
Abstract
The lung is a complex ecosystem of host cells and microbes often disrupted in pathological conditions. Although bacteria have been hypothesized as agents of carcinogenesis, little is known about microbiota profile of the most prevalent cancer subtypes: adenocarcinoma (ADC) and squamous cell carcinoma (SCC). To characterize lung cancer (LC) microbiota a first a screening was performed through a pooled sequencing approach of 16S ribosomal RNA gene (V3-V6) using a total of 103 bronchoalveaolar lavage fluid samples. Then, identified taxa were used to inspect 1009 cases from The Cancer Genome Atlas and to annotate tumor unmapped RNAseq reads. Microbial diversity was analyzed per cancer subtype, history of cigarette smoking and airflow obstruction, among other clinical data. We show that LC microbiota is enriched in Proteobacteria and more diverse in SCC than ADC, particularly in males and heavier smokers. High frequencies of Proteobacteria were found to discriminate a major cluster, further subdivided into well-defined communities’ associated with either ADC or SCC. Here, a SCC subcluster differing from other cases by a worse survival was correlated with several Enterobacteriaceae. Overall, this study provides first evidence for a correlation between lung microbiota and cancer subtype and for its influence on patient life expectancy.
Similar content being viewed by others

Introduction
Lung Cancer (LC) is the most common and lethal cancer worldwide with a reported incidence of 11.6%, a mortality rate of 18.4% and according to recent estimates - 2.1 million new cases1. From a histological point of view, non-small cell lung cancer (NSCLC) is the most prevalent type, which can be further subdivided into two major subtypes: adenocarcinoma (ADC) and squamous cell carcinoma (SCC). To date, diverse environmental factors besides cigarette smoking, like biomass burning, indoor and outdoor pollutants, are suggested to play a role in LC pathogenesis, as well as in chronic obstructive pulmonary disease (COPD)2. This later illness, characterized as the persistence of airflow limitation in result of bronchitis and/or emphysema, is also recognized as a critical comorbidity in LC, always associated to a worse prognosis3. Moreover, a mechanistic link between COPD and LC has been proposed partially due to the findings of chronic inflammation and its repeated cycles of injury and repair, which in COPD are known to promote tumorigenesis and malignant transformation4.
Notably and similarly to the model established for Helicobacter pylori in gastric cancer, Mycobacterium tuberculosis has been hypothesized as a LC agent, once it induces inflammatory markers and causes significant alterations in lung tissues5,6. However, this association to tuberculosis is hard to disentangle because of its co-occurrence with other risk factors such as smoking, emphysema and bronchitis5.
In this area of knowledge, the impact of lung microbiota, or the bacteria communities inhabiting the lung, has been more extensively studied in COPD than in LC. This is likely to result from COPD patients often suffering from acute exacerbations, which are considered to be of infectious nature and caused by bacteria and/or virus7.
Until now, studies aiming to characterize COPD and LC microbiota used different biological specimens, including sputum, bronchoalveaolar lavage fluid (BALF) and lung tissue. And, whereas in COPD, samples were collected during distinct phases of the disease (stable or exacerbation); in LC, those were gathered in some instances from both tumor and non-tumor regions. Moreover, although previous works were mainly based in the screening of 16S ribosomal RNA (16S rRNA) gene, their experimental approaches concerning sample size, hypervariable regions covered and sequencing technologies employed are quite diverse and consequently, their findings are not always consensual8,9,10,11,12,13,14,15,16,17,18,19,20.
Nonetheless, most studies seem to agree in the common core microbiota of both healthy and diseased subjects dominated by Firmicutes, Bacteriodetes, Proteobacteria and Actinobacteria phyla, and by Streptococcus, Haemophilus, Veilonella, Pseudomonas and Prevotella genera8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23. Interestingly, in COPD, microbiota tends to be relatively stable over time and affected during exacerbations, when it shifts toward Proteobacteria manly due to a Moraxella increase and a Streptococcus reduction8,22. Conversely, in LC, a Firmicutes switch was suggested in result of augmented Streptococcus, Granulicatella, Veillonella and Megasphaera prevalence14,16,21,24. Critical changes in microbiota composition were suggested to occur along with airway disease progression12,17,18,22. However, the use of certain bacterial taxa as potential biomarkers for improved patient stratification or even as treatable traits, is far from being a reality.
Here, we explore an association of lung microbiota with cancer, while addressing also the impact of COPD co-morbidity. Briefly, we performed a microbiota profiling in a small set of Portuguese samples, used later to guide the characterization of bacterial communities in an extended cohort of ADC and SCC cases from The Cancer Genome Atlas (TCGA) Research Network25. This allowed us to detect significant differences in microbiota diversity of cancer subtypes, as well as to identify among SCC a well-defined community (Enterobacteriaceae) connected to a worse patient survival mainly due to non-cancer complications. Furthermore, we identified Achromobacter as a gram-negative bacterium linked with both SCC and COPD.
Materials and Methods
Sample collection
BALF was collected from subjects undergoing bronchoscopy for evaluation of lung disease at three hospitals in Portugal: Centro Hospitalar São João (CHSJ), in Porto; Centro Hospitalar Baixo Vouga (CHBV), in Aveiro; and Hospital Pulido Valente - Centro Hospitalar Lisboa Norte (CHLN), in Lisbon. Informed consent was obtained for all participants and sample collection for Human Research was approved by hospital ethical committees: Comissão de Ética para a Saúde (CES) – CHSJ, Comissão de Ética – CHBV and Comissão de Ética para a Saúde (CES) – CHLN. The study was conducted in accordance with ethical guidelines and regulations for Human research and with Helsinki Declaration. Sample collection was targeted toward affected lung segments and done by bronchoscope wedging into subsegmental lung regions. In this study, we used only bronchoscope working channel washes, which were done twice with a minimum volume of 15 mL (0.9% saline solution). Samples were then stratified in LC (N = 49) or controls (N = 54) based in positive or negative cytology results (Supplementary Table 1). However, in a follow-up analysis carried out up to 2 years after BALF collection, two cases were found to be false positives and another four initially classified as negative, over time progressed to LC26. The pooled sample strategy prevented the reallocation of these cases to controls and vice-versa.
Lung microbiota 16S rRNA screening
DNA extraction from BALF (200 µL) was performed using DNA Mini kit (Qiagen) according to manufacture instructions for capturing bacterial DNA in body fluids. Two 16S rRNA fragments spanning hypervariable regions V3-V4 and V4-V6 were amplified using universal primers (Supplementary Table 2). Pooled samples containing PCR products (~200 ng/sample) were generated for LC (N = 49) and controls (N = 54) and processed as previously described27. Briefly, two libraries were constructed according to Ion Xpress™ Plus Fragment Library Kit protocol (Life Technologies) and ran in an Ion PGM™ System - 316™ chip (Life Technologies). The tools USEARCH, UCHIME, QIIME and Greengenes were used in the analysis of operational taxonomic units (≥97% nucleotide sequence identity cut-off) as previously described27.
TCGA dataset
Raw RNAseq reads from tumors and clinical data files corresponding to 515 ADC and 501 SCC cases from TCGA, were downloaded from Genomic Data Commons (GDC) Data Portal (https://gdc.cancer.gov/). To perform a quantitative analysis of lung microbiota we used reads not aligning with human reference sequence (unmapped reads) as input for QmihR28. This pipeline combines Trimmomatic, Bowtie2, and RSEM for a probabilistic inference of bacterial taxa abundances28. To instruct bacterial sequence surveys, we first defined a microbiota reference panel based in previous evidence of lung colonization in healthy and diseased patients. Exactly, we considered a total of 567 bacterial taxa according to the data available in: 1) the Human Microbiome Project (HMP) – airways; 2) specialized literature; and 3) our own 16S rRNA study; for which whole genome sequences could be collected from https://www.ncbi.nlm.nih.gov/genome/microbes/. Upon quality control, relative abundances of 112 genera were obtained for 509 ADC and 500 SCC cases. These samples were then stratified according to several variables including ancestry (European or African), gender, age at diagnosis (≤65 or >65), anatomic positioning (Upper or Lower lung), localization in lung parenchyma (Peripheral and Central Lung) and pathological tumor stage (Stages I, II and III + IV). In addition, post-bronchodilator forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) ratio were used to determine the presence (FEV1/FVC < 0.70) or absence of airflow obstruction3. Smoking history in pack per years (PPY) was considered using a first 20 PPY subdivision (data not shown). Given that most cases largely surpassed this value, a naïve 45 PPY split was used instead based on its proximity to average values (all cases 48; ADC 42 and SCC 53 PPY). Several patient follow-up variables were also considered in this study to evaluate clinical significance of collected data. These included vital (dead or alive) and cancer (tumor free or with tumor) status, days to death and primary therapy outcome.
Statistical analysis
Statistical analysis of microbiota diversity was performed in R studio (https://www.rstudio.com/; version 1.1.383) using phyloseq29. Alpha diversity was evaluated through inverse Simpson and Shannon indexes. Beta diversity, which integrates phylogenetic relationships of bacteria was calculated by weighted UniFrac. Distances matrixes were used in Principal coordinates analysis (PCoA) and in hierarchical clustering (complete linkage) of TCGA samples. The linear discriminant analysis (LDA) effect size (LEfSe) algorithm30 was used to detect taxa with differential abundances between TCGA cases. Survival analyses and log rank tests for pairwise comparisons of different case sets were carried out through the Cohort Comparison tool available at GDC Data Portal.
Results
Characterization of lung bacterial communities
In our pooled sequencing approach (16S rRNA V3–V6) of cases and controls using BALF samples (Supplementary Table 1), we were able to identify a total of 11 phyla and 54 genera with relative frequencies above 0.1% (Supplementary Tables 3, 4). The prevailing phyla in our dataset were Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes (Fig. 1a), as it could be expected from previous studies8,9,10,11,12,13,14,15,16,17,18,19,20,21,22. Some variation in phyla proportions were observed between pools for Proteobacteria, (38.7% in cases vs 49.2% in controls; Z-score P = 0.284) and Actinobacteria (16.5% in cases vs 8.0% in controls; Z-score P = 0.187). Among genera, Haemophilus (Proteobacteria); Streptococcus and Veillonella, (Firmicutes); Corynebacterium and Actinomyces (Actinobacteria) and Prevotella (Bacteroidetes) were the most common in the two pools. Again, our screening agreed with former reports of lung microbiota8,9,10,11,12,13,14,15,16,17,18,19,20,21,22, disclosing only non-significant changes between pools in relative abundances of Haemophilus (29.5% in cases vs 37.5% in controls; Z-score P = 0. 390) and Corynebacterium (8.2% in cases vs 1.3% in controls; Z-score P = 0.095). Overall, bacterial communities were both fairly diverse as indicated by Shannon index at genus level (2.69 in cases vs 2.53 in controls). However, for the inverse Simpson, LC cases were found to be considerably more diverse than controls (7.98 vs 5.74, respectively).

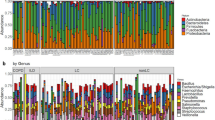
Characterization of lung cancer (LC) microbiota. (a) Relative abundance at the phylum and genus levels for Portuguese cases and controls. This data corresponds to the DNA pooling of 16S rRNA amplicons (V3-V6) of 49 and 54 individual samples, respectively. (b) Relative abundance of genera identified among tumor sections of 1009 lung cancer cases from The Cancer Genome Atlas (TCGA). ADC: adenocarcinoma (N = 509). SCC: Squamous cell carcinoma (N = 500).
In the TCGA series (tumor RNAseq) several discrepancies to former estimated proportions were observed. With an overall frequency of 59.4%, Proteobacteria surpassed by far the remaining phyla Actinobacteria, Firmicutes, and Bacteroidetes (23.4%, 12.0% and 4.4% respectively; Supplementary Table 5). Conversely, top genera comprised Pseudomonas, Acinetobacter, Sphingomonas, Methylobacterium and Enterobacter (Proteobacteria); Propionibacterium, Corynebacterium, and Micrococcus (Actinobacteria); and Streptococcus and Staphylococcus (Firmicutes); all with average prevalence above 3%. Taxa previously identified as abundant in 16S rRNA pooled sequencing were confirmed to be present in TCGA but at variable frequencies, ranging from 3.7% for Corynebacterium to 0.3% for Haemophilus (Fig. 1b; Supplementary Table 6). Concerning microbiota diversity, TCGA cases showed similar values to our BALF samples for Shannon index (2.86 ± 0.43) and higher statistics for inverse Simpson (11.54 ± 5.06).
Differentiation of ADC and SCC subtypes
The availability of clinical parameters for TCGA cases allowed an in-depth analysis of possible factors affecting lung microbiota. Aside from some variability in genera abundance per cancer subtype (Fig. 1b), we found that SCC tends to show higher diversity than ADC as indicated by inverse Simpson (Fig. 2a). This difference seems to be correlated with European ancestry, male gender, heavy smoking (PPY >45) and older ages at the time of diagnosis (>65 years) (Fig. 2b). Yet, we could not detect any effect on microbiota diversity when considering tumor localization, upper or lower lung and central or peripheral parenchyma (Fig. 2b).

Alpha diversity of lung microbiota from lung cancer (LC) cases of The Cancer Genome Atlas (TCGA). (a) Inverse Simpson and Shannon indexes for LC cases grouped by histological subtype. (b) Inverse Simpson index of LC subtypes grouped according to different clinical variables available at TCGA database (ancestry, gender, age at diagnosis, smoking history, lung region and lung parenchyma). Welch’s t-test was used to access statistical significance of pairwise comparisons (*P-value < 0.05; **P-value < 0.01, ***P-value < 0.001). ADC: adenocarcinoma. SCC: Squamous cell carcinoma.
We also used TCGA dataset to test the hypothesis of a loss of microbiota diversity along with disease progression. Nevertheless, no differences were observed across pairwise comparisons of cancer stages (I, II and III + IV) or COPD presence and absence. Stratification by cancer subtype did not alter the results (Supplementary Fig. 1).
To evaluate the similarity of microbiota profiles weighted UniFrac distances were calculated. As plotted in PCoA (Fig. 3), LC communities are quite variable across samples and overlap between ADC and SCC. Nonetheless, some individual profiles appear to cluster and to be correlated with either ADC (upper right quadrant) or SCC (lower right quadrant). No other variable was found to aggregate groups of cases (results not shown).

Beta diversity of lung microbiota from lung cancer (LC) cases of The Cancer Genome Atlas (TCGA). The Principal Coordinates Analysis (PCoA) plot was generated using weighted UniFrac distances. ADC: adenocarcinoma. SCC: Squamous cell carcinoma.
Then, to gain a better insight into LC microbiota profiles we performed a hierarchical clustering of TCGA samples, followed by a graphic display of their lung communities at two taxonomic ranks - phylum and genus (Fig. 4a–d). In the phylum analysis some heterogeneity among cases could be already witnessed, as indicated by upper tree clusters and subclusters. Higher abundances of Proteobacteria were connected with a first cluster (p_C1) and a second one (p_C2) could be divided into three major subclusters. Basically, these diverged in the relative proportions of common phyla: p_C2s1 was dominated by Actinobacteria; p_C2s2 had balanced frequencies of Proteobacteria, Actinobacteria, Firmicutes and Bacteriodetes and p_C2s3 was Firmicutes enriched.

Bacterial communities of lung cancer (LC) cases from The Cancer Genome Atlas (TCGA). (a) Hierarchical clustering of LC cases built using weighted UniFrac distances and complete linkage method. Major clusters identified are indicated in the tree. (b) Schematic representation of LC subtype and gender variables available for all samples. (c) Phyla relative abundance per each sample. (d) Genera relative abundance per each sample. Less frequent taxa are grouped in a single category and labeled as “Others”. ADC: adenocarcinoma. SCC: Squamous cell carcinoma.
The analysis at the genus level depicted a larger complexity of lung microbiota, where cases often clustered into small groups showing long terminal branches. Notably, inside p_C1 two clusters contrasted with the remaining tree by their shorter terminal branches (p_C1/g_C1 and p_C1/g_C2; Fig. 4a). Whereas p_C1/g_C1 could be characterized by a community composed by prevalent genera such as Sphingomonas, Brevundimonas, Acinetobacter and Methylobacterium; p_C1/g_C2 could be defined by Enterobacter, Morganella, Kluyvera and Capnocytophaga. Interestingly, p_C1/g_C1 contained only ADC cases (N = 32), all of them located in the upper right quadrant of PCoA plot (Supplementary Fig. 2). In contrast, p_C1/g_C2 included essentially SCC cases (89 in 94), this turn corresponding to the plot lower right quadrant (Supplementary Fig. 2). In the p_C2s2 a single cluster emerged as less heterogeneous (p_C2s2/g_C1), in this instance, this could be correlated with high frequencies of Propionibacterium and mostly linked to ADC cases (32 in 42).
In the LEfSE analysis of ADC and SCC cases a total of 37 genera were detected to display contrasting correlations between LC subtypes (Fig. 5a). Precisely, for ADC the genera with higher LDA scores (>3.5) and extreme P-values (P < 5 × 10−8) were Acinetobacter, Propionibacterium, Phenylobacterium, Brevundimonas and Staphylococcus. On the other hand, for SCC the genera fitting such requirements were Enterobacter, Serratia, Kluyvera, Morganella, Achromobacter, Capnocytophaga and Klebsiella (Supplementary Table 7). Interestingly, most of these bacteria could be correlated with previously identified clusters - p_C1/g_C1, p_C1/g_C2 and p_C2s2/g_C1. A similar approach was used to address a possible contribution of bacteria into COPD, as a common co-morbidity to both ADC and SCC (Fig. 5b). Among the 12 taxa identified Achromobacter was the one most strongly correlated with airflow obstruction (LDA scores >3.5 and P-values ≤0.010; Supplementary Table 7).

Microbial differentiation of The Cancer Genome Atlas (TCGA) cases according to disease status and linear discriminant analysis (LDA) effect size (LEfSe) algorithm. (a) lung cancer subtype. (b) COPD co-morbidity presence or absence. ADC: adenocarcinoma (N = 509). SCC: Squamous cell carcinoma (N = 500). W/o COPD: cases without COPD (N = 164); W/COPD: cases with COPD (N = 110).
Bacterial communities as prognostic biomarkers
To investigate if our findings could have clinical potential, especially in a better stratification of LC cases, we compared the survival curves of previously identified clusters. In a first step, no significant differences were detected between p_C1 (Proteobacteria dominated) and p_C2s2 (intermediate abundances of common phyla), not even when separated by ADC and SCC. Yet, in the global comparison, and among SCC cases p_C1 cluster appears to be associated with a slower decay of survival rates (P = 0.076 and P = 0.089, respectively; Fig. 6). Several analyses were performed also in ADC, for p_C1/g_C1 (Acinetobacter/Brevundimonas community), p_C2s2/g_C1 (Propionibacterium community) and other cases, but all failed to reach compelling results possibly due to their low sample sizes. On the other hand, among SCC the p_C1/g_C2 cluster (Enterobacter community) was found to departure from the remaining p_C1 cases with a worse survival (P = 0.011), closer to the one observed in p_C2s2 cluster. Still, the strongest divergence in SCC survival rates was observed for non-p_C1/g_C2 (Proteobacteria dominated without Enterobacter community) and p_C2s2 (P = 0.006; Fig. 6). Interestingly, p_C1/g_C2 was the cluster associated with the highest mortality rate during follow-up (approximately 5000 days’ maximum for SCC and 7500 days for ADC), and the one correlated with an increased number of deaths in tumor free patients (Table 1). Conversely, non-p_C1/g_C2 was shown to display a reduced mortality even when the disease progressed negatively after primary therapy and a period of complete remission (Table 1). The small size of p_C1/g_C1 and p_C2s2/g_C1 clusters prevented in ADC an accurate evaluation of the impact of these well-defined communities in patient outcome (Table 1).

Survival plots of The Cancer Genome Atlas (TCGA) cases organized according to different microbial clusters identified and lung cancer subtypes. ADC: adenocarcinoma. SCC: Squamous cell carcinoma.
Discussion
In this study, we performed a characterization of LC microbiota using two distinct datasets and methodological approaches: a pooled sequencing of 16S rRNA in BALF samples from Portuguese cases and controls; and a surveying of bacterial RNAseq reads made available through TCGA, for which tumor sections of hundreds of patients were collected. The main advantage of the first approach was to provide a preliminary and raw overview of lung microbiota at very low cost. However, this pooled approach has several limitations starting by its inability to address inter-individual variability and to accurately pinpoint bacterial communities to each individual. Another weakness is related to sample heterogeneity, which contains several cases lacking a complete histological classification and controls that include manly subjects with other pathologies. At last, surveying 16S rRNA can be considered also a shortcoming, once it is expected to introduce some ascertainment bias in taxa identification. This caveat is attributed, on one hand, to the differential annealing affinities of universal primers used in 16S rRNA amplification, and on the other, to the distinct resolving power of covered hypervariable regions27,31. Conversely, in the second approach, we could benefit from a larger cohort of ADC and SCC cases, for which detected RNAseq reads are more likely to represent an accurate composition of lung microbiota. The unique disadvantage of this strategy is that in order to maximize efficiency in mapping bacterial reads, we provided a database of reference genomes28. This was built using taxa identified in our 16S rRNA survey, combined with HMP data and published elsewhere8,9,10,11,12,13,14,15,16,17,18,19,20,21,22.
Overall, Proteobacteria emerged as the predominant LC phylum, a trend captured also in a large sample of cancer patients for which non-malignant tissue sections were collected17. Until now, increased frequencies of Proteobacteria were mostly correlated with asthma, COPD exacerbations and advanced COPD stages8,9,32,33. However, given current findings a Proteobacteria enrichment could also be a feature of cancerous lungs. In-depth surveys uncovered distinctive scores of Proteobacteria defining two major clusters: a first one truly dominated by Proteobacteria (p_C1); and another one displaying intermediate frequencies of Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes (p_C2s2). Altogether, these results are suggestive of substructure in lung microbiota that as far as we could investigate is not correlated with cancer subtype, or any other evaluated clinical variable.
Also, in a global perspective, Pseudomonas, Streptococcus, Staphylococus, Veillonella and Moraxella were identified among the top rank bacteria of cancer cases fitting the so-called lung core microbiome8,9,10,11,12,13,14,15,16,17,18,19,20,21,22. Nevertheless, individual distributions showed a different scenario, in which cases are generally quite divergent in their microbial composition. The exceptions to this rule are three specific communities displaying remarkable links to LC subtypes: Brevundimonas/Acinetobacter (p_C1/g_C1) for ADC; Enterobacter (p_C1/g_C2) for SCC, and Propionibacterium (p_C2/g_C1) for ADC.
Most impressively, p_C1/g_C2 cluster could be related with an overall poor survival if comparing SCC cases within p_C1 group. Furthermore, p_C1/g_C2 was characterized by several Enterobacteriaceae (Enterobacter, Morganella, Serratia, Klebesiela and Kluyvera), a taxon with recognized pathogenic potential causing airway infections in COPD34, colonizing bronchi of LC patients35 and underlying nosocomial infections with resistance to antimicrobial molecules36. This cluster comprised also Achromobacter, another multidrug resistant microbe previously found in airway infections of cystic fibrosis patients and among subjects with solid malignancies37.
Moreover, as gram-negative bacteria, Enterobacteriaceae and Achromobacter synthetize lipopolysaccharides capable of stimulating host inflammatory responses. In this respect, Enterobacteriaceae overgrowth has been described as a key event of gut dysbiosis in obesity, Crohn’s disease and colorectal cancer37,38. In asthma and cystic fibrosis there is also a growing body of evidence for a negative effect of Enterobacteriaceae39,40. Therefore, a contribution of p_C1/g_C2 community to an enhanced pro-inflammatory cancer microenvironment seems like a plausible hypothesis, once it is reported to foster tumorigenesis and promote lung cells malignant transformation4,41. This rational is supported by reports for a worse LC prognosis when patient bronchi are colonized by Enterobacteriaceae35.
Nonetheless, p_C1/g_C2 cluster was also linked with an increased mortality in absence of any tumor, which suggests an increased risk of this group to other non-cancer complications. Indeed, several studies already reported diverse pulmonary infections, septicemia and enhanced death rates after cancer resection and chemotherapy in LC subjects carrying potential pathogenic microorganisms in their airways35,42,43. Although we were unable to rigorously address the impact of p_C1/g_C2 community in the health status of a group of individuals probably debilitated by advanced age, co-morbidities (e.g. COPD, cardiovascular disease, diabetes, etc.) and inclusively cancer treatment, our findings advocate for a differentiated medical intervention in these patients, namely in the selection of antimicrobial therapies.
Still, the overall survival of p_C1/g_C2 does not differ from p_C2s2 cluster, which advances Actinobacteria, Firmicutes and/or Bacteriodetes as additional risk factors in SCC possibly through similar mechanisms of cancer progression. On the contrary, non-p_C1/g_C2 group appears to somehow tolerate new tumor events, which leads to a decoupling of its survival curve from the rapid decline of p_C1/g_C2, and from the continuous decrease of p_C2s2.
Less remarkable findings were obtained for Brevundimonas/Acinetobacter (p_C1/g_C1) community, which did not diverge from other cases concerning ADC outcomes. However, if compared with SCC p_C1/g_C2 and p_C2s2 groups those disclosed a trend for extended survival or lifetime prognosis.
We also provide additional pieces of information for a burden of microbiota in pulmonary disease. In the TCGA cohort, cases showing COPD co-morbidity were linked to Achromobacter, thus highlighting a negative effect of this taxon in SCC and in airflow obstruction. However, COPD was not associated to any identified bacterial community in particular, nor to augmented prevalence of Moraxella and Haemophilus genera as frequently observed among these patients8,12,22. Noticeably, only 27% of cases were presented with lung function tests, and for those with COPD (11%) their majority was classified as mild or moderate cases (FEV > 50%; GOLD 1-2 stages). These features could explain the similar alpha diversity scores obtained for patients with or without COPD, once previous reports for a microbiota loss were centered in advanced cases22 (for opposite results19,40). More striking is the lack of differentiation across LC stages if taking into account former reports for a loss of diversity between tumor and non-tumor samples17,18. Nonetheless, there is no proof so far for a gradual decline of microbiota with cancer progression. In contrary, previous works uncovered higher alpha diversity values in advanced cases (IIIB and IV) than in earlier disease stages18. In our study, SCC cases were in average more diverse than ADC, a result that can be related to a heavier smoking load of these patients since cigarette and air pollutants were found to positively affect airway microbiota richness17,23. However, other unknown factors must play a role in LC bacterial colonization to explain the significant differences observed between SCC and ADC in heavy smokers. Moreover and consistently with a recent study carried out in mild to moderate COPD12, we also reported similar diversity levels across distinct lung regions (bronchial and peripheral lung), contradicting earlier findings for a microbiota differentiation in disparate lung anatomical regions10.
To our knowledge this work represents the largest scrutiny of LC microbiota. Shortly, we uncovered a predominance of Proteobacteria among cancerous lungs a feature shared with other airway disorders. However, Proteobacteria abundance is not universal and rather dictates a microbiota substructure independently of LC subtype, COPD co-morbidity, smoking history, age or lung region. In SCC, we found evidence for a differential effect of bacterial communities in patient survival, particularly when stratified into an Enterobacteriaceae cluster. Given that this taxon has documented complications in pulmonary illnesses, we proposed a contribution of this cluster to an inflammatory cancer microenvironment, as well as, for other post-operative and/or therapeutic non-cancer complications. Finally, we believe that the discovery of such well-defined communities may shed light into bacteria as promising LC biomarkers for patient stratification and as future prognostic tools or even as therapeutic targets.
Data Availability
The data used in this study is included in this manuscript and in supplementary material files. Additional files used to generate data analysis are available from the corresponding author on reasonable request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424, https://doi.org/10.3322/caac.21492 (2018).
Meiners, S., Eickelberg, O. & Konigshoff, M. Hallmarks of the ageing lung. Eur Respir J 45, 807–827, https://doi.org/10.1183/09031936.00186914 (2015).
Vogelmeier, C. F. et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Eur Respir J 49, https://doi.org/10.1183/13993003.00214-2017 (2017).
Vermaelen, K. & Brusselle, G. Exposing a deadly alliance: novel insights into the biological links between COPD and lung cancer. Pulmonary pharmacology & therapeutics 26, 544–554, https://doi.org/10.1016/j.pupt.2013.05.003 (2013).
Liang, H. Y. et al. Facts and fiction of the relationship between preexisting tuberculosis and lung cancer risk: a systematic review. International journal of cancer. Journal international du cancer 125, 2936–2944, https://doi.org/10.1002/ijc.24636 (2009).
Mao, Q. et al. Interplay between the lung microbiome and lung cancer. Cancer Lett 415, 40–48, https://doi.org/10.1016/j.canlet.2017.11.036 (2018).
Acute Exacerbations of Pulmonary Diseases. (2017).
Wang, Z. et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J 47, 1082–1092, https://doi.org/10.1183/13993003.01406-2015 (2016).
Millares, L. et al. Functional Metagenomics of the Bronchial Microbiome in COPD. PLoS One 10, e0144448, https://doi.org/10.1371/journal.pone.0144448 (2015).
Erb-Downward, J. R. et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6, e16384, https://doi.org/10.1371/journal.pone.0016384 (2011).
Einarsson, G. G. et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax 71, 795–803, https://doi.org/10.1136/thoraxjnl-2015-207235 (2016).
Pragman, A. A. et al. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome 6, 7, https://doi.org/10.1186/s40168-017-0381-4 (2018).
Cameron, S. J. et al. Metagenomic Sequencing of the Chronic Obstructive Pulmonary Disease Upper Bronchial Tract Microbiome Reveals Functional Changes Associated with Disease Severity. PLoS One 11, e0149095, https://doi.org/10.1371/journal.pone.0149095 (2016).
Cameron, S. J. S. et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One 12, e0177062, https://doi.org/10.1371/journal.pone.0177062 (2017).
Lee, S. H. et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 102, 89–95, https://doi.org/10.1016/j.lungcan.2016.10.016 (2016).
Hosgood, H. D. 3rd et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environmental and molecular mutagenesis 55, 643–651, https://doi.org/10.1002/em.21878 (2014).
Yu, G. et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol 17, 163, https://doi.org/10.1186/s13059-016-1021-1 (2016).
Liu, H. X. et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. International journal of cancer. Journal international du cancer 142, 769–778, https://doi.org/10.1002/ijc.31098 (2018).
Sze, M. A. et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185, 1073–1080, https://doi.org/10.1164/rccm.201111-2075OC (2012).
Park, H., Shin, J. W., Park, S. G. & Kim, W. Microbial communities in the upper respiratory tract of patients with asthma and chronic obstructive pulmonary disease. PLoS One 9, e109710, https://doi.org/10.1371/journal.pone.0109710 (2014).
Yan, X. et al. Discovery and validation of potential bacterial biomarkers for lung cancer. American journal of cancer research 5, 3111–3122 (2015).
Mayhew, D. et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax, https://doi.org/10.1136/thoraxjnl-2017-210408 (2018).
Morris, A. et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187, 1067–1075, https://doi.org/10.1164/rccm.201210-1913OC (2013).
Lee, S. Y. et al. Polymorphisms in cancer-related pathway genes and lung cancer. Eur Respir J 48, 1184–1191, https://doi.org/10.1183/13993003.02040-2015 (2016).
Cancer Genome Atlas Research, N. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120, https://doi.org/10.1038/ng.2764 (2013).
Carvalho, A. S. et al. Bronchoalveolar Lavage Proteomics in Patients with Suspected. Lung Cancer. Scientific reports 7, 42190, https://doi.org/10.1038/srep42190 (2017).
Monteiro, C. et al. Characterization of microbiota in male infertility cases uncovers differences in seminal hyperviscosity and oligoasthenoteratozoospermia possibly correlated with increased prevalence of infectious bacteria. American journal of reproductive immunology. https://doi.org/10.1111/aji.12838 (2018).
Cavadas, B., Ferreira, J., Camacho, R., Fonseca, N. A. & Pereira, L. QmihR: Pipeline for Quantification of Microbiome in Human RNA-seq. 616, 173–179, https://doi.org/10.1007/978-3-319-60816-7_21 (2017).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60, https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
D’Amore, R. et al. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics 17, 55, https://doi.org/10.1186/s12864-015-2194-9 (2016).
Huang, Y. J. et al. The airway microbiome in patients with severe asthma: Associations with disease features and severity. J Allergy Clin Immunol 136, 874–884, https://doi.org/10.1016/j.jaci.2015.05.044 (2015).
Marri, P. R., Stern, D. A., Wright, A. L., Billheimer, D. & Martinez, F. D. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol 131, 346–352 e341–343, https://doi.org/10.1016/j.jaci.2012.11.013 (2013).
Renom, F. et al. Prognosis of COPD patients requiring frequent hospitalization: role of airway infection. Respir Med 104, 840–848, https://doi.org/10.1016/j.rmed.2009.12.010 (2010).
Laroumagne, S. et al. Bronchial colonisation in patients with lung cancer: a prospective study. Eur Respir J 42, 220–229, https://doi.org/10.1183/09031936.00062212 (2013).
Pitout, J. D. Multiresistant Enterobacteriaceae: new threat of an old problem. Expert Rev Anti Infect Ther 6, 657–669, https://doi.org/10.1586/14787210.6.5.657 (2008).
Swenson, C. E. & Sadikot, R. T. Achromobacter respiratory infections. Annals of the American Thoracic Society 12, 252–258, https://doi.org/10.1513/AnnalsATS.201406-288FR (2015).
Zeng, M. Y., Inohara, N. & Nunez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol 10, 18–26, https://doi.org/10.1038/mi.2016.75 (2017).
Li, N. et al. Sputum microbiota in severe asthma patients: Relationship to eosinophilic inflammation. Respir Med 131, 192–198, https://doi.org/10.1016/j.rmed.2017.08.016 (2017).
Zemanick, E. T. et al. Assessment of airway microbiota and inflammation in cystic fibrosis using multiple sampling methods. Annals of the American Thoracic Society 12, 221–229, https://doi.org/10.1513/AnnalsATS.201407-310OC (2015).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674, https://doi.org/10.1016/j.cell.2011.02.013 (2011).
Schussler, O. et al. Postoperative pneumonia after major lung resection. Am J Respir Crit Care Med 173, 1161–1169, https://doi.org/10.1164/rccm.200510-1556OC (2006).
Klastersky, J. et al. Bacteraemia in febrile neutropenic cancer patients. Int J Antimicrob Agents 30(Suppl 1), S51–59, https://doi.org/10.1016/j.ijantimicag.2007.06.012 (2007).
Acknowledgements
We would like to thank all patients for donating their samples and for collaborating in this study. IPATIMUP integrates the i3S Research Unit, which is partially supported by the Portuguese Foundation for Science and Technology (FCT). This work was supported by the Portuguese Foundation for Science and Technology (FCT), financed by the European Social Funds (COMPETE-FEDER) and National Funds through the FCT (projects PEstC/SAU/LA0003/2013 and POCI-01-0145-FEDER-007274, fellowships SFRH/BPD/77646/2011 and SFRH/BPD/120777/2016 to S.G. and P.I.M., respectively, grant PTDC/BEXGMG/0242/2012 to S.S. and by Programa Operacional Regional do Norte (ON.2 – O Novo Norte and Norte 2020), through FEDER funds under the Quadro de Referência Estratégico Nacional (QREN; projects NORTE-07-0162-FEDER-00018 and NORTE-070162-FEDER-000067, and NORTE-01-0145-FEDER-000029).
Author information
Authors and Affiliations
Contributions
S.G. and S.S. wrote the manuscript; S.G., L.P. and S.S. conceived the study; S.G. and C.M. performed laboratorial work; S.G., B.C., J.C.F., P.I.M. and S.S. carried out bioinformatics and statistical analyses; M.S., C.S., L.V.R., G.T., P.P., T.T.A., C.B., J.S., L.M., A.S.C., R.M. provided samples and managed clinical data, P.I.M., M.S., A.S.C., R.M. and L.P. critically revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gomes, S., Cavadas, B., Ferreira, J.C. et al. Profiling of lung microbiota discloses differences in adenocarcinoma and squamous cell carcinoma. Sci Rep 9, 12838 (2019). https://doi.org/10.1038/s41598-019-49195-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49195-w
This article is cited by
-
Tobacco use, self-reported professional dental cleaning habits, and lung adenocarcinoma diagnosis are associated with bronchial and lung microbiome alpha diversity
Respiratory Research (2024)
-
Lung microbiome: new insights into the pathogenesis of respiratory diseases
Signal Transduction and Targeted Therapy (2024)
-
Intratumoural microbiota: a new frontier in cancer development and therapy
Signal Transduction and Targeted Therapy (2024)
-
The dynamic lung microbiome in health and disease
Nature Reviews Microbiology (2023)
-
Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy
Signal Transduction and Targeted Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
