sequencing (NGS)2,15. They provide a tool for evaluation of a wide spectrum of genetic variants, but at a cost of greater sequencing error when compared to clonal sequencing16. Concomitantly, novel computational methods aimed at NGS error correction and variants reconstruction have been devised15,17.
The aim of the present study was to infer genetic variability of HVR1 (hypervariable region 1), a highly variable fragment of envelope 2 glycoprotein, which is a major target for specific humoral immune response18, in an unique collection of plasma samples from blood donors at an early seronegative stage of HCV genotype 1b or 3 infection (HCV-RNA positive, anti-HCV-negative) using a next-generation sequencing strategy. Samples from genotype matched, seropositive chronically infected patients were also analyzed. Our study showed that HCV genotype 3 displays significantly lower HVR1 genetic variability than genotype 1 during both early acute and chronic infection.
Results
In total, 388 202 HVR1 sequence reads were obtained, 4006.8 (mean per sample) for early infection seronegative subjects and 3595.7 (mean per sample) for chronically-infected patients. Similar number of reads (depth of sequencing) was obtained for genotype 1b and genotype 3 samples, both in early (4060.0 vs 3947.2) and chronic infection (3217.3 vs 4033.7).
The reads were further corrected for errors and reconstructed into haplotypes using ShoRAH. From one to 31 variants were identified per sample in early infection subjects (three to 31 for genotype 1b and one to 16 for genotype 3) and from five to 114 for chronically-infected patients (five to 114 for genotype 1b and five to 39 for genotype 3). After implementation of 0.5% cutoff to the reconstructed variants, from one to 14 variants were retained per sample in early infection subjects (one to 14 for genotype 1b and one to ten for genotype 3) and from one to 36 in chronically-infected patients (five to 36 for genotype 1b and one to 21 for genotype 3). Consequently, the mean number of nucleotide variants was significantly lower in genotype 3 infected subjects than in those infected with genotype 1b, both for early (3.5 vs 5.9; p = 0.002) and chronic (8.5 vs 19.5; p < 0.0001) infection (Fig. 1). Likewise, the number of intra-population nucleotide substitutions, when compared to consensus sequence, was significantly lower in case of genotype 3, both for early (7.0 vs 17.7 p = 0.02) and chronic (13.9 vs 43.8, p = 0.0004) infection. Similarly, the nucleotide diversity per site π was significantly lower in chronic genotype 3 than genotype 1b infection (0.0318 vs 0.0701; p < 0.0001). It was also lower in early genotype 3 infection, but this difference did not reach statistical significance (0.0184 vs 0.0439; p = 0.053). The summarized results are presented graphically on Fig. 1.

Nucleotide variability parameters of HVR1 HCV variants in genotype 1b and genotype 3 in early and chronic infection after application of 0.5% (dotted bars) and 1% (plain bars) frequency cutoff. (A) Number of HVR1 nucleotide variants, (B) number of nucleotide substitutions, (C) nucleotide diversity per site. The parameters shown in parts (B,C) were calculated with respect to consensus sequence (the most represented sequence within the sample). Numbers over the horizontal bars represent p-values.
When early and chronic infections were compared, the number of HCV variants, substitutions and the extent of nucleotide diversity were all significantly higher (p < 0.04) in the latter group for both genotype 1b and genotype 3.
In order to verify whether the observed effect is not caused by low frequency erroneous variants, we also re-assessed the variability parameters after increasing the cutoff to 1%. While the number of variants has decreased both for early and chronic infection, the statistical significance of difference between the two genotypes was still retained (p = 0.01 for early and p < 0.0001 for chronic infection, Fig. 1). Likewise, with the application of 1% cutoff, the number of substitutions and the extent of nucleotide diversity were still significantly lower in chronic genotype 3 compared to chronic genotype 1b infection (p = 0.0004 and p < 0.0001, respectively). In case of early genotype 3 infection these parameters were also lower, but not statistically significant (p = 0.08 and p = 0.16, respectively).
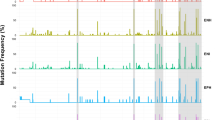
Comparison of intra-host populations revealed that genotype 3 HVR1 displayed higher frequency contribution of the dominant (i.e. the most frequent) variant than genotype 1b (mean 84.9% vs 72.4% for early infection, p = 0.004 and 62.2% vs 33.3% for chronic infection, p = 0.0003). The frequency distribution of major variants in genotype 1b and 3 infection using 0.5% cutoff is presented on Fig. 2.

Mean frequencies of two most prevalent HCV HVR1 variants in early infection with genotype 1b (A) and genotype 3 (B) and in chronic infection with genotype 1b (C) and genotype 3 (D). The frequency of each variant was determined by LStructure from ShoRAH suite and ranked from highest to lowest. Next, mean values were calculated for each of the two highest frequency variants in each patient (shown in blue and orange) and for the remaining ‘other’ variants (shown in gray). The applied frequency cutoff was 0.5%.
Discussion
Our study aimed to characterize in-depth intra-host genetic variability of HCV genotypes 1b and 3, both at the very early phase of infection prior to seroconversion, reflecting the natural complexity of the virus in the absence of humoral pressure and in a group of seropositive, treatment-naïve chronically infected patients. A unique opportunity to study very early virus-related events was provided by access to samples from HCV RNA positive, anti-HCV negative donors. Such samples represent very early infection stage and are detected at a frequency of 18.5 per 1 million donations19.
We found that during the early infection, the number of HVR1 variants was significantly lower in subjects infected with genotype 3 than in those infected with genotype 1b. Importantly, the effect observed was neither related to number of reads nor to viral titer. Furthermore, the results of analysis at two different cutoffs (0.5% and 1%) were concordant. The detected differences between HCV genotype 1b and 3 HVR1 molecular characteristics are striking and to our best knowledge no previous study addressed this issue.
To determine whether differences in HVR1 variability are affected by serologic response, a similar comparison was performed in a group of seropositive, chronically-infected patients. Since differences between the two genotypes were still maintained, it seems that genotype 3 HVR1 displays lower evolutionary dynamics than genotype 1b during the entire course of infection. Nevertheless, HVR1 variability parameters were significantly higher in chronic infection compared to early infection, which is concordant with earlier findings of bottleneck effect, due to selective transmission of only some HCV variants2,8,13. Subsequently, gradual expansion of the intra-host population occurs, which can be eventually disturbed by environmental pressures or fitness costs to the virus1,20.
Importantly, both during early and chronic genotype 3 infection, there was a different intra-host frequency structure of variants than during genotype 1b infection, with higher frequency contribution of a major (i.e. the most frequent) variant. This implies that genotype 3 has lower capacity to form minor frequency variants, which could provide fitness advantage under environmental pressures, such as exerted by the immune system or treatment. It can be speculated that this is the reason why genotype 3 infection is characterized by better response to immunomodulatory treatment with interferon alpha and ribavirin21. The differences in HVR1 genetic variability may also have clinical implications for the natural course of infection. Some observations imply that while genotype 3 is responsible for the majority of community-acquired HCV infections, is often eliminated at the early stage of infection and its potential to establish chronic infection is lower when compared to other HCV genotypes, particularly 1b19,22. In a study of Hwang et al., comprising 67 patients with acute post-transfusion hepatitis C, patients with genotype 1b were more likely to develop chronic hepatitis than patients infected with other genotypes (89.7% vs. 64.3%; p = 0.019)23. Similarly, Amoroso et al. showed, that the rate of progression to chronicity was 92% in patients exposed to HCV genotype 1b compared to 50% in patients exposed to genotype 36. These data provide evidence that HCV genotype may play an important role in the clinical course of infection following exposure to HCV. Lower genetic variability of genotype 3 observed by us during the seronegative early phase of infection may be related to higher chance of spontaneous elimination of the virus, since the immune system may be more efficient at targeting “uniform” viral population3.
While NGS is an extremely useful tool for the evaluation of genetic diversity, it is highly prone to sequencing errors. Because of that, we conducted an extensive assessment of the inherent raw sequencing error of the whole procedure using HVR1 plasmid insert as template24, applied subsequent error correction and frequency cutoff. Importantly, the differences were observed at two different cutoffs (i.e. 0.5% and 1%).
It should be noted that, while we found evident differences between the genotype 1b and genotype 3 variability, these observations are limited to immunologically important, yet single and small genomic region and may not extend to other hypervariable regions of HCV such as HVR2 and HVR325. Notably, novel hypervariable regions HVR495 and HVR575 have been found to be present in genotype 3, but not in genotype 1a, 1b, 2a or 6a genomes26. Since these are also subject to positive immune selection, further research is needed to determine their variability characteristics in early and chronic infection.
In conclusion, the HVR1 genetic variability parameters were lower in patients infected with genotype 3 than in patients infected with genotype 1b. Considering that these differences could be observed both in acute and chronic infection, it seems that the phenomenon is not due to immune pressure, but may rather represent an inherent property of particular viral genotypes.
Methods
Patients
The study encompassed plasma samples from 60 blood donors who were found to be HCV RNA positive and anti-HCV negative at the time of blood donation. Since their samples collected 3–6 months earlier were negative for the presence of HCV RNA and anti-HCV, they were assumed to be recently infected. Importantly, the time from the last blood donation, in which HCV-RNA and anti-HCV were negative, was identical for genotype 1b and genotype 3-infected donors (median 3 months, p = 0.838). These samples have been collected in the years 1999–2013 as part of a routine blood donor screening conducted by Polish blood donation centers and the Institute of Hematology.
Plasma samples from 41 HCV RNA–positive, anti-HCV-positive treatment-naïve chronic hepatitis C patients infected with genotype 1b or 3, presenting for treatment at the Hepatology Outpatient Clinic of the Warsaw Hospital for Infectious Diseases were also analyzed. Although the exact duration of infection was not known, the time from the initial diagnosis of HCV infection was similar (mean 3.9 years for genotype 1b vs 3.0 years for genotype 3).
The study protocol followed ethical guidelines of the 2013 Declaration of Helsinki and was approved by the Bioethical Committee of the Medical University of Warsaw (Approval Number KB/17/2013 and KB/107/2010). All patients provided written informed consent. Some clinical and virological characteristics of the study subjects are presented in Table 1.
HVR1 amplification
HVR1 amplification was performed as described previously27. In brief, total RNA was extracted from 250 μl of plasma by a modified guanidinium thiocyanate-phenol/chlorophorm method using Trizol (Life Technologies, Carlsbad, CA, USA). RNA was subjected to reverse transcription at 37 °C for 30 minutes using AccuScript High Fidelity Reverse Transcriptase (Agilent Technologies, Santa Clara, CA, USA) and random hexamers (Life Technologies). A gene fragment encompassing HVR1 was amplified in two-step PCR using FastStart High Fidelity Taq DNA Polymerase (Roche, Indianapolis, IN, USA) using target-specific primers (Table 2). Additionally, primers employed in the second round PCR contained tags recognized by GS Junior sequencing platform and standard 10-nucleotide multiplex identifiers27.
Pyrosequencing
The amount of DNA equivalent to 3 × 107 amplicons was subjected to emulsion PCR using GS Junior Titanium emPCR Lib-A Kit (454 Life Sciences, Branford, CT, USA). Pyrosequencing was carried out according to the manufacturer’s protocol for amplicons using GS Junior System (454 Life Sciences).
Data analysis
Sequencing errors (mismatches, insertions and deletions) were corrected and haplotypes inferred using the program diri_sampler from the ShoRAH software (https://www1.ethz.ch/bsse/cbg/software/shorah)17. It employs a local reconstruction model based on probabilistic clustering algorithm to correct errors28. This is accomplished by clustering all reads that overlap the same region of the genome of length approximately equal to the read length. The consensus sequence of each cluster represents the original haplotype from which the erroneous reads are obtained. ShoRAH does not feature a generative model for indels correction, but rather relies on the sequence alignment. Each read is aligned to the reference, and from this set of pairwise alignments a multiple one is built. If a given indel is present in a sufficient number of reads (i.e. if the model identifies it as real variation and not as a sequencing error), it will be present in one or more haplotypes.
The raw error rate of amplification and pyrosequencing of a clone-derived amplicon was shown in our previous study to be 1.5% (measured as the frequency of the most prevalent erroneous variant)24. However, error correction by ShoRAH reduced this error to 0.5% and this cutoff was thus implemented in our analysis. Nevertheless, in order to verify our results, the analysis was repeated using 1% cutoff.
Haplotypes that had posterior probability >95% and were represented by at least 10 reads were extracted with LStructure (https://github.com/ozagordi/LocalVariants/blob/master/src/LStructure.py). Subsequently, haplotypes were aligned to the HCV reference sequences (GenBank accession number AJ406073 for genotype 1b and EF442261.1 for genotype 3) and trimmed to be equal in length29. Genetic diversity parameters (i.e. intra-host number of variants, number of nucleotide substitutions and nucleotide diversity per site π) were assessed by DNA SP version 5 (http://www.ub.edu/dnasp/)30 and MEGA 5.029.
Statistical analysis
Significance of differences in HVR1 sequence diversity and clinical data (ALT levels, viral load) were tested using the Unpaired T test or Mann-Whitney U test where appropriate based on the Kolmogorov-Smirnov normality test. Differences in sex distribution between HCV genotypes were tested using Fisher’s exact test. All p-values were two tailed and considered significant when <0.05.
Data Availability
All the HVR1 sequences are publically available at the Zenodo repository (https://doi.org/10.5281/zenodo.3266795).
References
Bull, R. A. et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS pathogens 7, e1002243, https://doi.org/10.1371/journal.ppat.1002243 (2011).
Li, H. et al. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS pathogens 8, e1002880, https://doi.org/10.1371/journal.ppat.1002880 (2012).
Domingo, E., Sheldon, J. & Perales, C. Viral quasispecies evolution. Microbiology and molecular biology reviews: MMBR 76, 159–216, https://doi.org/10.1128/MMBR.05023-11 (2012).
Farci, P. & Purcell, R. H. Clinical significance of hepatitis C virus genotypes and quasispecies. Semin Liver Dis 20, 103–126 (2000).
Farci, P. et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288, 339–344 (2000).
Amoroso, P. et al. Correlation between virus genotype and chronicity rate in acute hepatitis C. J Hepatol 28, 939–944 (1998).
Duffy, S., Shackelton, L. A. & Holmes, E. C. Rates of evolutionary change in viruses: patterns and determinants. Nature reviews. Genetics 9, 267–276, https://doi.org/10.1038/nrg2323 (2008).
Haaland, R. E. et al. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS pathogens 5, e1000274, https://doi.org/10.1371/journal.ppat.1000274 (2009).
Smith, D. B. et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59, 318–327, https://doi.org/10.1002/hep.26744 (2014).
Okamoto, H. et al. Full-length sequence of a hepatitis C virus genome having poor homology to reported isolates: comparative study of four distinct genotypes. Virology 188, 331–341 (1992).
Liu, Z. et al. Accurate representation of the hepatitis C virus quasispecies in 5.2-kilobase amplicons. Journal of clinical microbiology 42, 4223–4229, https://doi.org/10.1128/JCM.42.9.4223-4229.2004 (2004).
Herring, B. L., Tsui, R., Peddada, L., Busch, M. & Delwart, E. L. Wide range of quasispecies diversity during primary hepatitis C virus infection. J Virol 79, 4340–4346, https://doi.org/10.1128/JVI.79.7.4340-4346.2005 (2005).
Laskus, T. et al. Analysis of hepatitis C virus quasispecies transmission and evolution in patients infected through blood transfusion. Gastroenterology 127, 764–776 (2004).
Keele, B. F. et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA 105, 7552–7557, https://doi.org/10.1073/pnas.0802203105 (2008).
Prosperi, M. C. et al. The threshold bootstrap clustering: a new approach to find families or transmission clusters within molecular quasispecies. PLoS One 5, e13619, https://doi.org/10.1371/journal.pone.0013619 (2010).
Barzon, L. et al. Next-generation sequencing technologies in diagnostic virology. J Clin Virol 58, 346–350, https://doi.org/10.1016/j.jcv.2013.03.003 (2013).
Zagordi, O., Bhattacharya, A., Eriksson, N. & Beerenwinkel, N. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics 12, 119, https://doi.org/10.1186/1471-2105-12-119 (2011).
Kurosaki, M., Enomoto, N., Marumo, F. & Sato, C. Rapid sequence variation of the hypervariable region of hepatitis C virus during the course of chronic infection. Hepatology 18, 1293–1299 (1993).
Brojer, E. et al. The hepatitis C virus genotype and subtype frequency in hepatitis C virus RNA-positive, hepatitis C virus antibody-negative blood donors identified in the nucleic acid test screening program in Poland. Transfusion 44, 1706–1710, https://doi.org/10.1111/j.0041-1132.2004.04156.x (2004).
Li, H. & Roossinck, M. J. Genetic bottlenecks reduce population variation in an experimental RNA virus population. J Virol 78, 10582–10587, https://doi.org/10.1128/JVI.78.19.10582-10587.2004 (2004).
Trepo, C. Genotype and viral load as prognostic indicators in the treatment of hepatitis C. J Viral Hepat 7, 250–257 (2000).
Flisiak, R. et al. Prevalence of HCV genotypes in Poland - the EpiTer study. Clinical and experimental hepatology 2, 144–148, https://doi.org/10.5114/ceh.2016.63871 (2016).
Hwang, S. J. et al. Hepatitis C viral genotype influences the clinical outcome of patients with acute posttransfusion hepatitis C. J Med Virol 65, 505–509 (2001).
Cortes, K. C. et al. Deep sequencing of hepatitis C virus hypervariable region 1 reveals no correlation between genetic heterogeneity and antiviral treatment outcome. BMC Infect Dis 14, 389, https://doi.org/10.1186/1471-2334-14-389 (2014).
Troesch, M. et al. Study of a novel hypervariable region in hepatitis C virus (HCV) E2 envelope glycoprotein. Virology 352, 357–367, https://doi.org/10.1016/j.virol.2006.05.015 (2006).
Humphreys, I. et al. Full-length characterization of hepatitis C virus subtype 3a reveals novel hypervariable regions under positive selection during acute infection. J Virol 83, 11456–11466, https://doi.org/10.1128/JVI.00884-09 (2009).
Caraballo Cortes, K. et al. Ultradeep pyrosequencing of hepatitis C virus hypervariable region 1 in quasispecies analysis. Biomed Res Int 2013, 626083, https://doi.org/10.1155/2013/626083 (2013).
Zagordi, O., Klein, R., Daumer, M. & Beerenwinkel, N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic acids research 38, 7400–7409, https://doi.org/10.1093/nar/gkq655 (2010).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution 28, 2731–2739, https://doi.org/10.1093/molbev/msr121 (2011).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452, https://doi.org/10.1093/bioinformatics/btp187 (2009).
Acknowledgements
This study was funded by grant UMO-2015/19/D/NZ6/01303 and UMO2013/11/B/NZ6/01679 from the National Science Center, Poland.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.C.C., T.L., P.G. Data curation: K.P., D.K.R. Data analysis: M.J., K.C.C., O.Z., S.O. Funding acquisition: K.C.C. Investigation: K.C.C., M.J., A.P. Methodology: M.J., R.P., I.B.O. Resources: P.G., H.B. Software: K.P., K.C.C. Supervision: T.L., R.P., P.G. Manuscript review: M.J., K.P., P.G., D.K.R., O.Z., H.B., S.O., A.P., I.B.O., R.P., T.L., K.C.C.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janiak, M., Perlejewski, K., Grabarczyk, P. et al. Hepatitis C virus (HCV) genotype 1b displays higher genetic variability of hypervariable region 1 (HVR1) than genotype 3. Sci Rep 9, 12846 (2019). https://doi.org/10.1038/s41598-019-49258-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49258-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
