
Abstract
The faecal microbiota plays a critical role in host health, with alterations in the human faecal microbial composition associated with various conditions, particularly diarrhoeal diseases. However, little is known about microbial changes during cryptosporidiosis, one of the most important diarrhoeal diseases caused by protozoa in cattle. In this study, alterations in the faecal microbiota of neonatal calves as a result of Cryptosporidium parvum infection were investigated on a C. parvum-positive farm. Comparisons were made among groups of C. parvum-infected, rotavirus-infected, and the pathogen-negative calves. A specific increase in the abundance of Fusobacterium was observed in the faecal microbiota of C. parvum-infected animals. Diarrhoea severity increased in accordance with the abundance of C. parvum and Fusobacterium. Moreover, the specific increase of Fusobacterium appeared to be a universal feature of C. parvum infection, since neonatal calves from geographically separated areas showed the same result. These observations indicated that the growth of Fusobacterium may be an important aggravating factor of cryptosporidiosis.
Similar content being viewed by others


Introduction
The gut microbiota plays a critical role in the gut health of the host. It protects against enteropathogens, extracts nutrients and energy from food and contributes to normal immune function. Disruption of the normal composition of the gut microbiota has been associated with obesity, malnutrition, inflammatory bowel disease, neurological disorders and several cancers1. Alterations of the normal human gut microbiota have been documented, particularly in diarrhoeal diseases such as antibiotic-associated diarrhoea2, inflammatory bowel disease3, acute post radiotherapy diarrhoea4 and irritable bowel syndrome5,6.
In a previous study of the faecal microbiota of neonatal calves, Firmicutes was the most abundant phylum, with a prevalence ranging from 63.84–81.90%, followed by Bacteroidetes (8.36–23.93%), Proteobacteria (3.72–9.75%), Fusobacteria (0.76–5.67%) and Actinobacteria (1.02–2.35%)7. Changes in the digestive tract microbiome have also been identified in cattle exhibiting diarrhoea8,9. While a shift in the faecal microbiota of cattle infected with Mycobacterium avium subsp. paratuberculosis (Johne’s disease) has been reported10, limited information is available about the effects of other infectious diarrhoeal pathogens on the faecal microbiota of cattle.
Cryptosporidium parvum is a coccidian protozoan parasite that causes enteric infection and diarrhoeal disease in many mammals, including both immunocompetent and immunocompromised humans11. The parasite is widely distributed and is a common cause of severe neonatal diarrhoea among calves, with cryptosporidiosis being one of the most important infectious diarrhoeal diseases caused by protozoa for the cattle industry. While cryptosporidiosis is usually mild and self-limiting in immunocompetent humans, individuals with various immune disorders, including acquired immune deficiency syndrome, often contract chronic, life-threating infections12. While nitazoxanide has been licensed for the treatment of Cryptosporidium-induced diarrhoea in humans13, its efficacy in calves remains unclear14. Although one study reported beneficial effects of nitazoxanide in the treatment of cryptosporidiosis in experimentally-infected neonatal calves15, another study revealed no prophylactic or therapeutic efficacy16. Therefore, clinical disease control in calves has been hampered by the lack of drugs and vaccines that are effective for either treatment or prevention of cryptosporidiosis17,18.
The gut microbiota is thought to affect resistance to C. parvum infection because germfree adult immunocompetent mice showed high susceptibility to infection19. Moreover, a study using severe combined immunodeficient (SCID) mice supported the hypothesis that resistance of adult mice to C. parvum infection does not require a specific immune response but can be mediated by nonspecific mechanisms associated with the presence of intestinal microflora20. This hypothesis was based on results showing that C. parvum was not readily detected in flora-bearing adult SCID mice, while germfree SCID mice were heavily infected following challenge with the parasite20. These observations indicate that some interaction occurs between C. parvum and the faecal microbiota in infected hosts.
Administration of live Lactobacillus bacterial cell-free supernatants reduces the viability of C. parvum oocysts in vitro21,22. Furthermore, probiotics can limit C. parvum infection in immunocompromised individuals in mouse models of the disease23,24, with similar results observed for human cases25. However, probiotic treatment of calves infected with C. parvum did not result in a significant decrease in the incidence of diarrhoea or oocyst shedding compared with the controls26.
At present, nothing is known about the faecal microbiota of neonatal calves infected with C. parvum. Interactions between the faecal microbiota and C. parvum must be analysed to understand pathophysiological changes that occur during disease progression. To address this knowledge gap, we examined the faecal microbiota profiles of neonatal calves from a C. parvum-endemic farm. Metagenomic sequencing analysis was conducted using the IonPGM, and the composition of the microbiota revealed by the high-throughput sequencing was re-examined and confirmed by quantitative polymerase chain reaction (qPCR) analysis. Moreover, neonatal calves from different regions of Japan were examined to evaluate the universality of observed alterations in the faecal microbiota caused by C. parvum infection.
Results
Specific increase of Fusobacterium abundance in C. parvum-only infected calves revealed by metagenomic analysis
Metagenomic analysis based on 16 S rRNA gene sequences was performed for a total of 120 faecal samples collected at six time points from 20 neonatal Holstein calves (Table S1). The calves were aged between 0 and 15 days old and were located on a farm (farm #A) in Iwate Prefecture, Japan. All of the calves were female and were born in identical cattle sheds. Birth weights ranged from 35–45 kg. All treatments after birth, including colostrum practices, were identical for all calves.
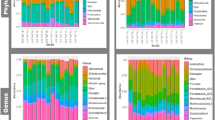
The distribution of the relative abundance of the different bacterial genera is shown in Fig. 1a for each of the three groups: C. parvum-only-infected (n = 8), rotavirus-only-infected (n = 5) and the pathogen-negative group (n = 7) determined by the commercial immunochromatographic test (ICT) strips (Bio-X Diagnostics SPRL, Jemelle, Belgium). None of the animals tested positive for coronavirus infection.

Distribution of bacterial relative abundance among the three groups of faecal samples collected at farm #A: Cryptosporidium parvum-only-infected (n = 8), rotavirus-only-infected (n = 5) and pathogen-negative (n = 7). An average value for the relative abundance across the six sampling points for each genus is shown using a gradient scale. (a) The 261 analysed genera (Table S2) were plotted as rows in which the most abundant genus in the C. parvum-only infected group is shown at the top. The next most abundant genera are shown sequentially in order of abundance. (b) The 10 most abundant genera (shown on the left-hand side) in the C. parvum-only-infected group in comparison with the corresponding abundances in the rotavirus-only-infected and pathogen-negative groups. The top 10 genera were obtained from (a). A specific increase of Fusobacterium in the C. parvum-only-infected group was suggested. (c–e) Time-dependent change of bacterial relative abundance among C. parvum-only-infected, rotavirus-only-infected and pathogen-negative groups. The column titles are the numbers of faecal samples collected in each age in days (see Table S1).
The most abundant genus in C. parvum-only-infected calves was Fusobacterium (14.1%, average of all samples), followed by Escherichia-Shigella (12.8%). The relative ratio of Fusobacterium was low in the rotavirus-only-infected (0.7%) and pathogen-negative (2.0%) groups, while the relative ratios of Escherichia-Shigella in the two groups (11.9% and 17.0%, respectively) were similar to that observed in the C. parvum-infected group (Fig. 1b, Table S2).
The abundance of Fusobacterium appeared to be increased in the faecal microbiota of C. parvum-only-infected calves (Figs 1c and 2a). A significant increase was found in the abundance of the bacteria between the 1st (0–1 day old) and the 6th (10–15 days old) sampling points (Fig. 2a, Wilcoxon test, P < 0.05). No significant increase was detected in rotavirus-only-infected (n = 5) and the pathogen-negative calves (n = 7) (Fig. 2a). No other genus-specific increase was observed among the top 10 genera identified in the C. parvum-only-infected samples (listed in Fig. 1b–e).

Comparisons of Fusobacterium loads obtained by metagenomic analysis among the three groups: Cryptosporidium parvum-only-infected (n = 8), rotavirus-only-infected (n = 5), and the pathogen-negative (n = 7) samples collected from farm #A. (a) The relative ratios of Fusobacterium for all the calves in relation to their age (upper). Wilcoxon test was performed to reveal the significant differences between the 1st (0–1 day old) and the 6th (10–15 days old) sampling points in the three groups (lower). The increase of Fusobacterium was detected in C. parvum-only-infected group (*P < 0.05). (b) The specific increase in the abundance of Fusobacterium in C. parvum-only-infected calves at the 6th sampling point was confirmed by quantitative PCR (Kruskal-Wallis test, *P < 0.05).
The results of the metagenomic analysis were reproduced by the qPCR assays at the 6th sampling point (10–15 days old) (Fig. 2b). A specific increase in the abundance of Fusobacterium was detected in C. parvum-only-infected calves (n = 8) in comparison with the rotavirus-only-infected (n = 5) and the pathogen-negative calves (n = 7). A significant difference was observed between the C. parvum-positive group and the other groups (Kruskal-Wallis test, P < 0.05).
Abundance of C. parvum and Fusobacterium in the samples from other locations as determined by qPCR
An increase in the abundance of Fusobacterium was also detected in 11–15 days old C. parvum-positive calves from the farms located in Okinawa (n = 7), Kagoshima (n = 1), Iwate (n = 9) and Hokkaido (n = 6) prefectures (Supplementary Fig. S1). The ages of these calves were very similar to those of calves from farm #A at the 6th sampling point (10–15 days old). The increase in abundance of Fusobacterium was only observed in C. parvum-positive calves (n = 9), with no increase detected in the C. parvum-negative calves (n = 14) (Fig. 3). Moreover, the Fusobacterium load showed a strong positive correlation with that of C. parvum (r = 0.61, P < 0.05) (Fig. 3).

Fusobacterium loads in samples from Cryptosporidium parvum-positive or -negative calves aged 11–15 days from the farms in Okinawa (n = 7), Kagoshima (n = 1), Iwate (n = 9) and Hokkaido (n = 6) prefectures. The increase in abundance of Fusobacterium in C. parvum-positive samples (n = 9) was detected in six samples by quantitative PCR (qPCR). No increase was observed in C. parvum-negative samples (n = 14). A highly significant correlation was observed (r = 0.61, *P < 0.05).
Association between the presence of C. parvum and Fusobacterium in faecal samples from the different locations
Fisher’s exact test revealed that the number of Fusobacterium-positive/C. parvum-positive samples was significantly greater than the number of Fusobacterium-positive/C. parvum-negative samples from the different locations (Okinawa, Kagoshima, Iwate and Hokkaido prefectures) (P < 0.05) (Table 1).
Oocyst numbers and faecal scores during C. parvum infections in relation to the relative ratios of Fusobacterium
The number of C. parvum oocysts detected in the infected neonatal calves from farm #A increased at day 8 (Fig. 4a). Again, the relative ratios of Fusobacterium began to increase at the same time point (Fig. 4a). The faecal scores, indicating severity of diarrhoea, increased in accordance with the abundance of C. parvum and Fusobacterium (Fig. 4a). A moderately positive correlation was observed between the number of oocysts and the relative ratio of Fusobacterium (r = 0.47, P < 0.05) (Fig. 4b).

Oocyst numbers and faecal scores during Cryptosporidium parvum infections in relation to the relative ratios of Fusobacterium. (a) Average numbers of C. parvum oocysts, relative ratios of Fusobacterium, and faecal scores for each sampling day for the C. parvum-positive samples from farm #A. (b) Correlation between the number of C. parvum oocysts and the relative ratios of Fusobacterium for the C. parvum-positive samples from farm #A. A moderately positive correlation was observed (r = 0.47, *P < 0.05).
Discussion
The metagenomic analysis conducted in this study revealed that the abundance of Fusobacterium was particularly increased in C. parvum-only-infected calves (Figs 1 and 2a). Although the Fusobacterium species detected in this study was not identified, the specific increase in Fusobacterium among C. parvum-positive samples from farm #A was confirmed by qPCR analysis (Fig. 2b).
Fusobacterium species are anaerobic, elongated, Gram-negative rods. While there are multiple species of Fusobacterium, the species most commonly associated with human and animal disease is F. necrophorum, an opportunistic pathogen that causes numerous necrotic conditions (necrobacillosis) and both specific and non-specific infections in a variety of animals. Bovine liver abscesses and foot rot caused by F. necrophorum are significant concerns in the cattle industry27.
How factors such as the environment and diet shape the human faecal microflora remains unclear. Nonetheless, studies have revealed increased levels of Prevotella or Bacteroides in response to a high-fibre diet or a long-term diet rich in animal proteins, respectively1. In calves, probiotic administration or the use of lactic acid bacteria has been identified as a tool to maintain the intestinal microbial balance and to prevent the establishment of opportunistic pathogens28. Therefore, dietary differences between farms, especially in the presence or absence of probiotic supplementation, will likely affect the composition of the faecal microbiota of calves. However, in this study, the specific increase of Fusobacterium was observed not only in animals from farm #A, but also in calves from completely different locations (Table 1, Fig. 3). This suggests that the growth of Fusobacterium is a common feature among C. parvum-positive calves, regardless of differences in diet and environmental conditions between farms.
The oocyst shedding pattern of C. parvum observed in this study (Fig. 4a) was similar to those recorded in previous reports29,30. Interestingly, the initiation of oocyst shedding (at 8 days old) appeared to coincided with the increase of Fusobacterium (at 8 days old) (Fig. 4a). The protozoan infection occurred prior to the increase in Fusobacterium growth because the oocyst incubation period in dairy cattle is 3 to 6 days prior to shedding11. Moreover, the relative ratios of the bacteria tended to increase in accordance with increasing numbers of oocysts (Fig. 4b). These observations suggest that the increase in C. parvum may benefit the growth of Fusobacterium. The beneficial interaction between these two microbes appeared to be universal based on the strong positive correlation between C. parvum and Fusobacterium observed in faecal samples from different origins (Fig. 3).
Previous studies have revealed that cryptosporidiosis symptoms are strongly correlated with the load of C. parvum in host faecal samples. Challenge of human volunteers with C. parvum showed that the number of oocysts excreted in faeces was significantly higher in subjects with diarrhoea than in those with enteric symptoms but no diarrhoea31. In addition, a previous study on risk factors for neonatal calf diarrhoea caused by C. parvum reported a significant association between high oocyst number and the occurrence of diarrhoea32. In the present study, faecal scores appeared to worsen (Fig. 4a) with corresponding increases in the abundance of both C. parvum (Fig. 4a) and Fusobacterium (Figs 2a and 4a). Therefore, the increase of Fusobacterium appears to be an important aggravating factor for cryptosporidiosis because the growth of the bacterium increases the C. parvum load in neonatal calves (Figs 3b and 4b). An animal model using neonatal calves for C. parvum challenge will be required in the future to gather direct evidence of the co-increase of C. parvum and Fusobacterium.
We propose that the specific increase in Fusobacterium may be caused by the following mechanism. Damage to microvilli on the surface of intestinal epithelial cells caused by the growth of C. parvum increases susceptibility to Fusobacterium infection. Fusobacterium then becomes a dominant genus and grows effectively because of its ability to adhere to and invade intestinal cells33,34,35, which may cause severe diarrhoea in cattle. Because this hypothesis remains unconfirmed, a pathological analysis of intestinal cells is needed to understand the beneficial interaction between C. parvum and Fusobacterium.
Clinical studies on human diarrhoeal diseases35,36 have shown that Fusobacterium is found in colonic tissues of patients with inflammatory bowel disease. Fusobacterium varium is a known causative agent of ulcerative colitis37. A previous study showed that a 2-week course of combination antibiotic therapy (amoxicillin, 1,500 mg; tetracycline, 1,500 mg; and metronidazole, 750 mg/day) reduces the density of F. varium in the mucosa, resulting in a higher remission rate in the treatment group compared with the control group38. The present study suggests that this kind of antibiotic therapy targeting Fusobacterium may also relieve the severity of diarrhoea in cryptosporidiosis cases.
This is the first study to reveal a significant correlation between Fusobacterium and C. parvum in cases of neonatal calf diarrhoea. The increase of Fusobacterium in the faecal microbiota is likely to be an important aggravating factor of cryptosporidiosis in calves. This novel finding may contribute to the development of improved control strategies for cryptosporidiosis.
Methods
Ethics statement
This study was performed in strict accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of Ministry of Educations, Culture Sports, Science and Technology, Japan. The protocol was approved by the Committee on the Ethics of Animal Experiments of Iwate University (Permit number A201536).
Faecal sample collection from farm #A for metagenomic analysis
Faecal samples were collected from 20 neonatal Holstein calves at a farm (farm #A) in Iwate Prefecture, Japan, where cryptosporidiosis caused by C. parvum had consistently been reported in neonatal calves39. To achieve consistent sampling quality, all of the calves were housed and handled in the same way as much as possible. All of the calves were female and were born in identical cattle sheds. Birth weights ranged from 35–45 kg. Premature calves were excluded from the sampling. No vaccination programme was implemented for neonatal calves from farm #A. Within 2 h of birth, calves were fed 2 litres of colostrum, followed by 3 litres of colostrum twice daily for 4 days. An equivalent amount of a milk substitute (Pote-Mow Milk; NOSAN, Yokohama, Japan) was used thereafter. A calf starter (Bio calf Neo, NOSAN) was fed to all calves during this period.
Faecal samples were collected from each calf every two or three days between birth and 15 days of age, resulting in a total of 120 faecal samples from farm #A across six sampling points. Each sample was divided between two plastic tubes. From the first tube, half of each sample was analysed for the presence of C. parvum, rotavirus, coronavirus, and Escherichia coli K99 antigens using commercial immunochromatographic test (ICT) strips (Bio-X Diagnostics SPRL, Jemelle, Belgium). The remaining half of each faecal sample from the first tube was used to calculate the abundance of oocysts, as described below. The faecal score for each sample was recorded at the same time point, with the severity of diarrhoea scored according to the appearance of faecal material: normal (0), loose (1), muddy (2), and watery (3). The second tube from each sample was preserved at −80 °C for DNA extraction.
Faecal sample collection from farms in different locations
To examine the correlation between C. parvum and the faecal microbiome in the neonatal calves from different origins, 23 faecal samples were collected from calves aged 11–15 days born at farms located in Okinawa, Kagoshima, Iwate and Hokkaido prefectures, Japan (Supplementary Fig. S1). The ages of the calves were very similar to those of calves from farm #A at the 6th sampling point (10–15 days old). The faecal samples were preserved at −20 °C for DNA extraction.
DNA extraction
A Powersoil DNA Isolation Kit (MoBio Laboratories Inc., Carlsbad, CA, USA) was used to isolate DNA from the faecal samples from farm #A and from the different locations in Iwate and Hokkaido prefectures. A QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) was used for DNA extraction from the samples from Okinawa and Kagoshima prefectures.
Metagenomic analysis of samples from farm #A
Each library was prepared with a primer set (784 F:5′-AGGATTAGATACCCTGGTA-3′ and 1061 R: 5′-CRRCACGAGCTGACGAC-3′3′) targeting the V5-V6 region of the 16 S rRNA genes of bacterium and an Ion Plus Fragment Library Kit (Life Technologies). Sequencing was performed using a 318 chip and an Ion PGM Sequencing 400 Kit (Life Technologies) on the Ion PGM sequencer (Life Technologies). Raw sequences were demultiplexed and quality-trimmed using the following procedures: (i) trimming of bases with a quality less than Q15 from the 3ʹ end of each read, (ii) removal of reads with an average quality less than Q20, (iii) removal of reads without primer sequences at both ends and (iv) removal of reads with a total length less than 260 bp. These steps were carried out using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html) and BBtrim (http://bbmap.sourceforge.net/). Subsequently, 10,000 reads per sample were randomly sampled, using the random_sequence_sample.pl (https://www.ualberta.ca/~stothard/software.html) algorithm for taxonomic assignment. These sequences were then clustered into operational taxonomic units (OTU) defined at 97% similarity cutoff using UCLUST version 1.2.22q. Representative sequences for each OTU were classified taxonomically using RDP Classifier version 2.2 with the greengenes database (gg_13_8).
qPCR analysis
qPCR assays were carried out to determine the abundance of C. parvum and Fusobacterium sequences in each of the samples. Faecal samples collected at the 6th sampling point from farm #A along with the 23 samples from different origins were used for qPCR analyses.
To determine the abundance of C. parvum sequences, a qPCR targeting the 18 S rRNA gene was performed with the following primers: forward, 5′-CTCGACTTTATGGAAGGGTTG-3′; reverse, 5′-CAGAAACTTGAATGATATGTCACATTTAA-3′40. Reaction mixtures were prepared to a final volume of 20 µl and contained 0.2 µM of each primer, 0.4 µl of 50 × ROX reference dye, 10 µl of SYBR qPCR Mix (THUNDERBIRD SYBR qPCR Mix, TOYOBO Co., Osaka, Japan) and template DNA. The qPCR was performed using a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with an initial denaturation step of 95 °C for 60 s, followed by 35 cycles of 95 °C for 15 s and 64 °C for 30 s.
To determine the abundance of Fusobacterium sequences, a qPCR targeting the 16 S rRNA gene was performed using the following primers: forward, 5′-C(A/T)AACGCGATAAGTAATC-3′; reverse, 5′-TGGTAACATACGA(A/T)AGGG-3′41. The composition of the reaction mixture was identical to that described above. The thermal cycler conditions consisted of an initial denaturation step of 95 °C for 60 s followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s.
The amount of template DNA varied according to the quality of DNA: 1.6 ng for samples from farm #A, 4.0 ng for samples from Okinawa and Kagoshima prefectures and 40 ng for the samples from Iwate and Hokkaido prefectures.
Control plasmids containing the 18 S rRNA and 16 S rRNA gene sequences from C. parvum and Fusobacterium, respectively, were constructed and used to generate standard curves for the qPCR assays. Briefly, each PCR product from the respective targets was cloned into pUC118 using a Mighty Cloning Reagent Set (Blunt End) (TaKaRa Bio Inc., Otsu, Japan). The resultant plasmids were then linearised by digestion with EcoRI.
The C. parvum and Fusobacterium loads in each sample (copy number/ng DNA) were determined based on a standard calibration curve generated from qPCR assays carried out using a dilution series (103–108 copies/μl) of each of the control plasmids. qPCR assays were repeated at least twice to confirm the reproducibility of the results.
Calculation of oocyst numbers in faecal samples from farm #A
Oocyst numbers in the C. parvum-positive samples identified by ICT strips were determined using the sugar flotation method, as described previously42. Briefly, 1 g of faecal sample was resuspended in water and centrifuged at 2,000 × g for 10 min. The supernatant was discarded, and the pellet resuspended in sucrose solution (1.2 g/ml) before being centrifuged using the same conditions as above. Following centrifugation, oocysts present in the sample floated to the top of the supernatant and could be counted to give the approximate number of oocysts per gram (OPG). The average number of oocysts in 10 fields at a magnification of 400× was calculated under an optical microscope, with the average number converted into OPG by using the total number of fields under the microscope. The total number of fields for the optical microscope used in this study (BX51; Olympus, Tokyo, Japan) was 1,089.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 7.04 (GraphPad Software Inc.).
Wilcoxon test was performed to compare the distribution of bacterial relative abundance in samples from in farm #A. The differences between the 1st and the 6th sampling points were examined respectively in the three groups: C. parvum-only-infected, rotavirus-only-infected, and the pathogen negative samples.
Correlation coefficients were calculated to examine the relationship between the number of C. parvum oocysts and the relative ratios of Fusobacterium, as well as between the DNA copy numbers of C. parvum and Fusobacterium.
The associations between C. parvum and Fusobacterium, based on the number of positive and negative samples for each determined by qPCR analysis of the faecal samples from different origins, were examined by Fisher’s exact test.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information Files).
References
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature. 489, 220–230 (2012).
Young, V. B. & Schmidt, T. M. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin Microbiol. 42, 1203–1206 (2004).
Manichanh, C., Borruel, N., Casellas, F. & Guarner, F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 9, 599–608 (2012).
Manichanh, C. et al. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J. Gastroenterol. 103, 1754–1761 (2008).
Krogius-Kurikka, L. et al. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol. 9, 95, https://doi.org/10.1186/1471-230X-9-95 (2009).
Rigsbee, L. et al. Quantitative profiling of gut microbiota of children with diarrhea-predominant irritable bowel syndrome. Am J. Gastroenterol. 107, 1740–51 (2012).
Oikonomou, G. et al. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS One. 8, e63157 (2013).
Xie, G. et al. Alteration of digestive tract microbiome in neonatal Holstein bull calves by bacitracin methylene disalicylate treatment and scours. J. Anim. Sci. 91, 4984–4990 (2013).
Zeineldin, M., Aldridge, B. & Lowe, J. Dysbiosis of the fecal microbiota in feedlot cattle with hemorrhagic diarrhea. Microb. Pathog. 115, 123–130 (2018).
Fecteau, M. E. et al. Dysbiosis of the Fecal Microbiota in Cattle Infected with Mycobacterium avium subsp. paratuberculosis. PLoS One 11, e0160353 (2016).
Fayer, R., Speer, C. A. & Dubey, J. P. General biology of Cryptosporidium in Cryptosporidiosis of man and animals (ed. Dubey, J. P., Speer, C. A. & Fayer, R.) 1–29 (CRC Press, 1990).
Crawford, F. G. & Vermund, S. H. Human cryptosporidiosis. Crit. Rev. Microbiol. 16, 113–159 (1988).
Fox, L. M. & Saravolatz, L. D. Nitazoxanide: a new thiazolide antiparasitic agent. Clin. Infect. Dis. 40, 1173–1180 (2005).
Nyadam, D. & Peregrine, A. S. Present and future control of cryptosporidiosis in cattle. AABP PROCEEDINGS 38, 15–18 (2005).
Ollivett, T. L. et al. Effect of nitazoxanide on cryptosporidiosis in experimentally infected neonatal dairy calves. J. Dairy Sci. 92, 1643–1648 (2009).
Schnyder, M., Kohler, L., Hemphill, A. & Deplazes, P. Prophylactic and therapeutic efficacy of nitazoxanide against Cryptosporidium parvum in experimentally challenged neonatal calves. Vet. Parasitol. 160, 149–154 (2009).
Fayer, R. & Ungar, B. L. P. Cryptosporidium spp. and cryptosporidiosis. Microbiol. Rev. 50, 458–483 (1986).
Soave, R. & Armstrong, D. Cryptospoiidium and cryptosporidiosis. Rev. Infect. Dis. 8, 1012–1023 (1986).
Harp, J. A., Wannemuehler, M. W., Woodmansee, D. B. & Moon, H. W. Susceptibility of germfree or antibiotic-treated adult mice to Cryptosporidium parvum. Infect Immun. 56, 2006–2010 (1988).
Harp, J. A., Chen, W. & Harmsen, A. G. Resistance of severe combined immunodeficient mice to infection with Cryptosporidium parvum: the importance of intestinal microflora. Infect Immun. 60, 3509–3512 (1992).
Foster, J. C. et al. Effect of Lactobacillus and Bifidobacterium on Cryptosporidium parvum oocyst viability. Food Microbiol. 20, 351–357 (2003).
Glass, M. D., Courtney, P. D., LeJeune, J. T. & Ward, L. A. Effects of Lactobacillus acidophilus and Lactobacillus reuteri cell-free supernatants on Cryptosporidium viability and infectivity in vitro. Food Microbiol. 21, 423–429 (2004).
Alak, J. I. et al. Effect of Lactobacillus reuteri on intestinal resistance to Cryptosporidium parvum infection in a murine model of acquired immunodeficiency syndrome. J. Infect Dis. 175, 218–221 (1997).
Alak, J. I. et al. Supplementation with Lactobacillus reuteri or L. acidophilus reduced intestinal shedding of Cryptosporidium parvum oocysts in immunodeficient C57BL/6 mice. Cell Mol Biol 45, 855–863 (1999).
Pickerd, N. & Tuthill, D. Resolution of cryptosporidiosis with probiotic treatment. Postgrad Med J. 80, 112–113 (2004).
Harp, J. A. et al. Field testing of prophylactic measures against Cryptosporidium parvum infection in calves in a California dairy herd. Am J. Vet Res. 57, 1586–1588 (1996).
Nagaraja, T. G., Narayanan, S. K., Stewart, G. C. & Chengappa, M. M. Fusobacterium necrophorum infections in animals: pathogenesis and pathogenic mechanisms. Anaerobe. 11, 239–246 (2005).
Signorini, M. L. et al. Impact of probiotic administration on the health and fecal microbiota of young calves: a meta-analysis of randomized controlled trials of lactic acid bacteria. Res. Vet. Sci. 93, 250–258 (2012).
Uga, S. et al. Prevalence of Cryptosporidium parvum infection and pattern of oocyst shedding in calves in Japan. Vet Parasitol. 94, 27–32 (2000).
Nydam, D. V., Wade, S. E., Schaaf, S. L. & Mohammed, H. O. Number of Cryptosporidium parvum oocysts or Giardia spp cysts shed by dairy calves after natural infection. Am J. Vet Res. 62, 1612–1615 (2001).
Chappell, C. L., Okhuysen, P. C., Sterling, C. R. & DuPont, H. L. Cryptosporidium parvum: intensity of infection and oocyst excretion patterns in healthy volunteers. J. Infect Dis. 173, 232–236 (1996).
Trotz-Williams, L. A. et al. Calf-level risk factors for neonatal diarrhea and shedding of Cryptosporidium parvum in Ontario dairy calves. Prev Vet Med. 82, 12–28 (2007).
Ohkusa, T., Okayasu, I., Tokoi, S. & Ozaki, Y. Bacterial invasion into the colonic mucosa in ulcerative colitis. J. Gastroenterol Hepatol. 8, 116–118 (1993).
Ohkusa, T. et al. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: a possible pathogenic mechanism of ulcerative colitis. J. Med Microbiol. 58, 535–545 (2009).
Strauss, J. et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 17, 1971–1978 (2011).
Ohkusa, T. et al. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J. Gastroenterol Hepatol. 17, 849–853 (2002).
Ohkusa, T. et al. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut. 52, 79–83 (2003).
Ohkusa, T. et al. Effectiveness of antibiotic combination therapy in patients with active ulcerative colitis: a randomized, controlled pilot trial with long-term follow-up. Scand J. Gastroenterol. 40, 1334–1342 (2005).
Aita, J. et al. Molecular characterization of Cryptosporidium parvum detected in Japanese black and Holstein calves in Iwate Prefecture and Tanegashima Island, Kagoshima Prefecture, Japan. J. Vet Med Sci. 77, 997–999 (2015).
Stroup, S. E. et al. Real-time PCR detection and speciation of Cryptosporidium infection using Scorpion probes. J. Med Microbiol. 55, 1217–1222 (2006).
Rinttilä, T. et al. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl Microbiol. 97, 1166–1177 (2004).
Ichikawa-Seki, M. et al. Molecular characterization of Cryptosporidium parvum from two different Japanese prefectures, Okinawa and Hokkaido. Parasitol Int. 64, 161–166 (2015).
Acknowledgements
This study was supported in part by a Joint Research Grant from the National Research Center for Protozoan Diseases, Obihiro University of Agriculture and Veterinary Medicine (27-joint-2, 28-joint-6), and by a Grant for Joint Research Project of the Research Institute for Microbial Diseases, Osaka University.
Author information
Authors and Affiliations
Contributions
Y.N. designed the study. M.I.-S., D.M., J.A., K.H. and A.T. performed experiments. A.K., F.M. and Y.T. collected samples. M.I.-S., D.N., S.N., T.I., T.H. and Y.N. discussed the results. M.I.-S. and Y.N. drafted the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ichikawa-Seki, M., Motooka, D., Kinami, A. et al. Specific increase of Fusobacterium in the faecal microbiota of neonatal calves infected with Cryptosporidium parvum. Sci Rep 9, 12517 (2019). https://doi.org/10.1038/s41598-019-48969-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48969-6
This article is cited by
-
Specific pathway abundances in the neonatal calf faecal microbiome are associated with susceptibility to Cryptosporidium parvum infection: a metagenomic analysis
Animal Microbiome (2023)
-
Pectin modulates intestinal immunity in a pig model via regulating the gut microbiota-derived tryptophan metabolite-AhR-IL22 pathway
Journal of Animal Science and Biotechnology (2023)
-
Fecal microbiota dynamics and its relationship to diarrhea and health in dairy calves
Journal of Animal Science and Biotechnology (2022)
-
Cohesive prediction model for analyzing the recurrent attributes of infectious cryptosporidiosis disease
Journal of Ambient Intelligence and Humanized Computing (2021)
-
A probiotic treatment increases the immune response induced by the nasal delivery of spore-adsorbed TTFC
Microbial Cell Factories (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
