
Abstract
Unlike other tumours, TP53 is rarely mutated in melanoma; however, it fails to function as a tumour suppressor. We assume that its functions might be altered through interactions with several families of proteins, including p53/p73, NME and GLI. To elucidate the potential interplay among these families we analysed the expression profiles of aforementioned genes and proteins in a panel of melanoma cell lines, metastatic melanoma specimens and healthy corresponding tissue. Using qPCR a higher level of NME1 gene expression and lower levels of Δ40p53β, ΔNp73, GLI1, GLI2 and PTCH1 were observed in tumour samples compared to healthy tissue. Protein expression of Δ133p53α, Δ160p53α and ΔNp73α isoforms, NME1 and NME2, and N′ΔGLI1, GLI1FL, GLI2ΔN isoforms was elevated in tumour tissue, whereas ∆Np73β was downregulated. The results in melanoma cell lines, in general, support these findings. In addition, we correlated expression profiles with clinical features and outcome. Higher Δ133p53β and p53α mRNA and both GLI1 mRNA and GLI3R protein expression had a negative impact on the overall survival. Shorter overall survival was also connected with lower p53β and NME1 gene expression levels. In conclusion, all examined genes may have implications in melanoma development and functional inactivity of TP53.
Similar content being viewed by others


Introduction
Malignant melanoma remains the most aggressive and treatment-resistant form of skin cancer with increasing incidence1. Although recent advances in melanoma therapy improve the overall patient survival, they are still hampered by rapid and pervasive treatment resistance. Thus, novel molecular approaches that would contribute to a better understanding of the disease should be deployed.
p53 plays a substantial role in the response to stress by coordinating diverse signalling pathways, thereby preventing the tumour formation. Unlike other cancers, in metastatic melanoma TP53 gene is relatively rarely mutated. However, it fails to function as a tumour suppressor and reduced levels of p53 contribute to aggressiveness and resistance to therapy2. Several diverse mechanisms of p53 inactivation in melanomagenesis have been proposed. The most common mechanisms include mutations of cyclin-dependent kinase inhibitor CDKN2A (encoding for both p16INK4A and p14ARF, which inactivates MDM2) and MDM2 (negative regulator of p53) overexpression, activation of iASPP (inhibitor of apoptosis stimulating protein of p53) or silencing of the TP53 gene by epigenetic mechanisms2,3,4,5. However, the later phenomenon has not been fully understood.
The p53 family comprises of p53, p73 and p63. Transcription from alternative promoters, alternative splicing and diverse translation initiation sites contribute to the family complexity6,7, and twelve protein isoforms with different N- and C-termini are encoded by the single TP53 gene (Fig. 1)8. The diversity in structure leads to diversity in subcellular localization, and consequently in various biochemical/biological activities which are cell-type dependent. Finally, p53-mediated cell response is the sum of the activities which results from co-expressed p53 isoforms8. Currently, the evidence explaining the involvement of p53 isoforms in tumour formation is still limited.

Scheme of the TP53 gene (a) and the protein isoforms (b). The colour of the protein domains matches the corresponding exon. Black boxes represent noncoding sequences, whereas coding sequences are in grayscale. The TP53 gene is composed of 11 classical exons and alternative exons (9 β and γ). Furthermore, it contains an internal promoter (P2), alternative splice variants (Δ40, β, γ) and internal initiation of translation sites (ATG1, ATG40, ATG133, ATG160). The p53 protein is composed of several domains - transactivation domain (TAD, which actually include two different domains, TAD1 and TAD2), the DNA-binding domain (DBD) and the nuclear localization signal (NLS). Twelve protein isoforms with different N- and C-termini are encoded by the human TP53 gene – alternative splicing of human intron 2 gives rise to Δ40p53 (truncated transactivation domain, TAD) and intron 9 to α, β and γ isoforms; the usage of the alternative promoter produces Δ133p53 and Δ160p53 (both lacking the entire TAD), while alternative translation initiation site produces Δ40p53 and Δ160p53.
Likewise, two main groups of p73 isoforms with different N-termini are produced: the TAp73 isoforms with the entire TAD and the N-terminally truncated isoforms, ΔNp73, lacking TAD and acting, mainly, as dominant-negative inhibitors towards p53 and TAp739. In contrast to p53, p73 is essentially never mutated in cancer, but it is overexpressed6,10.
Most of the p53 family isoforms possess the ability to interact between themselves forming heterotetramers and compete for DNA binding, so the overall activity of p53/p73 proteins is a result of the ratio between the different isoforms. Thus, in melanoma it has been found that p63 interacts with p53 influencing its tumour suppressive role11, while the Δ40p53 isoform forms heterotetramers with p53 modifying downstream p53 target genes and promoting apoptosis over cell cycle arrest12. However, we assume that p53 function in metastatic melanoma might be altered through interactions with some other proteins, like NME and GLI families of proteins.
The NME (NM23, NDPK, Awd) gene/protein family consists of ten members named after the first one identified, NME1/NM23-H1/NDPKA13, which is considered to be a metastasis suppressor gene. Besides its enzymatic activity (nucleoside-diphosphate kinase), the NME proteins have been assigned several additional biochemical functions such as transcription regulators, protein kinases and DNases14. A connection between NME expression and melanoma formation or metastasis has been described in several reports15,16,17. Although the investigations were primarily focused on NME1 as a primary metastasis suppressor, some data on NME2 in murine and human melanoma are also available18,19,20. Lately, several independent studies suggest there is either a direct or indirect connection between p53 and NME gene/protein family members21,22,23.
The Hedgehog-GLI (HH-GLI) signalling pathway plays a vital role during embryonic development, stem and progenitor cell maintenance, and carcinogenesis. Binding of the Hedgehog (HH) ligand to the receptor Patched (PTCH1) triggers the cascade leading to the activation of transcription factors GLI1, GLI2 and GLI3. They regulate cell proliferation, cell cycle regulation, adhesion, epithelial-mesenchymal transition, self-renewal and pathway autoregulation (PTCH1 and GLI1 genes)24. GLI1 is the exclusive activator, while GLI2 and GLI3 are cleaved into repressor forms when the HH signal is not present25. PTCH1, GLI1 and GLI2 overexpressions have been associated with melanoma progression and invasive and metastatic phenotype26,27. The HH-GLI pathway is also governed by non-HH signalling pathways by interactions with other pathways, including p5328,29,30. p53 negatively inhibits GLI1-driven neural stem cell self-renewal, cell proliferation and tumour growth and, in turn, GLI1 represses p5331,32. The balance between p53 and GLI1 activities appears to be critical: loss of p53 in cancer could enable activities of GLI1 and GLI232,33, while repression of GLI3 activity potentiates p53-dependent cell growth inhibition in colon cancer cells34.
The aim of this work is to study the potential interplay between these three pathways involved in melanoma development and progression. To gain this knowledge, it was necessary to determine the expression pattern of a range of isoforms of the above mentioned proteins, which have not been systematically investigated in melanoma so far. In all the studies performed so far on metastatic melanoma, p53 isoforms’ expression was determined in cell lines, not in surgical tissue samples, while GLI isoforms have not been examined at all. Therefore, we investigated the expression of three groups of genes and proteins and their correlation to clinicopathological characteristics.
Results and Discussion
Gene expression in metastatic melanoma tissue samples and cell lines
Gene expression analysis was performed on 32 tumour tissue samples and 19 healthy skin samples from the same patients. The TP53 isoforms were pre-amplified in two separate pre-amplification reactions, giving a “long” and “short” template for quantitative real-time PCR (qPCR) amplification and analysis. The mean relative expression of “long” TP53 isoforms in tumour tissue, ranked from the highest to the lowest, was as follows: p53α > p53β > p53ɣ > ∆40p53α > ∆40p53β > ∆40p53ɣ. Ranking of the mean relative expression of “short” TP53 isoforms was: ∆133p53α > ∆133p53β > ∆133p53ɣ. Only one out of nine TP53 isoforms was significantly downregulated in tumour tissue (∆40p53β, p = 0.017) (Fig. 2). A similar trend was observed for the ∆40p53ɣ isoform but without statistical significance (p = 0.054). The expression levels of the studied genes were confirmed using a panel of eight melanoma cell lines. We have observed almost a perfect match of the mean gene expression levels in cell lines and metastatic melanoma tissues with a small deviation of ∆40p53β and ∆40p53ɣ and the shortest, Δ133p53γ. The role of ∆40p53β was not particularly studied so far. However, based on obtained data, it has to be taken into account.

Relative expression levels of TP53 isoforms analysed by pre-amplification followed by qPCR using SYBR Green dye. The expression was normalized to the expression level of total TP53. Results are presented in a log scale, the bars represent interquartile range and the black triangle represents median value. *Denotes p < 0.05.
Previous clinical studies have reported the expression of TP53 isoforms in several tumour types, confirming that small molecular weight p53 isoforms might play an important role in tumorigenesis12,35,36,37,38,39,40. Avery-Kiejda and collaborators described that p53β and ∆40p53 mRNAs were expressed at higher levels than p53α in most melanoma cell lines examined, compared to fibroblasts and melanocytes, suggesting that their expression may play a role in melanoma development41. However, none of these aforementioned studies applied a sophisticated approach in which qPCR was performed as a nested reaction following initial RT-PCR amplification.
The remaining genes were analysed using standard TaqMan assays. We were able to determine the gene expression of two cancer-relevant TP73 isoforms, TAp73 and ∆Np73. Unexpectedly, the expression of full length isoform TAp73 was higher than ∆Np73. Further, ∆Np73 expression was significantly downregulated in metastatic melanoma tissue (p < 0.0001) (Fig. 3). There is a paucity of studies that have analysed the p73 isoforms’ gene expression in melanoma. The only detailed study of the expression and effect of particular p73 isoforms in metastatic melanoma showed overexpression of TAp73, Ex2p73 and Ex2/3p73 (spliced transcripts derived from the first promoter), whereas ΔNp73 was the predominant isoform in benign nevi42, which is in line with our findings.

Relative expression of genes analysed by TaqMan assays. The expression was normalized to the expression level of GUSB and TBP (2−ΔCt). Results are presented in a log scale, the bars represent interquartile range and black triangle represents median value. *Denotes p < 0.05.
In our study a significant downregulation of GLI1 (p = 0.001), GLI2 (p < 0.0001) and PTCH1 (p = 0.006) in tumour tissue was observed. Several studies have shown upregulation of HH-GLI signalling in melanoma. A study on clinical samples showed that melanoma cells express SHH, GLI1 and PTCH1 mRNA, but the surrounding stroma does not26. Another study showed upregulation of SMO, GLI2 and PTCH1 mRNA in a subset of melanoma cell lines43. GLI2 was found to be associated with invasive and metastatic phenotype in melanoma. Tumours with high GLI2 expression metastasize to the bone more quickly than tumours with low GLI2 expression27.
Several groups of authors analysed the correlation between the RNA and protein levels of NME and their metastatic potential with heterogenous results15,16,17,44,45. Although there has been evidence that the NME1 RNA levels can be elevated while the protein levels are low due to protein degradation in invasive melanoma cells46, in our experiments NME1, as well as NME2, was significantly up-regulated on both, the RNA and protein level in tumours compared to healthy tissue (p = 0.041) (Figs 3, 5 and Supplementary Fig. S1).

Comparison of relative expression values for tumour samples (solid line) and melanoma cell lines (dashed line) normalized to the expression level of GUSB and TBP. (a) Mean expression values of TP53 isoforms analysed by pre-amplification and SYBR Green RT-qPCR. (b) Mean expression of other tested genes analysed by TaqMan assays.

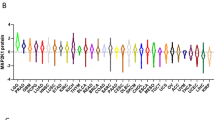
Expression of p53, p73, NME and GLI proteins. Proteins were analysed by PAGE, Western blot and densitometry with samples normalized to actin staining, followed by Wilcoxon paired sample analysis. P-values for proteins with significant difference in expression between tumour and healthy tissue are marked in bold. *Denotes p < 0.05.
The mean expression of TP73, NME and HH-GLI signalling pathway genes in a panel of eight melanoma cell lines almost completely match the mean expression in metastatic melanoma tissue (Fig. 4a). The expression levels of TP53 isoforms in melanoma cell lines showed some differences. It can be observed that ∆40p53 isoforms, particularly the shortest Δ133p53γ, showed lower expression levels in cell lines compared to tumour tissue samples (Fig. 4b). TP53 isoforms were not expressed at all in LM6 (4405 P) cell line probably due to the lack of TP53 locus47.
Protein expression in metastatic melanoma tissue samples and cell lines
In 30 paired tumour and healthy skin tissue samples we quantified the expression of 18 proteins in total: six p53 isoforms differing by both N- and C-termini (p53α, p53β, ∆40p53α, ∆133p53α, ∆133p53β and ∆160p53α); four p73 isoforms (TAp73α, TAp73β, ∆Np73α and ∆Np73β); NME1 and NME2; two GLI1 isoforms (N′ΔGLI1 and GLI1FL, molecular weights 130 and 160 kDa, respectively); two GLI2 isoforms (GLI2ΔN and GLI2FL, molecular weights 133 and 250 kDa, respectively), GLI3 activator (GLI3A, 190 kDa) and GLI3 repressor (GLI3R, 83 kDa). Summary of the results comprising the number and percentage of positive healthy and tumour samples is shown in Supplementary Table S1. When compared with healthy tissue, eight proteins showed statistically significant higher expression in tumours: both GLI1 isoforms (N′ΔGLI1, p = 0.030; GLI1FL, p = 0.002), GLI2ΔN isoform (p < 0.0001), both NME1 and NME2 (p = 0.0001 and p < 0.0001, respectively), ∆Np73α (p = 0.020) and two p53 isoforms - ∆133p53α and ∆160p53α (p = 0.0002 and p = 0.001, respectively). Conversely, the expression of ∆Np73β isoform was significantly downregulated in tumour tissue (p = 0.005) (Fig. 5). Further, the mean expression levels in a panel of eight melanoma cell lines were investigated (Supplementary Fig. S1) and when compared with metastatic melanoma tissue, almost perfect match of mean expression levels was observed with exception of Δ133p53β, ΔNp73 and GLI3R (Fig. 6).

Comparison of mean normalized protein expression values for tumour samples (solid line) and melanoma cell lines (dashed line). Protein expression was determined by densitometry and normalized to actin.
Due to the different site of the translation initiation, the presence of all known protein isoforms can be determined only by Western blot using specific antibodies that recognize a particular set of isoforms and p53/p73 standards (cells transfected with the specific isoform) helped to identify every single isoform (Supplementary Fig. S1). Previous studies reported wild type p53 overexpression in melanoma cell lines and tissue samples48,49. However, in these studies the authors used immunohistochemical analysis in which it was impossible to distinguish the specific isoforms. Therefore, our study is the first detailed study of p53 isoform expression in metastatic melanoma tissue.
Using this approach, ΔNp73α protein was significantly more expressed in the metastatic melanoma than in healthy tissue as compared to gene expression analysis. These inconsistencies between gene and protein expression are not unusual, since post-transcriptional regulation plays an important role in modification of proteins. Also, the experimental approach we used enabled us to distinguish the isoforms more precisely. Overexpression of total p73 in invasive cutaneous melanoma was recently shown using immunohistochemistry50. Overexpression of ΔNp73 is reasonable considering that ΔNp73 drives migration and invasion of nonmetastatic melanoma cells51.
As mentioned earlier, our results show elevated NME1 and NME2 mRNA and protein levels in metastatic melanoma although we would expect them to drop in metastasis due to the proposed role of NME as a metastasis suppressor. However, the mechanism by which NME contributes to tumour progression seems to be rather complex and does not rely solely on the rate of their expression. It has been reported that the NME1 content significantly varies in different cell populations within a tumour tissue sample. As an example, it has been suggested by Martinez and co-workers that NME1 could be less expressed in the invasion front compared to other cellular populations within the tumour mass52 which would probably be possible to detect immunohistochemically but not using Western blot, as was the case in our study.
Again, we found a discrepancy between gene and protein expression regarding HH-GLI pathway components. In recent years it has become clear that the GLI code is more complex than previously thought, with five known isoforms of GLI1, five of GLI2 (two of which with repressive activity), and two of GLI3, the GLI3A and GLI3R (reviewed in53) which can be distinguished only by Western blot analysis. Interestingly, Roessler and collaborators have demonstrated that GLI2ΔN is up to 30-fold more potent than the full length GLI254. GLI2ΔN can induce genomic instability by interfering with the cell cycle, acting through downregulation of mitosis regulators 14-3-3σ and p21WAF1/CIP1 55.
Correlation of gene and protein expression
Several TP53 isoforms were found to be correlated on the level of gene expression. A very strong correlation (ρ > 0.8) was found between Δ40p53γ and Δ40p53β, Δ133p53γ and Δ133p53α, and Δ133p53γ and Δ133p53β isoforms. A strong correlation (0.8 > ρ > 0.6) was found between Δ133p53β and Δ133p53α, TAp73 and Δ133p53β and a moderate correlation (0.6 > ρ > 0.4) between p53γ and p53α, Δ40p53β and Δ40p53α. For other genes tested, a strong correlation was determined for GLI1 and Δ133p53α, and GLI1 and GLI2, a moderate correlation for NME1 and NME2, GLI2 and NME2, PTCH1 and NME2, and PTCH1 and ΔNp73. A weak correlation (ρ < 0.4) was found between GLI2 and TAp73, and GLI3 and ΔNp73 (Supplementary Table S2).
At the protein level, a strong correlation was detected between TAp73β and TAp73α, and TAp73β and p53α. The remaining correlations between p53/p73 isoforms were mostly moderate (p53α with Δ133p53β, Δ160p53α, TAp73α and ΔNp73α; Δ133p53α with Δ133p53β, Δ160p53α with TAp73β; Δ160p53α with TAp73α; TAp73α with TAp73β and ΔNp73α). Interestingly, the expression of Δ133p53β was only negatively correlated with other isoforms.
NME1 protein was weakly correlated with ΔNp73α, but strongly with NME2 and GLI2ΔN. A mutual correlation was found between the two GLI1 isoforms, while GLI1FL isoform was negatively correlated with GLI2FL isoform. Unlike GLI1, where both isoforms were similarly correlated with other proteins, for the correlations between GLI3A and GLI3R the correlations were completely different: GLI3A correlated weakly with TAp73α and moderately with N′ΔGLI1, while GLI3R correlated strongly with p53α, moderately with TAp73β and weakly with GLI3A (Supplementary Table S3).
Accordingly, a strong correlation was found primarily between the family members, which is somehow expected because isoforms often share common transcriptional regulation. However, observed correlations between members of different families, e.g. negative correlation between GLI1 and TP53 isoforms or positive correlations between either TP73 isoforms or NME2 and HH-GLI pathway genes, are much more interesting since they can indicate a potential interplay among those families of genes and proteins.
There was almost no correlation between gene and protein expression, with NME1 gene being the only exception showing a moderate correlation between gene and protein expression (ρ = 0.43, p = 0.031) (data not shown). We assume this is a consequence of using different translation initiation sites and numerous translational modifications happening within p53 and GLI families of proteins.
Determination of BRAF and TP53 mutation status
Twenty out of 38 samples (52.6%) were positive for BRAF mutation. The most frequent BRAF mutation found in melanoma, c.1799T > A (p.V600E), was found in 19 cases (95%), while c.1798_1799delGTinsAA (p.V600K) was found in the remaining mutation positive sample. This is in line with the available literature56.
Conversely, only 3 out of 30 samples were positive for TP53 mutations (Supplementary Table S4). This observation is consistent with previously studies demonstrating the mutation of TP53 locus as a rare event during melanomagenesis56; between 15–20% according to the cBioPortal online tool of TCGA Consortium (http://www.cbioportal.org/).
Levels of gene/protein expression and their relation to clinicopathological characteristics
We analysed the association of various patients’ characteristics mutually and in relation to gene/protein expression. It was observed that BRAF mutation-positive patients tend to develop metastases at a younger age (median 63.5 years vs. 69.5 years, p = 0.019) (Fig. 7a). This was in accordance with Ekedahl and colleagues who in a clinic-based metastatic melanoma cohort found that BRAF mutation was associated with a younger age at primary diagnosis57. In tumour tissue from BRAF mutation-positive patients has been noticed a trend of higher GLI3 expression (p = 0.058) (data not shown). Although it was found that the expression of GLI1 is higher in human primary melanoma harbouring BRAF p.V600E mutation58, we did not detect such correlation in our studies. Interestingly, tumour samples from patients who had two or more previous metastases had significantly higher Δ133p53α protein expression levels compared to those with only one previous metastasis (p = 0.039) (Fig. 7b). In addition, it was observed that location of a specific metastatic tissue has an impact on the expression of various genes and proteins. For instance, skin metastases had the lowest gene expression of p53α compared to metastases from lymph nodes or other locations (p = 0.033) (Fig. 7c). The same was observed for ΔNp73α protein (p = 0.039) (Fig. 7d), while the opposite trend (higher expression in skin metastases) was determined for ΔNp73 gene expression (p = 0.057) (data not shown). Interestingly, the expression of Δ133p53β gene was significantly lower in tumour samples from patients who developed subsequent metastases (p = 0.013) (Fig. 7e).

Association of patients’ characteristics and gene/protein expression. (a) BRAF mutation status is associated with age of onset (p = 0.019). (b) Occurrence of previous metastases is associated with Δ133p53α protein expression (p = 0.039). (c) Location of sampled tissue is associated with the expression of p53α gene (p = 0.033) and (d) ΔNp73α protein (p = 0.039). (e) Development of subsequent metastasis is associated with Δ133p53β gene expression (p = 0.013). Black rectangle represents the median value and bars represent the interquartile range. *Denotes p < 0.05.
Impact of gene/protein expression and clinicopathological characteristics on patients’ survival
Nineteen patients (50%) died during the follow-up period. The median survival time of our patients was 19 months. To examine the impact of clinicopathological characteristics of patients and gene/protein expression profiles on overall survival (OS), a univariate analysis was performed. Among all patients’ characteristics, only the development of new metastasis has shown to have a significant impact on OS. Interestingly, patients who did not develop subsequent metastases had almost four times higher chances to die compared to those with at least one new metastasis (hazard ratio (HR) 3.88, 95% confidence interval (CI) 1.54–9.78, p = 0.009) (Fig. 8a). Although counterintuitive, it could be easily assumed that those patients from the start had a more severe form of disease, and in consequence, did not live long enough to develop new metastases.

Kaplan-Meier survival curves showing significant impact of clinicopathological characteristic and gene expression levels on overall survival (OS) for metastatic melanoma patients. (a) OS according appearance of new metastasis. (b) OS according p53α gene expression. (c) OS according Δ133p53β gene expression. Tick marks indicate censored cases.
Furthermore, patients with high (above median value) p53α gene expression tended to live shorter compared to those with low p53α expression (HR 3.16, 95% CI 0.99–10.09, p = 0.037) (Fig. 8b). In addition, patients with high Δ133p53β gene expression had more than four times higher risk to die compared to those with low Δ133p53β expression (HR 4.30, 95% CI 1.16–15.98, p = 0.042) (Fig. 8c). This could be connected with patients who did not develop subsequent metastases which have particularly higher Δ133p53β expression (Fig. 7e), and both have a negative impact on OS of patients with metastatic melanoma (Fig. 8a,c). Recently, it was also shown that the elevated expression of Δ133p53β causes tumour cells to spread to other organs regardless of the TP53 mutation status and increases the risk of cancer recurrence and death in patients with breast cancer59. Therefore, it is worthwhile to further explore the prognostic significance of this isoform as a marker for shorter OS in metastatic melanoma patients.
When we dichotomized expression data based on the receiver operating characteristic (ROC) curve analyses, expression of several additional genes and one protein showed a significant impact on OS. Opposite to p53α, patients with low p53β gene expression had shorter OS (HR 5.36, 95% CI 1.52–18.85, p = 0.009) (Supplementary Fig. S2a). Few studies investigated the prognostic value of p53β and reported the association of p53β expression with longer disease-frees survival in TP53 mutant breast cancer37; in cholangiocarcinoma p53β downregulation combined with Δ133p53 upregulation was associated with shorter OS38. The tumour suppressive role of p53β is supported with findings which show that p53β enhances both p53 and TAp73-mediated apoptosis60,61. Also, p53β was shown to enhance, p53-dependent transcription of p21 and PUMA in melanoma cell line41. Low NME1 expression was also associated with shorter OS in our cohort of metastatic melanoma patients (HR 3.41, 95% CI 1.15–10.11, p = 0.027) (Supplementary Fig. S2b) which is in accordance with its role as a metastasis suppressor. Several groups reported the correlation between low NME1 expression and worse patient prognosis and lower overall survival15,16,45. Further support comes from the study where the lowest NME1 protein expression was found in primary skin melanoma samples which metastasized in lymph nodes62. However, the same study also showed that there was no difference in patient survival between NME1 high- or low-expressing primary melanomas. On the contrary, in an animal model it was shown that tumours expressing low levels of NME1 mRNA had significantly reduced survival rates and times63. We also observed that patients with high GLI3 expression showed shorter OS compared to patients with lower expression, which was observed for both GLI3 gene (HR 2.88, 95% CI 1.03–8.02, p = 0.043) (Supplementary Fig. S2c) and GLI3R protein (HR 3.28, 95% CI 1.07–10.00, p = 0.037) (Supplementary Fig. S2d). This is in contrast with one previous study where it was observed that higher expression of GLI3 mRNA was associated with better survival of metastatic melanoma patients43.
Our study again showed that assessment of the association between continuous variables such as gene expression and survival time markedly depends on the method used for binarization of potential biomarker data64.
Conclusion
In summary, in this study we have examined a large number of isoforms of protein families involved in melanoma development, progression and metastasis. Gene expression has proven to be less reliable in the detection of specific isoforms, so protein levels should be examined when discussing protein activity. It has been demonstrated that many isoforms positively or negatively affect the activities of wild type proteins, so specific isoforms should be further examined to establish their role in tumour progression.
Materials and Methods
Patients
Metastatic melanoma tissues and matched adjacent healthy skin were obtained from 38 patients (Supplementary Table S4). All patients were treated at the Sestre milosrdnice University Hospital Center and clinical data were available. The study complied with the Helsinki Declaration and was approved by the Ethics Review Committee of Sestre milosrdnice UHC and the Bioethical committee of Ruđer Bošković Institute. Informed consent according to the World Medical Association Declaration of Helsinki was obtained from all patients. The tissues were collected during surgery, frozen immediately in dry ice and stored at −80 °C until extraction. Survival time was measured from the date of surgery to the time of death or the last follow-up observation. The median follow-up of patients at the time of analysis was 16 months (range 1–38 months). The information on mortality was obtained from Croatian National Cancer Registry, Croatian Institute of Public Health.
RNA extraction, RT and qPCR analysis
RNA was extracted using TRIzol Reagent and/or PureLink RNA Mini spin columns, and reverse transcribed using High Capacity cDNA Reverse Transcription Kit according to the manufacturers’ instructions. qPCR analysis was performed using TaqMan Gene Expression Master Mix and TaqMan Gene Expression Assays (all Thermo Fisher Scientific, MA, USA). Gene expression analyses were performed according to protocol suggested by the manufacturer on 7300 Real-Time PCR System (Thermo Fisher Scientific). To distinguish the different TP53 isoforms, a nested qPCR approach was used and qPCR was performed on CFX96 Real-Time PCR Detection System (Bio-Rad, CA, USA) using Takyon Low Rox SYBR MasterMix dTTP Blue (Eurogentec, Belgium). All details with primer sequences and positions are provided in Supplementary Materials and Methods, Tables S5 and S6, Fig. S3.
Protein extraction and western blot analysis
Proteins were extracted from frozen tumour tissues and corresponding healthy skin and separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Merck Millipore, USA). The list of antibodies used and all details are available in Supplementary Materials and Methods. Densitometric quantification of protein levels was determined using ImageJ software (https://imagej.net).
Extraction of DNA and BRAF mutation analysis
DNA was extracted from frozen tumour tissues. Exon 15 of BRAF gene was amplified by PCR using F-TTCATGAAGACCTCACAGTAAAAA and R-CCACAAAATGGATCCAGACA primers. PCR products were denatured (HR-1 High-Resolution Melter, Idaho Technology, UT, USA) at 0.2 °C/s ramp rate and melting curves were analysed using HR-1 software. Samples with aberrant melting patterns were sequenced in both directions using Big Dye Terminator 1.1 Cycle Sequencing kit (Thermo Fisher Scientific) on ABI PRISM 310 Genetic Analyzer (Thermo Fisher Scientific).
TP53 mutation analysis
Using the RNA extracted and converted into cDNA as described above, we performed the well-established yeast functional assay (FASAY) to determine the TP53 status in melanoma patients. The TP53 coding sequence was amplified with a two-step nested PCR using Go-Taq G2 Master Mix (Promega). More details are available in Supplementary Materials and Methods.
Cell culture
A panel of melanoma cell lines (A375M, CHL1, LM6 (4405 P), Mel224, Mel501, Mel505, WM793B and WM983B) was kindly provided by Dr. Bergamaschi (Barts and The London School of Medicine and Dentistry, London, UK). The cells were grown in DMEM (Sigma Aldrich, USA) or RPMI 1640 (Lonza, Switzerland) supplemented with 10% FBS, 1 mM sodium pyruvate (both Thermo Fisher Scientific), 1% streptomycin - penicillin, and 2 mM L-glutamine (both Sigma Aldrich) and maintained at 37 °C with 5% CO2. All cell lines were tested to be mycoplasma free.
Statistical analysis
Is provided in Supplementary Materials and Methods.
Data Availability
All patients’ data including clinical characteristics and the results of gene and protein expressions are available as Supplementary Data file.
References
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA. Cancer J. Clin. 63, 11–30 (2013).
Palmieri, G. et al. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 5, 183 (2015).
Box, N. F. & Terzian, T. The role of p53 in pigmentation, tanning and melanoma. Pigment Cell Melanoma Res. 21, 525–533 (2008).
Palmieri, G. et al. Main roads to melanoma. J. Transl. Med. 7, 86 (2009).
Lu, M. et al. Restoring p53 Function in Human Melanoma Cells by Inhibiting MDM2 and Cyclin B1/CDK1-Phosphorylated Nuclear iASPP. Cancer Cell 23, 618–633 (2013).
Moll, U. M. & Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2, 371–386 (2004).
Murray-Zmijewski, F., Lane, D. P. & Bourdon, J.-C. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 13, 962–972 (2006).
Joruiz, S. M. & Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 6, a026039 (2016).
Zaika, A. I. et al. δNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J. Exp. Med. 196, 765–780 (2002).
Slade, N. & Horvat, A. Targeting p73 -a Potential Approach in Cancer Treatment. Curr. Pharm. Des. 17, 591–602 (2011).
Matin, R. N. et al. P63 Is an Alternative P53 Repressor in Melanoma That Confers Chemoresistance and a Poor Prognosis. J. Exp. Med. 210, 581–603 (2013).
Takahashi, R. et al. p53 isoform profiling in glioblastoma and injured brain. Oncogene 32, 3165–3174 (2013).
Steeg, P. S. et al. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 80, 200–204 (1988).
Bosnar, M. H., Bago, R. & Cetković, H. Subcellular localization of Nm23/NDPK A and B isoforms: a reflection of their biological function? Mol. Cell. Biochem. 329, 63–71 (2009).
Caligo, M. A. et al. A low NM23.H1 gene expression identifying high malignancy human melanomas. Melanoma Res. 4, 179–184 (1994).
McDermott, N. C. et al. Immunohistochemical expression of nm23 in primary invasive malignant melanoma is predictive of survival outcome. J. Pathol. 190, 157–162 (2000).
Bodey, B., Bodey, B. J., Groeger, A. M., Siegel, S. E. & Haiser, H. E. Nm23/nucleoside diphosphate (NDP) kinase expression in human malignant melanomas: significance and implications in tumor biology. Anticancer Res. 17, 505–511 (1997).
Baba, H. et al. Two isotypes of murine nm23/nucleoside diphosphate kinase, nm23-M1 and nm23-M2, are involved in metastatic suppression of a murine melanoma line. Cancer Res. 55, 1977–1981 (1977).
Novak, M., Jarrett, S. G., Mccorkle, J. R., Mellon, I. & Kaetzel, D. M. Multiple mechanisms underlie metastasis suppressor function of NM23-H1 in melanoma. Naunyn. Schmiedebergs. Arch. Pharmacol. 384, 433–438 (2011).
Jarrett, S. G. et al. Metastasis suppressor NM23-H1 promotes repair of UV-induced DNA damage and suppresses UV-induced melanomagenesis. Cancer Res. 72, 133–143 (2012).
An, R. et al. Over-expression of nm23-H1 in HeLa cells provides cells with higher resistance to oxidative stress possibly due to raising intracellular p53 and GPX1. Acta Pharmacol. Sin. 29, 1451–1458 (2008).
Jung, H., Seong, H.-A. & Ha, H. NM23-H1 tumor suppressor and its interacting partner STRAP activate p53 function. J. Biol. Chem. 282, 35293–35307 (2007).
Polanski, R. et al. MDM2 interacts with NME2 (non-metastatic cells 2, protein) and suppresses the ability of NME2 to negatively regulate cell motility. Carcinogenesis 32, 1133–1142 (2011).
Levanat, S., Sabol, M., Musani, V., Ozretić, P. & Trnski, D. Hedgehog Signaling Pathway as Genetic and Epigenetic Target in Ovarian Tumors. Curr. Pharm. Des. 23, 73–94 (2017).
Hui, C. & Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 (2011).
Stecca, B. et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 104, 5895–5900 (2007).
Alexaki, V.-I. et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 102, 1148–1159 (2010).
Dennler, S., André, J., Verrecchia, F. & Mauviel, A. Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J. Biol. Chem. 284, 31523–31 (2009).
Gu, D., Xie, J., Gu, D. & Xie, J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers (Basel). 7, 1684–1698 (2015).
Katoh, Y. & Katoh, M. Integrative genomic analyses on GLI2: Mechanism of Hedgehog priming through basal GLI2 expression, and interaction map of stem cell signaling network with P53. Int. J. Oncol. 33, 881–886 (1992).
Abe, Y. et al. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 105, 4838–4843 (2008).
Stecca, B. & Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 28, 663–676 (2009).
Ho, L. et al. Gli2 and p53 cooperate to regulate IGFBP-3- mediated chondrocyte apoptosis in the progression from benign to malignant cartilage tumors. Cancer Cell 16, 126–136 (2009).
Kang, H. N., Oh, S. C., Kim, J. S. & Yoo, Y. A. Abrogation of Gli3 expression suppresses the growth of colon cancer cells via activation of p53. Exp. Cell Res. 318, 539–549 (2012).
Hofstetter, G. et al. Alternative splicing of p53 and p73: the novel p53 splice variant p53δ is an independent prognostic marker in ovarian cancer. Oncogene 29, 1997–2004 (2010).
Bourdon, J.-C. et al. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 13, R7 (2011).
Avery-Kiejda, K. A., Morten, B., Wong-Brown, M. W., Mathe, A. & Scott, R. J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 35, 586–596 (2014).
Nutthasirikul, N., Limpaiboon, T., Leelayuwat, C., Patrakitkomjorn, S. & Jearanaikoon, P. Ratio disruption of the Δ133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 42, 1181–1188 (2013).
Anensen, N. et al. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. 12, 3985–3992 (2006).
Florijan, M. K. et al. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. Urologic Oncology: Seminars and Original Investigations 37(9), 578.e1–578.e10 (2019).
Avery-Kiejda, K. A. et al. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. 14, 1659–1668 (2008).
Tuve, S., Wagner, S. N., Schittek, B. & Pützer, B. M. Alterations of DeltaTA-p 73 splice transcripts during melanoma development and progression. Int. J. Cancer 108, 162–166 (2004).
O’Reilly, K. E. et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel). 6, 1429–1450 (2013).
Easty, D. J. et al. Expression of NM23 in human melanoma progression and metastasis. Br. J. Cancer 74, 109–114 (1996).
Flørenes, V. A. et al. Levels of nm23 messenger RNA in metastatic malignant melanomas: inverse correlation to disease progression. Cancer Res. 52, 6088–91 (1992).
Fiore, L. S. et al. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene 33, 4508–4520 (2014).
Daniotti, M. et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene 23, 5968–5977 (2004).
Houben, R. et al. High-level expression of wild-type p53 in melanoma cells is frequently associated with inactivity in p53 reporter gene assays. PLoS One 6, e22096 (2011).
Gwosdz, C. et al. Comprehensive analysis of the p53 status in mucosal and cutaneous melanomas. Int. J. Cancer 118, 577–582 (2006).
Ganzetti, G. et al. IL-17, IL-23, and p73 expression in cutaneous melanoma: A pilot study. Melanoma Res. 25, 232–238 (2014).
Steder, M. et al. DNp73 Exerts Function in Metastasis Initiation by Disconnecting the Inhibitory Role of EPLIN on IGF1R-AKT/STAT3 Signaling. Cancer Cell 24, 512–527 (2013).
Martinez, J. A. et al. Overexpression of nm23-H1 and nm23-H2 genes in colorectal carcinomas and loss of nm23-H1 expression in advanced tumour stages. Gut 37, 712–720 (1995).
Sabol, M., Trnski, D., Musani, V., Ozretić, P. & Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 19, 2562 (2018).
Roessler, E. et al. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 14, 2181–2188 (2005).
Pantazi, E. et al. GLI2 induces genomic instability in human keratinocytes by inhibiting apoptosis. Cell Death Dis. 5, e1028 (2014).
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012).
Ekedahl, H. et al. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br. J. Dermatol. 169, 1049–1055 (2013).
Jalili, A. et al. NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo. PLoS One 8, e69064 (2013).
Gadea, G. et al. TP53 drives invasion through expression of its Δ133p53β variant. eLife 5, e14734d (2016).
Zorić, A., Horvat, A. & Slade, N. Differential Effects of Diverse p53 Isoforms on TAp73 Transcriptional Activity and Apoptosis. Carcinogenesis 34, 522–529 (2012).
Bourdon, J.-C. et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 (2005).
Döme, B., Somlai, B. & Tímár, J. The loss of NM23 protein in malignant melanoma predicts lymphatic spread without affecting survival. Anticancer Res. 20, 3971–3974 (2000).
Ma, D., Luyten, G. P., Luider, T. M., Jager, M. J. & Niederkorn, J. Y. Association between NM23-H1 gene expression and metastasis of human uveal melanoma in an animal model. Invest. Ophthalmol. Vis. Sci. 37, 2293–2301 (1996).
Clark, T. G., Bradburn, M. J., Love, S. B. & Altman, D. G. Survival Analysis Part IV: Further concepts and methods in survival analysis. Br. J. Cancer 89, 781–786 (2003).
Acknowledgements
This work was supported by Croatian Science Foundation grant IP-11-2013-1615 to NS. The authors thank to Mrs. Martina Pehar for technical assistance.
Author information
Authors and Affiliations
Contributions
N.S., M.H.B. and S.L. designed the study and contributed unpublished essential data or reagents, I.M. and Z.P. enrolled the participants and collected the samples, P.O., N.H., B.P., M.S., D.T., M.R., V.M. and Y.C. collected the data, P.O., N.H. and M.S. analysed the data, P.O., N.H., M.S., M.H.B., S.L. and N.S. drafted the main manuscript text and all authors contributed to and approved the final version of manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ozretić, P., Hanžić, N., Proust, B. et al. Expression profiles of p53/p73, NME and GLI families in metastatic melanoma tissue and cell lines. Sci Rep 9, 12470 (2019). https://doi.org/10.1038/s41598-019-48882-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48882-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
